Beneficial Pharmacokinetic Drug Interactions: A Tool to Improve the Bioavailability of Poorly Permeable Drugs



Abstract

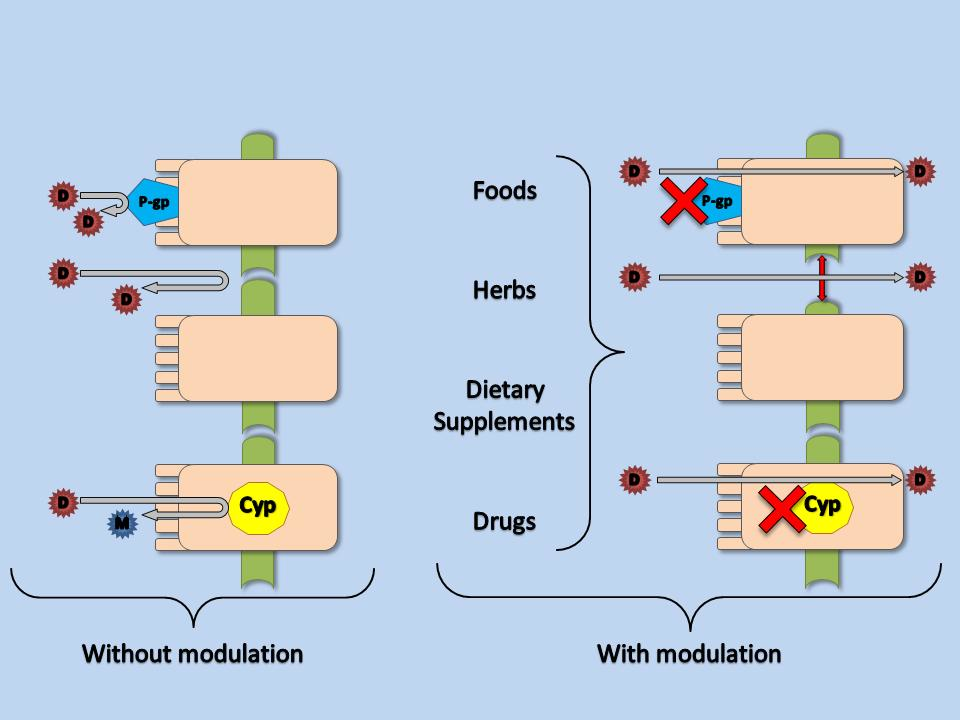
1. Introduction
2. Mechanisms of Pharmacokinetic Drug Interactions
2.1. Carrier-Mediated Transporters
2.2. Metabolism
3. Potential Beneficial Pharmacokinetic Interactions between Drugs and Other Substances
3.1. Food-Drug Interactions
3.1.1. Black Pepper
3.1.2. Orange Juice
3.1.3. Resveratrol
3.2. Herb-Drug Interactions
3.2.1. Aloe Leaf Materials
3.2.2. Salvia Miltiorrhiza
3.2.3. Andrographis Paniculata
3.2.4. Emodin
3.3. Dietary Supplement-Drug Interactions
3.3.1. Licorice (Glycyrrhiza Species)
3.3.2. Carotenoids
3.3.3. Green Coffee Beans
3.4. Drug-Drug Interactions
3.4.1. Ivermectin
3.4.2. Chlorambucil
3.4.3. Telaprevir
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Edwards, D.J. Beneficial pharmacokinetic drug interactions. Adv. Pharmacoepidemiol. Drug Saf. 2012, 1, 2. [Google Scholar] [CrossRef]
- Li, X.; Hu, J.; Wang, B.; Sheng, L.; Liu, Z.; Yang, S.; Li, Y. Inhibitory effects of herbal constituents on P-glycoprotein in vitro and in vivo: Herb-drug interactions mediated via P-gp. Toxicol. Appl. Pharmacol. 2014, 275, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Che, C.; Wang, Z.J.; Chow, M.S.S.; Lam, C.W.K. Herb-herb combination for therapeutic enhancement and advancement: Theory, practice and future perspectives. Molecules 2013, 18, 5125–5141. [Google Scholar] [CrossRef] [PubMed]
- Mouly, S.; Lloret-Linares, C.; Sellier, P.; Sene, D.; Bergmann, J. Is the clinical relevance of drug-food and drug-herb interactions limited to grapefruit juice and Saint-John’s Wort? Pharmacol. Res. 2017, 118, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Liu, Z.; Wang, C.; Meng, Q.; Huo, X.; Liu, Q.; Sun, H.; Sun, P.; Yang, X.; Ma, X.; et al. P-gp, MRP2 and OAT1/OAT3 mediate the drug-drug interaction between resveratrol and methotrexate. Toxicol. Appl. Pharmacol. 2016, 306, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, A.H.S.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Vasiliou, K.; Nebert, D.W. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genom. 2009, 3, 195–206. [Google Scholar] [CrossRef]
- Hediger, M.A.; Romero, M.F.; Peng, J.B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins. Eur. J. Physiol. 2004, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Ballent, M.; Maté, L.; Virkel, G.; Sallovitz, J.; Viviani, P.; Lanusse, C.; Lifschitz, A. Intestinal drug transport: Ex vivo evaluation of the interactions between ABC transporters and anthelmintic molecules. J. Vet. Pharmacol. Ther. 2014, 37, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Peng, Y.; Li, X.; Liu, L.; Liu, F.; He, L. A novel delocalized lipophilic cation-chlorambucil conjugate inhibits P-glycoprotein in HepG2/ADM cells. Bioorg. Med. Chem. 2017, 25, 5461–5467. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Uehera, S.; Tanaka, K.; Tadokoro, S.; Kusamori, K.; Katsumi, H.; Sakane, T.; Yamamoto, A. Effects of polyoxyethylene alkyl ethers on the intestinal transport and absorption of rhodamine 123: A P-glycoprotein substrate by in vitro and in vivo studies. J. Pharm. Sci. 2016, 105, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Huwyler, J.; Camenisch, G.; Gutmann, H. Interaction of the antiviral drug telaprevir with renal and hepatic drug transporters. Biochem. Pharmacol. 2012, 84, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y.; Ozeki, Y.; Isaka, T.; Honjo, H.; Iwamoto, K. Inhibitory effect of caffeic acid in human organic anion transporters hOAT1 and hOAT3: A novel candidate for food-drug interaction. Drug Metab. Pharmacokinet. 2011, 26, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sweet, D.H. Interaction of natural dietary and herbal anionic compounds and flavonoids with human organic anion transporters 1 (SLC22A6), 3 (SLC22A8), and 4 (SLC22A11). Evid. Based Complement. Alternat. Med. 2013, 2013, 612527. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wu, J.; Ning, J.; Hou, J.; Xin, H.; He, Y.; Ge, G.; Xu, X. Inhibition of human cytochrome P450 enzymes by licochalcone A, a naturally occurring constituent of licorice. Toxicol. In Vitro 2015, 29, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Coughtrie, M.; Tukey, R.H. Drug Metabolism. In Goodman & Gillman’s The Pharmacological Bases of Therapeutics, 12th ed.; Brunton, L.L., Chabner, B.A., Knollman, B.C., Eds.; McGraw Hill: New York, NY, USA, 2011; pp. 124–143. ISBN 978–0071624428. [Google Scholar]
- Li, G.; Simmler, C.; Chen, L.; Nikolic, D.; Chen, S.; Pauli, G.F.; van Breemen, R.B. Cytochrome P450 inhibition by three licorice species and fourteen licorice constituents. Eur. J. Pharm. Sci. 2017, 109, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Husni, Z.; Ismail, S.; Zulkiffli, M.H.; Afandi, A.; Haron, M. In vitro inhibitory effects of Andrographis paniculata, Gynura procumbens, Ficus deltoidea, and Curcuma xanthorrhiza extracts and constituents on human liver glucuronidation activity. Pharmacogn. Mag. 2017, 13, 236–243. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Min, J.S.; Kim, D.; Park, J.B.; Choi, S.; Lee, E.S.; Na, K.; Bae, S.K. In vitro inhibition of human UDP-glucuronosyl-transferase (UGT) isoforms by astaxanthin, β-cryptoxanthin, canthaxanthin, lutein and zeaxanthin: Prediction of in vivo dietary supplement-drug interactions. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. Roles of human UDP-glucuronosyltransferases in clearance and homeostasis of endogenous substrates, and functional implications. Biochem. Pharmacol. 2015, 96, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Coughtrie, M.W.H. Revisiting the latency of uridine diphosphate-glucuronosyltransferases (UGTs)—How does the endoplastic reticulum membrane influence their function? Pharmaceutics 2017, 9, 32. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Frye, R.F. Inhibitory effects of commonly used herbal extracts on UDP-Glucuronosyltransferase 1A4, 1A6, and 1A9 enzyme activities. Drug Metab. Dispos. 2011, 39, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Fugh-Berman, A. Herb-drug interactions. Lancet 2000, 355, 134–138. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, X.; Ho, P.C.L.; Chan, S.Y.; Heng, P.W.S.; Chan, E.; Duan, W.; Koh, H.L.; Zhou, S. Herb-drug interactions a literature review. Drugs 2005, 65, 1239–1282. [Google Scholar] [CrossRef] [PubMed]
- Merriam-Webster’s Medical Dictionary. Available online: https://www.merriam-webster.com/dictionary/food (accessed on 23 August 2017).
- Baily, D.G.; Spence, J.D.; Munoz, C.; Arnold, M.O. Interaction of citrus juices with felodipine and nifedipine. Lancet 1991, 337, 268–269. [Google Scholar] [CrossRef]
- Bhardwaj, K.R.; Glaeser, H.; Becquemont, L.; Klotz, U.; Gupta, S.K.; Fromm, M.F. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J. Pharmacol. Exp. Ther. 2002, 302, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Wang, X.; Di, X.; Liu, Y. Effect of piperine on the bioavailability and pharmacokinetics of emodin in rats. J. Pharm. Biomed. Anal. 2015, 115, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Q.; Ren, T.; Zhang, Y.; Lam, C.W.K.; Chow, M.S.S.; Zuo, Z. Non-linear pharmacokinetics of piperine and its herb-drug interactions with docetaxel in Sprague-Dawley rats. J. Pharm. Biomed. Anal. 2016, 128, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Cui, C.; Ji, H.; Tang, J.; Wang, Z.; Liu, H.; Qin, M.; Li, X.; Wu, L. Enhanced oral bioavailability of piperine by self-emulsifying drug delivery systems: In vitro, in vivo and in situ intestinal permeability studies. Drug Deliv. 2015, 22, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, B.; Unum, J.; Shao, J.; An, G. Ingredients in fruit juices interact with Dasatinib through inhibition of BCRP: A new mechanism of beverage-drug interaction. J. Pharm. Sci. 2014, 104, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yuan, Z.; Yao, X.; Ma, W.; Liu, L.; Liu, Z.; Xie, Y. Tangeretin, a citrus pantamethoxyflavone, antagonizes ABCB1-mediated multidrug resistance by inhibiting its transport function. Pharmacol. Res. 2016, 110, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bedada, S.K.; Neerati, P. Resveratrol pretreatment affects CYP2E1 activity of chlorzoxazone in healthy human volunteers. Phytother. Res. 2016, 30, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bedada, S.K.; Yellu, N.R.; Neerati, P. Effect of resveratrol treatment on the pharmacokinetics of diclofenac in healthy human volunteers. Phytother. Res. 2016, 30, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Merriam-Webster’s Medical Dictionary. Available online: https://www.merriam-webster.com/dictionary/herb (accessed on 23 August 2017).
- Beneke, C.; Viljoen, A.; Hamman, J. Modulation of drug efflux by aloe materials: An in vitro investigation across rat intestinal tissue. Pharmacogn. Mag. 2013, 9, S44–S48. [Google Scholar] [CrossRef]
- Wallis, L.; Malan, M.; Gouws, C.; Steyn, D.; Ellis, S.; Abay, E.; Wiesner, L.; Otto, D.P.; Hamman, J. Evaluation of isolated fractions of Aloe vera gel materials on indinavir pharmacokinetics: In vitro and in vivo studies. Curr. Drug Deliv. 2016, 13, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Akaberi, M.; Sobhani, Z.; Javadi, B.; Sahebkar, A.; Emami, S.A. Therapeutic effects of Aloe spp. in traditional and modern medicine: A review. Biomed. Pharmacother. 2016, 84, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Machado, D.I.; López-Cervantes, J.; Sendón, R.; Sanches-Silva, A. Aloe vera: Ancient knowledge with new frontiers. Trends Food Sci. Technol. 2017, 61, 94–102. [Google Scholar] [CrossRef]
- Chen, W.; Lu, Z.; Viljoen, A.; Hamman, J. Intestinal drug transport enhancement by Aloe vera. Planta Med. 2009, 75, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Beneke, C.; Viljoen, A.; Hamman, J. In vitro drug absorption enhancement effects of Aloe vera and Aloe ferox. Sci. Pharm. 2012, 80, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Lebitsa, T.; Viljoen, A.; Lu, Z.; Hamman, J. In vitro drug permeation enhancement potential of aloe gel materials. Curr. Drug Deliv. 2012, 9, 297–304. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, S.; Willers, C.; Steyn, D.; Steenekamp, J.; Hamman, S. Development and evaluation of a double-phase multiple-unit dosage form for enhanced insulin intestinal delivery. Drug Deliv. Lett. 2018, 8, 52–60. [Google Scholar] [CrossRef]
- Vinson, J.A.; Al Kharrat, H.; Andreoli, L. Effect of Aloe vera preparations on the human bioavailability of vitamins C and E. Phytomedicine 2005, 12, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Ojewole, E.; Mackraj, I.; Akhundov, K.; Hamman, J.; Viljoen, A.; Olivier, E.; Wesley-Smith, J.; Govender, T. Investigating the effect of Aloe vera gel on the buccal permeability of didanosine. Planta Med. 2012, 78, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.T.; Gerber, M.; du Preez, J.L.; du Plessis, J.; Hamman, J.H. Skin permeation enhancement effects of the gel and whole-leaf materials of Aloe vera, Aloe marlothii and Aloe ferox. J. Pharm. Pharmacol. 2014, 67, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Cao, H.; Zhu, S.; Lu, Y.; Shang, Y.; Wang, M.; Tang, Y.; Zhu, L. Investigation of the binding of Salvianolic acid B to human serum albumin and the effect of metal ions on the binding. Spectrochim. Acta Mol. Biomol. Spectrosc. 2011, 81, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Qi, W.; Huang, R.; Su, R.; He, Z. Elucidating the influence of gold nanoparticles on the binding of Salvianolic acid B and rosmarinic acid to bovine serum albumin. PLoS ONE 2015, 10, e0118274. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Ai, N.; Xu, D.; Fan, X. Exploring the interaction between Salvia miltiorrhiza and human serum albumin: Insights from herb-drug interaction reports, computional analysis and experimental studies. Spectrochim. Acta Mol. Biomol. Spectrosc. 2016, 161, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, U.R.; Ray, S.; Micard, V.; Ghosh, D.; Ghosh, K.; Bandyopadhyay, S.S.; Ray, B. Interaction with bovine serum albumin of anti-oxidative pectic arabinogalactan from Andrographis paniculata. Carbohydr. Polym. 2014, 101, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Merriam-Webster’s Medical Dictionary. Available online: https://www.merriam-webster.com/dictionary/dietary%20supplement (accessed on 24 August 2017).
- Teng, Y.; Sheu, M.; Hsieh, Y.; Wang, R.; Chiang, Y.; Hung, C. β-carotene reverses multidrug resistant cancer cells by selectively modulating human P-glycoprotein function. Phytomedicine 2016, 23, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Caterina, P.; Antonello, D.P.; Chiara, G.; Chiara, C.; Giacomo, L.; Antonio, S.; Giovambiattista, D.S.; Luca, G. Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 2013, 18, 600–609. [Google Scholar]
- Dalzell, A.M.; Mistry, P.; Wright, J.; Williams, F.M.; Brown, C.D.A. Characterization of multidrug transporter-mediated efflux of avermectins in human and mouse neuroblastoma cell lines. Toxicol. Lett. 2015, 235, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kigen, G.; Edwards, G. Drug-transporter mediated interactions between anthelminthic and antiretroviral drugs across the Caco-2 cell monolayers. BMC Pharmacol. Toxicol. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Marada, V.V.V.R.; Flörl, S.; Kühne, A.; Burckhardt, G.; Hagos, Y. Interaction of human organic anion transporter polypeptides 1B1 and 1B3 with antineoplastic compounds. Eur. J. Med. Chem. 2015, 92, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Valdez, B.C.; Hassan, M.; Andersson, B.S. Development of an assay for cellular efflux of pharmaceutically active agents and its relevance to understanding drug interactions. Exp. Hematol. 2017, 52, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Noguchi, K.; Suzuki, T.; Katayama, K.; Sugimoto, Y. Biochemical interaction of anti-HCV telaprevir with the ABC transporters P-glycoprotein and breast cancer resistance protein. BMC Res. Notes 2013, 6, 445. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Becker, J.P.; Haefeli, W.E. Telaprevir is a substrate and moderate inhibitor of P-glycoprotein, a strong inductor of ABCG2, but not an activator of PXR in vitro. Int. J. Antimicrob. Agents 2014, 43, 184–188. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Extract | Cytochrome P450 iso-enzymes 1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | |
| 18β-Glycyrrhetinic acid | + | + | + | ++ | + | + | + | + | ++ |
| Glabridin | + | + | + | ++ | + | + | + | + | ++ |
| Glycycoumarin | ++ | + | ++ | +++ | +++ | +++ | ++ | + | + |
| Isoliquiritigen | + | + | + | +++ | +++ | + | + | ++ | ++ |
| Licochalcone A | ++ | + | + | +++ | +++ | +++ | ++ | ++ | ++ |
| Licoricidin | + | + | +++ | +++ | +++ | +++ | ++ | ++ | ++ |
| Type of Study | Additive Agent | Drug/Marker | Mechanism | Effect | Ref. |
|---|---|---|---|---|---|
| Ex vivo | Ivermectin | R123 | P-gp | Inhibited | [10] |
| Danofloxacin | BCRP | ||||
| Selected aloe components | Cimetidine | P-gp | Inhibited | [37] | |
| Insulin | Tight junctions | Opened | [44] | ||
| Didanosine | Buccal Absorption | Increased | [46] | ||
| Ketoprofen | Transdermal delivery | Increased | [47] | ||
| In vitro | DAG | Digoxin | P-gp | Inhibited | [2] |
| Emodin | |||||
| Resveratrol | Methotrexate | P-gp, MRP2, OAT1 and OAT3 | Inhibited | [5] | |
| Chlorambucil | Adriamycin | P-gp | Inhibited | [11] | |
| Telaprevir | MPP+ | OCT2 | Inhibited | [13] | |
| Metformin | MATE1 | ||||
| Caffeic acid | PAH and ES | OAT1 and OAT3 | Inhibited | [14] | |
| Dicaffeoylquinic acid | PAH and ES | OAT1 and OAT3 | Inhibited | [15] | |
| 18β-Glycyrrhetinic acid | PAH | OAT1 | Inhibited | ||
| ES | OAT4 | Induced | |||
| Selected Glycyrrhiza species | Selected isoform-selective markers | CYP450 | Inhibited | [16,18] | |
| Aqueous Andropgraphis paniculata extract | 4-methylumbelliferone | UGTs | Inhibited | [19] | |
| β-Carotene | Selected isoform-selective markers | UGTs | Inhibited | [20] | |
| R123 | P-gp | Inhibited | [53] | ||
| Mitoxantrone | BCRP | ||||
| In vitro | Piperine | Digoxin and Cyclosporin A | P-gp | Inhibited | [28] |
| Verapamil | CYP3A4 | ||||
| Tangeretin | Dasatinib | BCRP and P-gp | Inhibited | [32] | |
| Doxorubicin | P-gp | Inhibited | [33] | ||
| Nobiletin | Dasatinib | BCRP and P-gp | Inhibited | [32] | |
| Selected aloe leaf material | Indinavir | CYP | Inhibited | [38] | |
| Insulin | Tight junctions | Opened | [41] | ||
| FITC-dextran | Tight junctions | Opened | [43] | ||
| Salvianolic acid B | Human serum albumin | Albumin | Competitive binding | [48,50] | |
| Bovine serum albumin | [49] | ||||
| Arabinogalactan | Bovine serum albumin | Albumin | Competitive binding | [51] | |
| Ivermectin | H33342 dye | P-gp and BCRP | Inhibited | [55] | |
| Lopinavir | P-gp posited | Inhibited | [56] | ||
| Chlorambucil | [3H]-Cholecystokinin octapeptide | OAT1B3 | Inhibited | [57] | |
| 5-Carboxyfluorescein | MRP1 | Inhibited | [58] | ||
| Telaprevir | [3H]-Estrone 3-sulfate | P-gp | Inhibited | [59] | |
| Calcein assay | [60] | ||||
| In vivo | DAG | Ketoconazole | CYP3A4/5 | Inhibited | [2] |
| Resveratrol | Methotrexate | P-gp,OAT1 and OAT3 | Inhibited | [5] | |
| Chlorzoxazone | CYP2E1 | Inhibited | [34] | ||
| Diclofenac | CYP2C9 | Inhibited | [35] | ||
| Piperine | Emodin | UGT | Inhibited | [29] | |
| Docetaxel | P-gp and CYP3A4 posited | Inhibited | [30] | ||
| Selected aloe leaf material | Indinavir | CYP | Inhibited | [38] | |
| Vitamins C and E | Intestinal absorption | Increased | [45] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerber, W.; Steyn, J.D.; Kotzé, A.F.; Hamman, J.H. Beneficial Pharmacokinetic Drug Interactions: A Tool to Improve the Bioavailability of Poorly Permeable Drugs. Pharmaceutics 2018, 10, 106. https://doi.org/10.3390/pharmaceutics10030106
Gerber W, Steyn JD, Kotzé AF, Hamman JH. Beneficial Pharmacokinetic Drug Interactions: A Tool to Improve the Bioavailability of Poorly Permeable Drugs. Pharmaceutics. 2018; 10(3):106. https://doi.org/10.3390/pharmaceutics10030106
Chicago/Turabian StyleGerber, Werner, Johan D. Steyn, Awie F. Kotzé, and Josias H. Hamman. 2018. "Beneficial Pharmacokinetic Drug Interactions: A Tool to Improve the Bioavailability of Poorly Permeable Drugs" Pharmaceutics 10, no. 3: 106. https://doi.org/10.3390/pharmaceutics10030106
APA StyleGerber, W., Steyn, J. D., Kotzé, A. F., & Hamman, J. H. (2018). Beneficial Pharmacokinetic Drug Interactions: A Tool to Improve the Bioavailability of Poorly Permeable Drugs. Pharmaceutics, 10(3), 106. https://doi.org/10.3390/pharmaceutics10030106