Polymer–Surfactant System Based Amorphous Solid Dispersion: Precipitation Inhibition and Bioavailability Enhancement of Itraconazole


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equilibrium Solubility
2.3. Preparation of Solid Dispersions
2.4. Supersaturation Dissolution Test with pH-Shift
2.5. Nucleation Induction Time Measurement
2.6. Characterization of Optimized ASD
2.6.1. Scanning Electron Microscopy (SEM)
2.6.2. Differential Scanning Calorimetry (DSC)
2.6.3. Powder X-ray Diffraction (PXRD)
2.6.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.7. In Vivo Bioavailability Study
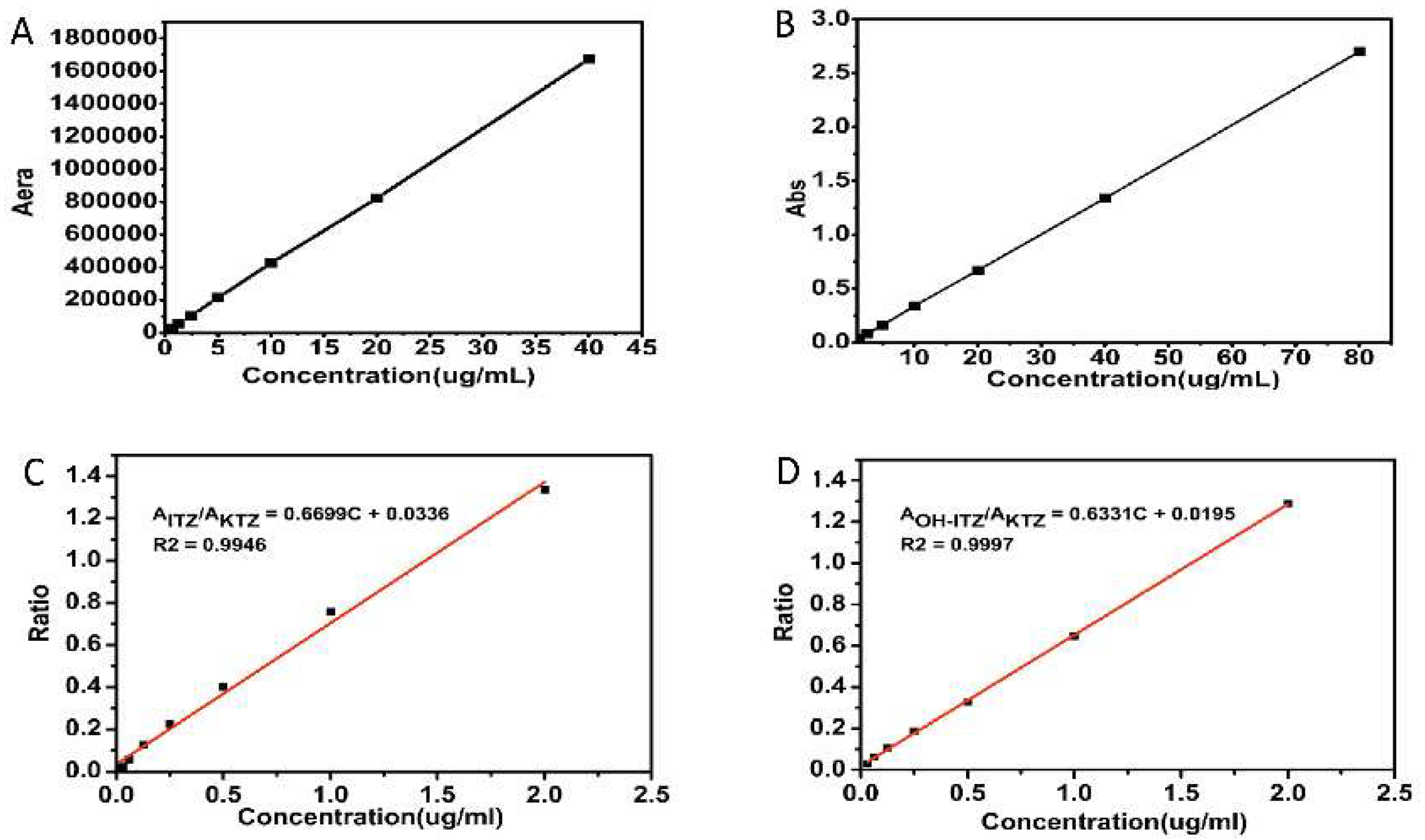
2.7.1. Quantitative Analysis of ITZ and OH-ITZ in Plasma
2.7.2. Pharmacokinetic Data Analysis and Statistical Analysis
3. Results
3.1. Selection of Surfactants
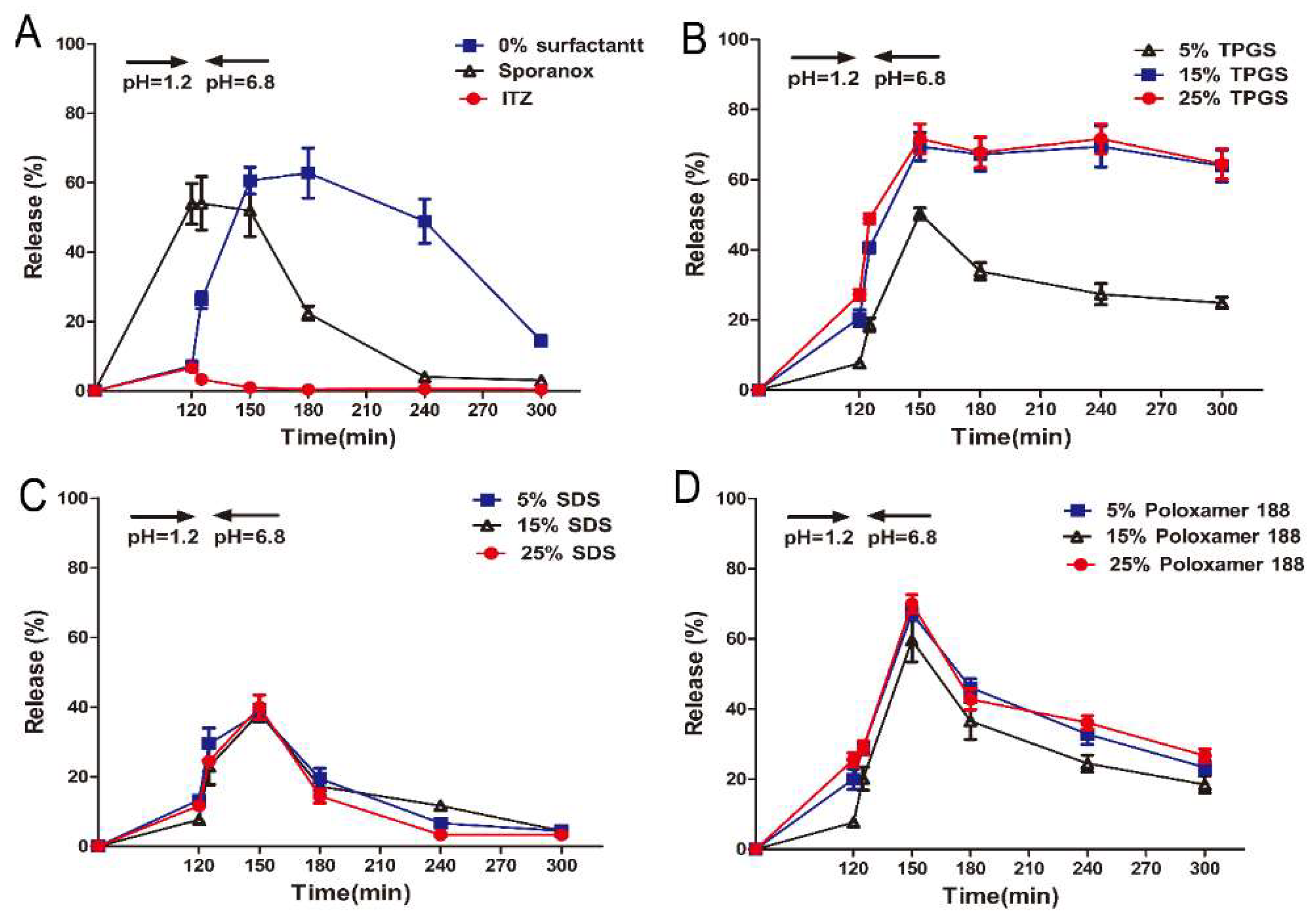
3.2. In Vitro Supersaturation Dissolution with pH-Shift
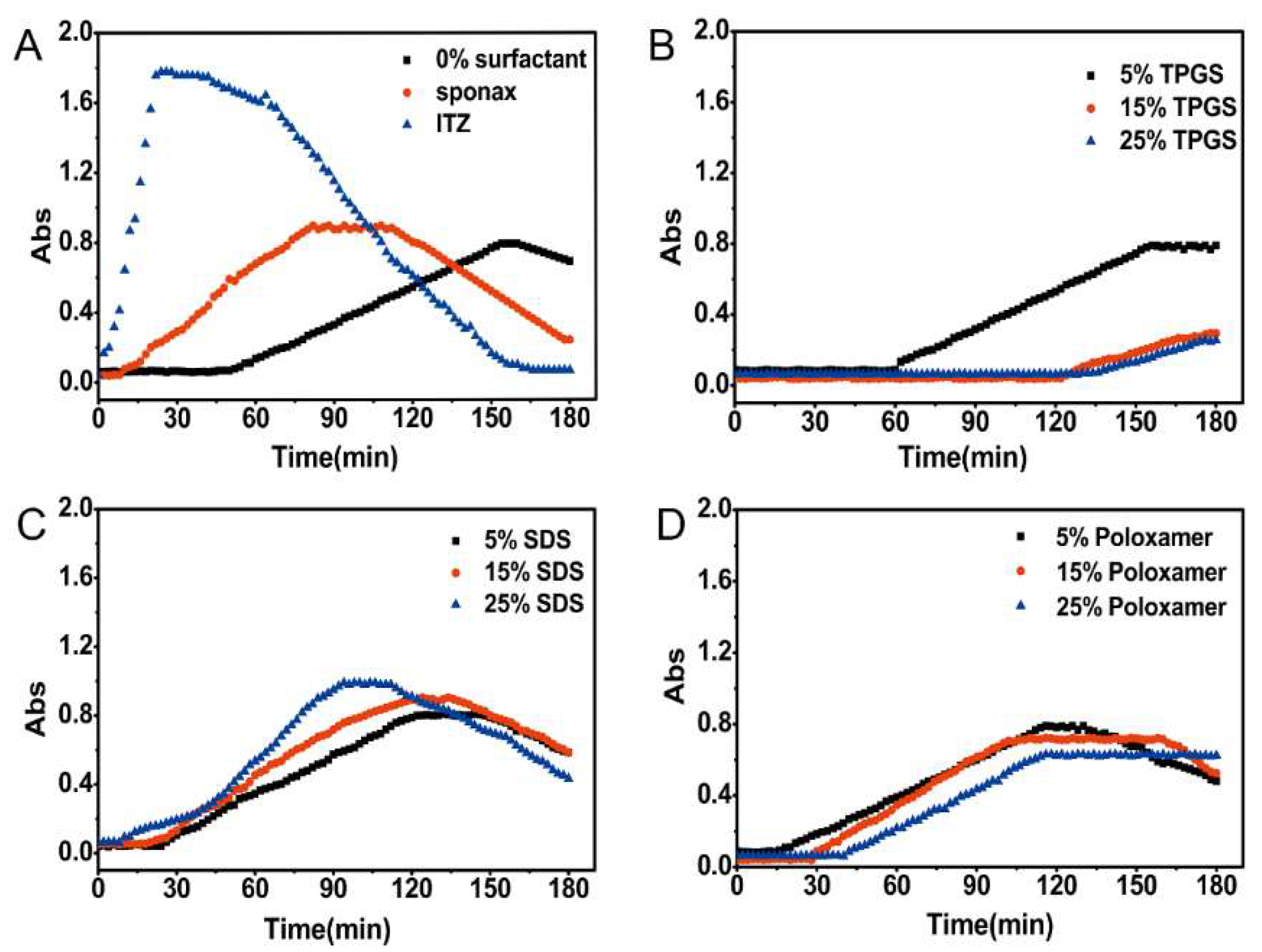
3.3. Nucleation Induction Time Measurement
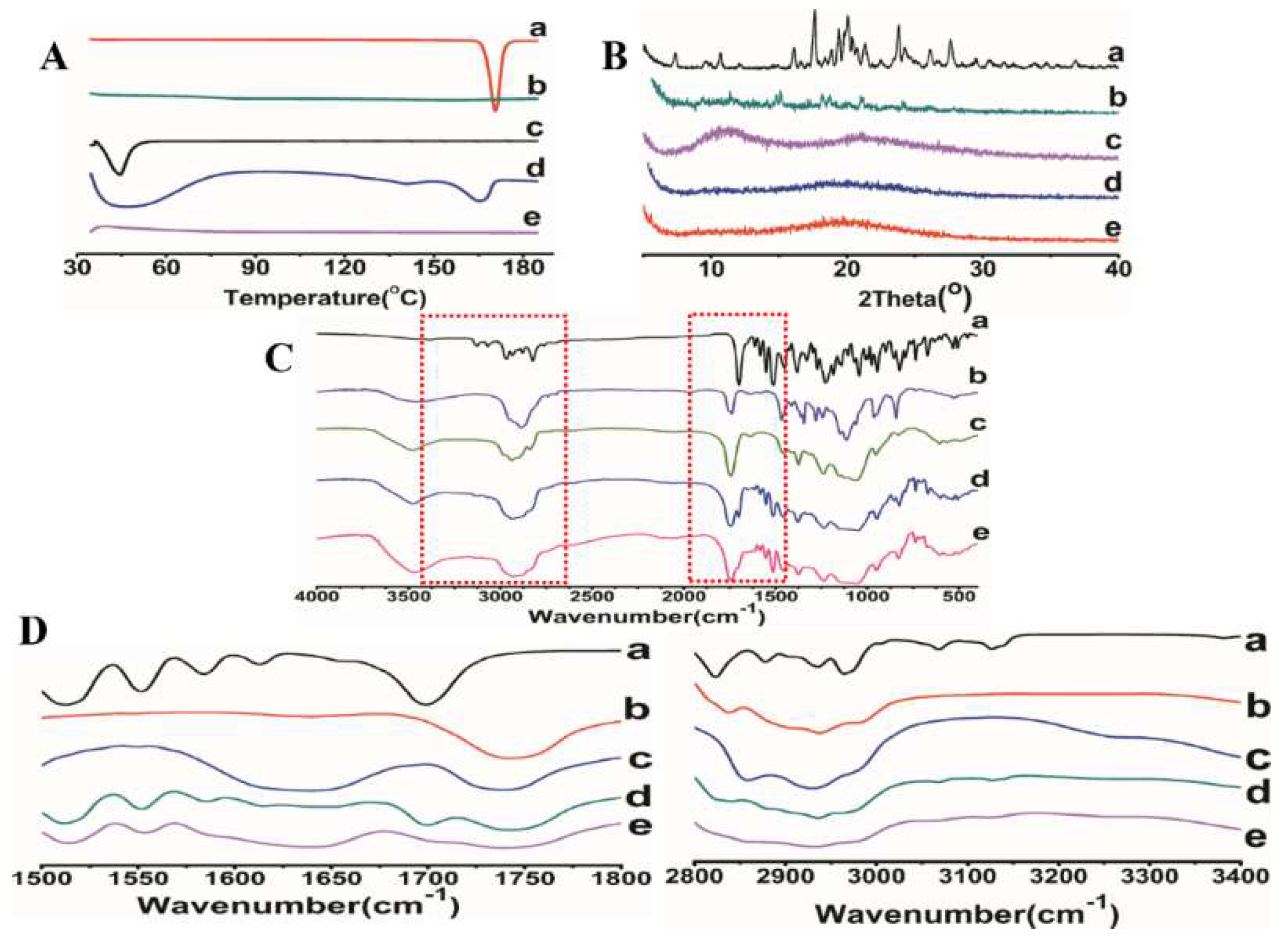
3.4. Characterization of Optimized Formulation
3.4.1. Scanning Electron Microscope Analysis (SEM)
3.4.2. Differential Scanning Calorimeter (DSC)
3.4.3. Powder X-ray Diffraction (XRD)
3.4.4. Fourier Transform Infrared Spectroscopy (FTIR)
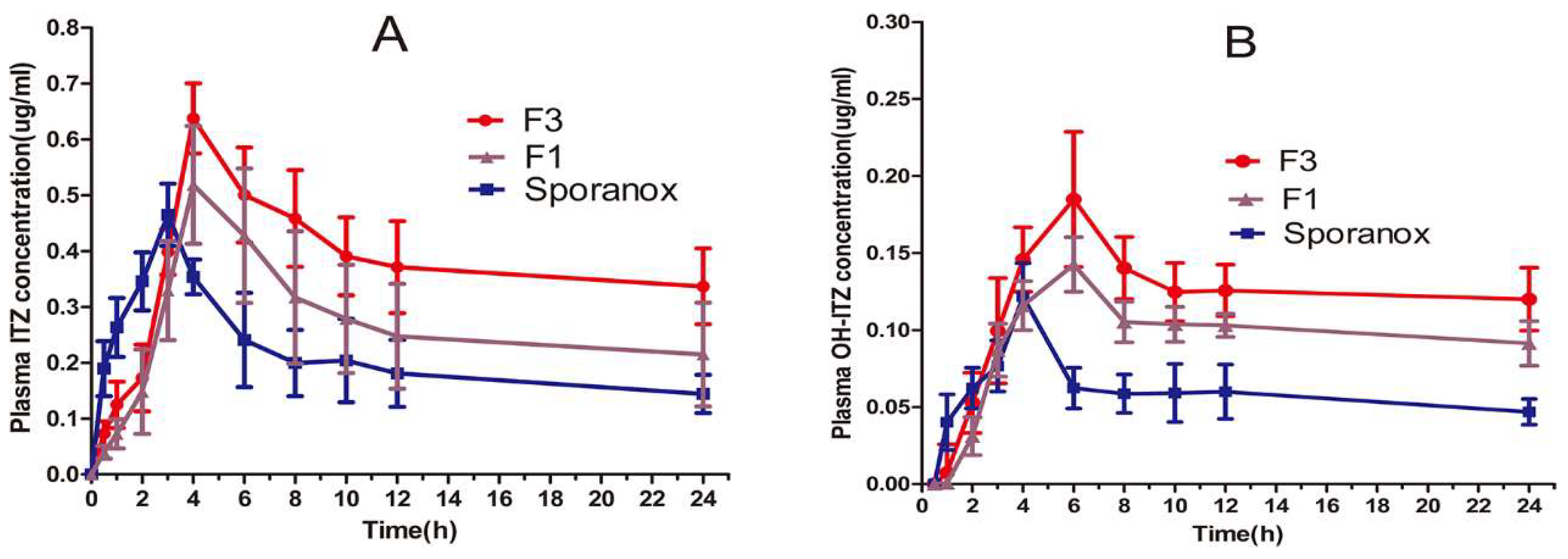
3.5. In Vivo Pharmacokinetic Study in Beagle Dogs
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Han, H.K.; Lee, B.J.; Lee, H.K. Enhanced dissolution and bioavailability of biochanin A via the preparation of solid dispersion: In vitro and in vivo evaluation. Int. J. Pharm. 2011, 415, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Vo, A.; Patil, H.; Tiwari, R.V.; Alshetaili, A.S.; Pimparade, M.B.; Repka, M.A. The effects of polymer carrier, hot melt extrusion process and downstream processing parameters on the moisture sorption properties of amorphous solid dispersions. J. Pharm. Pharmacol. 2016, 68, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Beig, A.; Carr, R.A.; Spence, J.K.; Dahan, A. A win-win solution in oral delivery of lipophilic drugs: Supersaturation via amorphous solid dispersions increases apparent solubility without sacrifice of intestinal membrane permeability. Mol. Pharm. 2012, 9, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Pikal, M.J. Calorimetric investigation of the structural relaxation of amorphous materials: Evaluating validity of the methodologies. J. Pharm. Sci. 2005, 94, 948–965. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Higashi, K.; Suzuki, T.; Tomono, K.; Moribe, K.; Yamamoto, K. Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with EUDRAGIT((R)) EPO. Pharm. Res. 2012, 29, 2777–2791. [Google Scholar] [CrossRef] [PubMed]
- Piao, Z.Z.; Choe, J.S.; Oh, K.T.; Rhee, Y.S.; Lee, B.J. Formulation and in vivo human bioavailability of dissolving tablets containing a self-nanoemulsifying itraconazole solid dispersion without precipitation in simulated gastrointestinal fluid. Eur. J. Pharm. Sci. 2014, 51, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.; Tran, T.T.; Piao, Z.Z.; Vo, T.V.; Park, J.B.; Lim, J.; Oh, K.T.; Rhee, Y.S.; Lee, B.J. Physical properties and in vivo bioavailability in human volunteers of isradipine using controlled release matrix tablet containing self-emulsifying solid dispersion. Int. J. Pharm. 2013, 450, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining Supersaturation in Aqueous Drug Solutions: Impact of Different Polymers on Induction Times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Bevernage, J.; Forier, T.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Excipient-mediated supersaturation stabilization in human intestinal fluids. Mol. Pharm. 2011, 8, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, H.; Hui-Gu, C.; Atef, E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J. Pharm. Sci. 2013, 102, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Dai, W.G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Effect of Binary Additive Combinations on Solution Crystal Growth of the Poorly Water-Soluble Drug, Ritonavir. Cryst. Growth Des. 2012, 12, 6050–6060. [Google Scholar] [CrossRef]
- Zhong, Y.; Jing, G.; Tian, B.; Huang, H.; Zhang, Y.; Gou, J.; Tang, X.; He, H.; Wang, Y. Supersaturation induced by Itraconazole/Soluplus® micelles provided high GI absorption in vivo. Asian J. Pharm. Sci. 2016, 11, 255–264. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Pollock-Dove, C.; Dong, L.C.; Chen, J.; Creasey, A.A.; Dai, W.G. Enhanced bioavailability of a poorly water-soluble weakly basic compound using a combination approach of solubilization agents and precipitation inhibitors: A case study. Mol. Pharm. 2012, 9, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Yeom, D.W.; Song, Y.S.; Cho, H.R.; Choi, Y.S.; Kang, M.J.; Choi, Y.W. Improved oral absorption of dutasteride via Soluplus(R)-based supersaturable self-emulsifying drug delivery system (S-SEDDS). Int. J. Pharm. 2015, 478, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ormes, J.D.; Higgins, J.D.; Taylor, L.S. Impact of surfactants on the crystallization of aqueous suspensions of celecoxib amorphous solid dispersion spray dried particles. Mol. Pharm. 2015, 12, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Ahn, H.I.; Park, J.Y.; Ho, M.J.; Lee, D.R.; Cho, H.R.; Park, J.S.; Choi, Y.S.; Kang, M.J. Improved oral absorption of tacrolimus by a solid dispersion with hypromellose and sodium lauryl sulfate. Int. J. Biol. Macromol. 2016, 83, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Roser, S.; Edler, K.J.; Pigliacelli, C.; Rogerson, M.; Weuts, I.; Van Dycke, F.; Stokbroekx, S. Insights into the role of polymer-surfactant complexes in drug solubilisation/stabilisation during drug release from solid dispersions. Pharm. Res. 2013, 30, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Vandecruys, R.; Peeters, J.; Verreck, G.; Brewster, M.E. Use of a screening method to determine excipients which optimize the extent and stability of supersaturated drug solutions and application of this system to solid formulation design. Int. J. Pharm. 2007, 342, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Gesenberg, C.; Levons, J.; Hubert, M.; Raghavan, K. A high-throughput spectrophotometric approach for evaluation of precipitation resistance. J. Pharm. Biomed. Anal. 2011, 56, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Ghazal, H.S.; Dyas, A.M.; Ford, J.L.; Hutcheon, G.A. In vitro evaluation of the dissolution behaviour of itraconazole in bio-relevant media. Int. J. Pharm. 2009, 366, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.A.; McConville, J.T.; Yang, W.; Williams, R.O., 3rd; McGinity, J.W. Hot-melt extrusion for enhanced delivery of drug particles. J. Pharm. Sci. 2007, 96, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.A.; DiNunzio, J.C.; Yang, W.; McGinity, J.W.; Williams, R.O., 3rd. Enhanced in vivo absorption of itraconazole via stabilization of supersaturation following acidic-to-neutral pH transition. Drug Dev. Ind. Pharm. 2008, 34, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Tajarobi, F.; Larsson, A.; Matic, H.; Abrahmsen-Alami, S. The influence of crystallization inhibition of HPMC and HPMCAS on model substance dissolution and release in swellable matrix tablets. Eur. J. Pharm. Biopharm. 2011, 78, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, W.; Nightingale, J.A.; Herbig, S.M. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm. Res. 2009, 26, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.G.; Dong, L.C.; Li, S.; Deng, Z. Combination of Pluronic/Vitamin E TPGS as a potential inhibitor of drug precipitation. Int. J. Pharm. 2008, 355, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Guzman, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.P.; Almarsson, O.; Remenar, J.F. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef] [PubMed]
- Alexandridis, P.; Holzwarth, J.F.; Hatton, T.A. Micellization of poly (ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) triblock copolymers in aqueous solutions: Thermodynamics of copolymer association. Macromolecules 1994, 27, 2414–2425. [Google Scholar] [CrossRef]
- Dintaman, J.M.; Silverman, J.A. Inhibition of P-glycoprotein by d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm. Res. 1999, 16, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Prasad, Y.R.; Puthli, S.; Eaimtrakarn, S.; Ishida, M.; Yoshikawa, Y.; Shibata, N.; Takada, K. Enhanced intestinal absorption of vancomycin with Labrasol and d-α-tocopheryl PEG 1000 succinate in rats. Int. J. Pharm. 2003, 250, 181–190. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ITZ | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| HPMC AS | 80 | 75 | 65 | 55 | 75 | 65 | 55 | 75 | 65 | 55 |
| TPGS | - | 5 | 15 | 25 | - | - | - | - | - | - |
| SDS | - | - | - | - | 5 | 15 | 25 | - | - | - |
| Poloxamer 188 | - | - | - | - | - | - | - | 5 | 15 | 25 |
| Surfactants | ITZ Concentration in SIF (μg/mL) |
|---|---|
| Blank group | 0.008 ± 0.002 |
| TPGS | 4.83 ± 0.81 |
| SDS | 1.66 ± 0.53 |
| Poloxamer 188 | 0.92 ± 0.27 |
| Soluplus | 0.10 ± 0.06 |
| Sodium cholate | 0.07 ± 0.02 |
| Tween 80 | 0.03 ± 0.01 |
| Pharmacokinetic Parameter | F3 | F1 | Sporanox® |
|---|---|---|---|
| Cmax (μg/mL), ITZ | 0.61 ± 0.08 * | 0.51 ± 0.12 | 0.45 ± 0.06 |
| Cmax (μg/mL), OH-ITZ | 0.18 ± 0.07 | 0.14 ± 0.03 | 0.12 ± 0.04 |
| Tmax (h), ITZ | 3.89 ± 0.41 * | 4.13 ± 0.57 | 2.92 ± 0.37 |
| Tmax (h), OH-ITZ | 5.64 ± 0.97 | 6.40 ± 0.59 * | 4.60 ± 0.63 |
| AUC0–24h (μg h/mL), ITZ | 8.98 ± 1.76 ** | 6.40 ± 1.12 * | 5.06 ± 1.43 |
| AUC0–24h (μg h/mL), OH-ITZ | 2.88 ± 0.76 * | 2.27± 0.94 * | 1.41± 0.37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, D.; Peng, T.; Huang, Z.; Singh, V.; Shi, Y.; Wen, T.; Lu, M.; Quan, G.; Pan, X.; Wu, C. Polymer–Surfactant System Based Amorphous Solid Dispersion: Precipitation Inhibition and Bioavailability Enhancement of Itraconazole. Pharmaceutics 2018, 10, 53. https://doi.org/10.3390/pharmaceutics10020053
Feng D, Peng T, Huang Z, Singh V, Shi Y, Wen T, Lu M, Quan G, Pan X, Wu C. Polymer–Surfactant System Based Amorphous Solid Dispersion: Precipitation Inhibition and Bioavailability Enhancement of Itraconazole. Pharmaceutics. 2018; 10(2):53. https://doi.org/10.3390/pharmaceutics10020053
Chicago/Turabian StyleFeng, Disang, Tingting Peng, Zhengwei Huang, Vikramjeet Singh, Yin Shi, Ting Wen, Ming Lu, Guilan Quan, Xin Pan, and Chuanbin Wu. 2018. "Polymer–Surfactant System Based Amorphous Solid Dispersion: Precipitation Inhibition and Bioavailability Enhancement of Itraconazole" Pharmaceutics 10, no. 2: 53. https://doi.org/10.3390/pharmaceutics10020053
APA StyleFeng, D., Peng, T., Huang, Z., Singh, V., Shi, Y., Wen, T., Lu, M., Quan, G., Pan, X., & Wu, C. (2018). Polymer–Surfactant System Based Amorphous Solid Dispersion: Precipitation Inhibition and Bioavailability Enhancement of Itraconazole. Pharmaceutics, 10(2), 53. https://doi.org/10.3390/pharmaceutics10020053