Orally Disintegrating Tablets Containing Melt Extruded Amorphous Solid Dispersion of Tacrolimus for Dissolution Enhancement

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
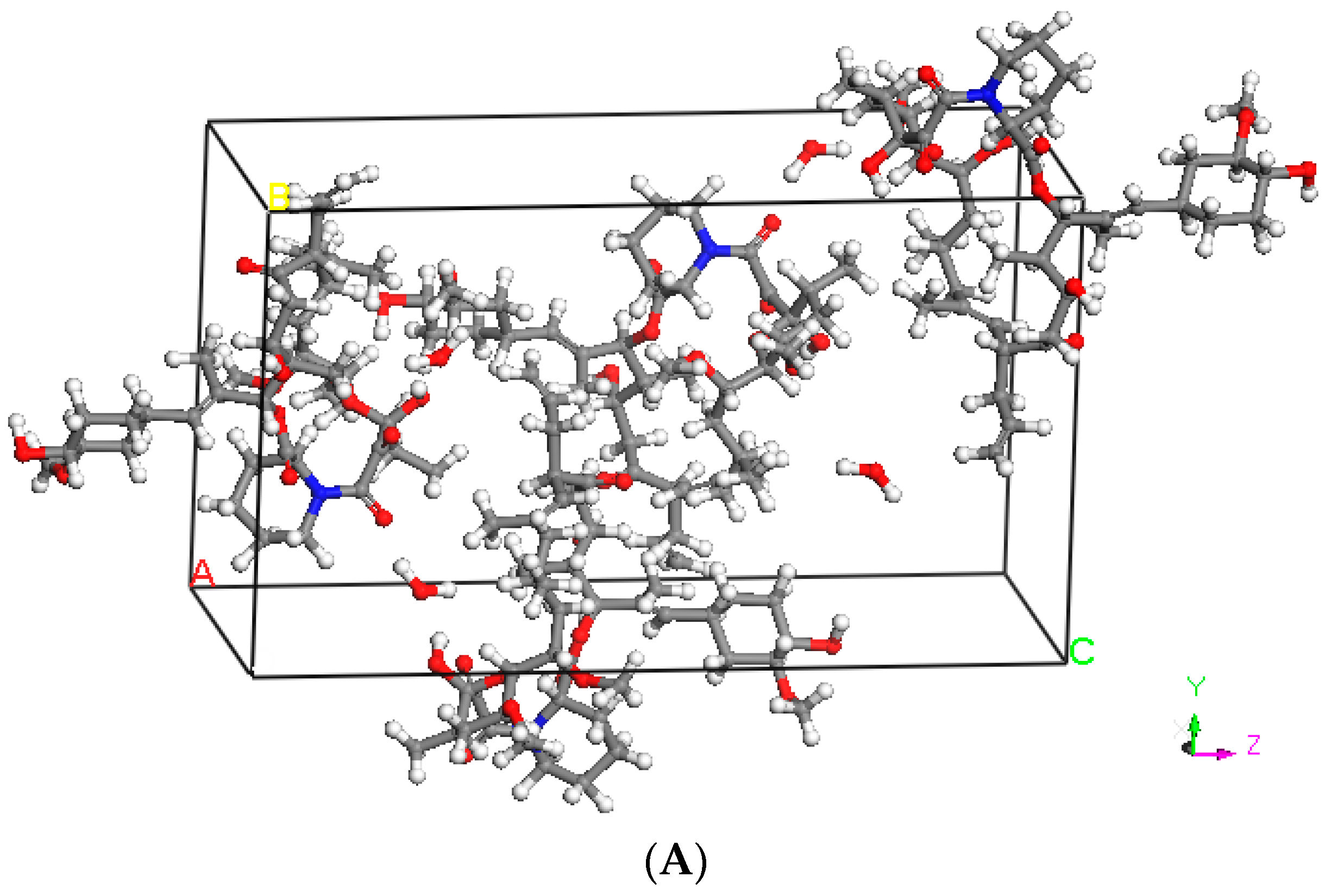
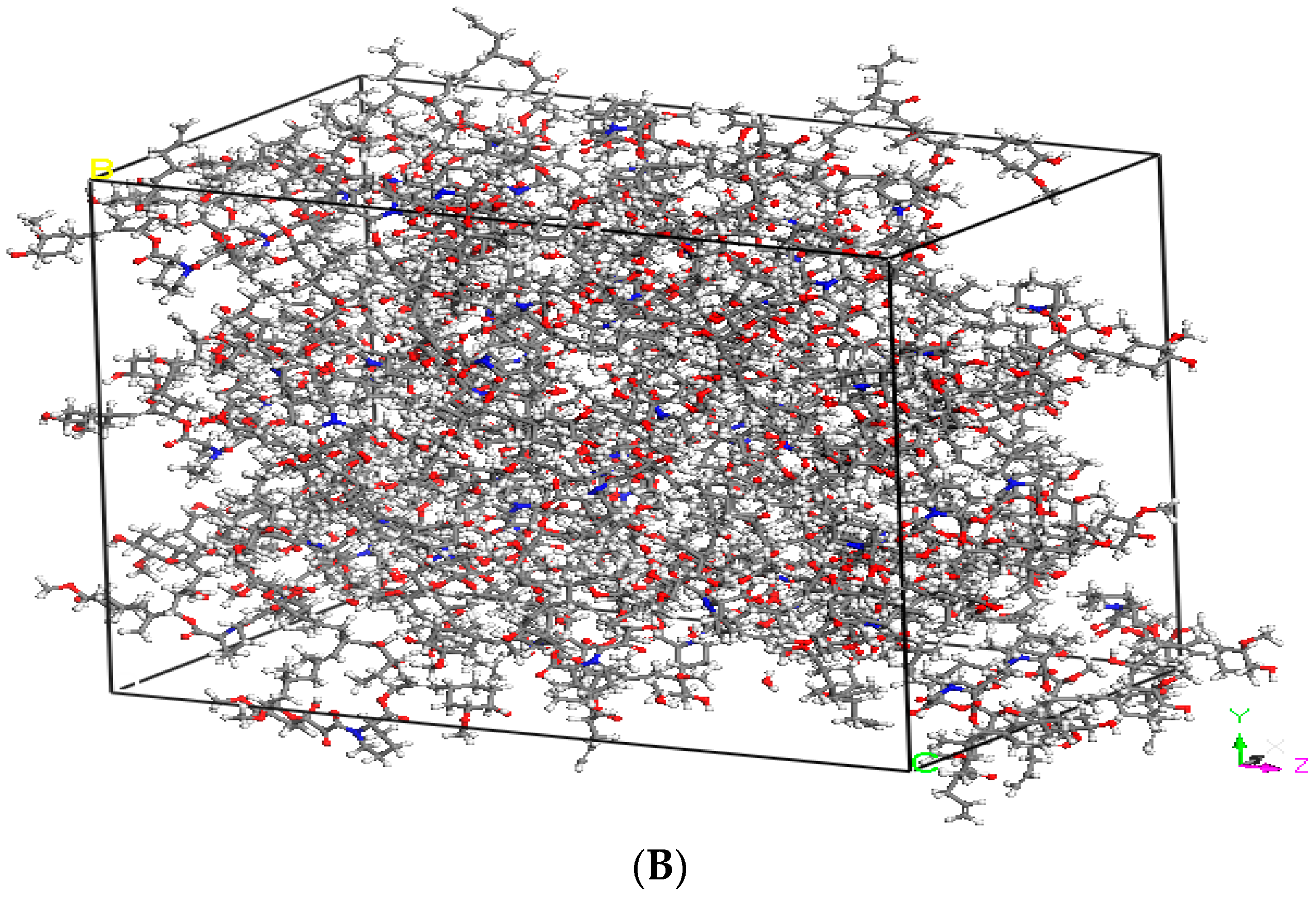
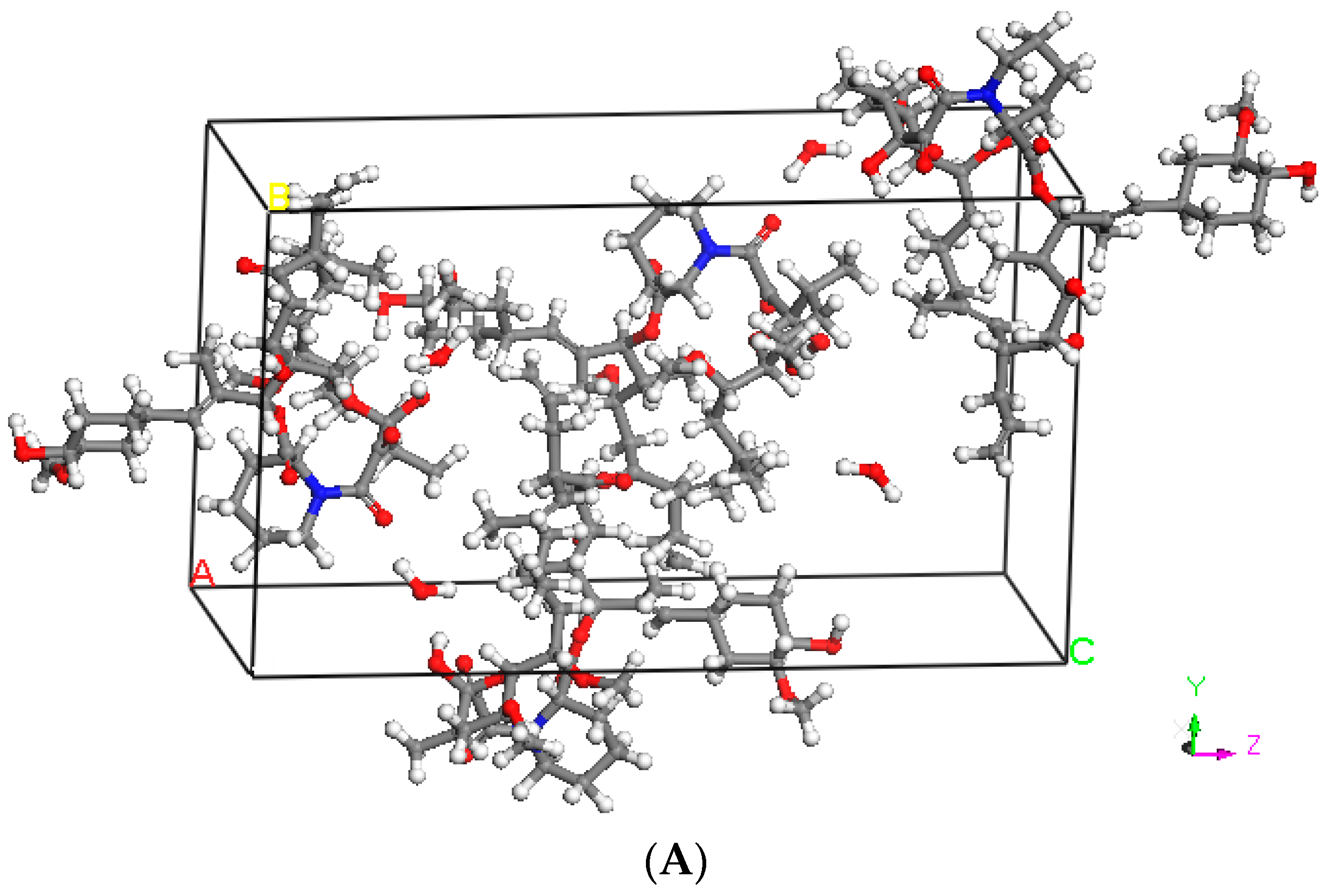
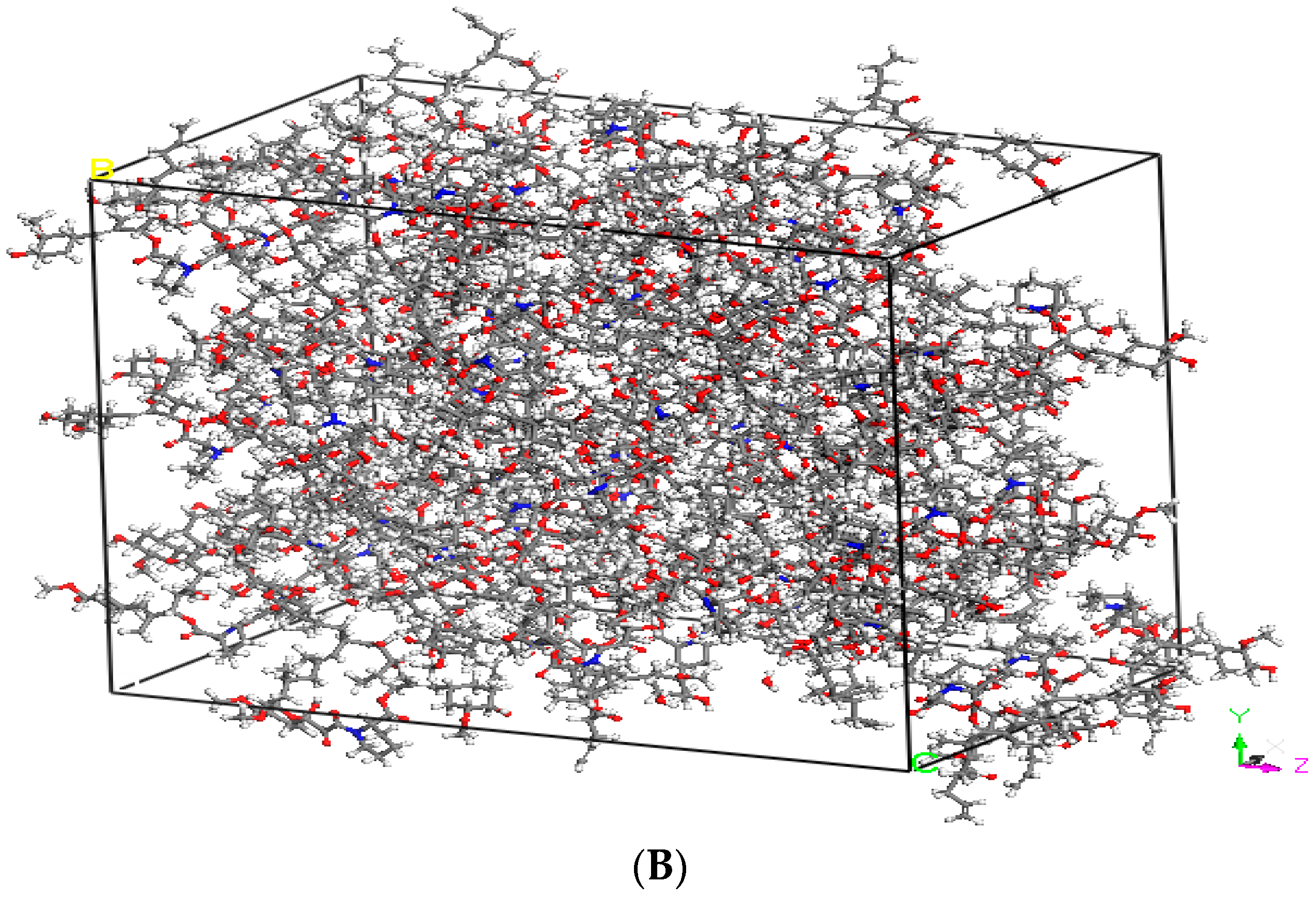
2.2. Hansen Solubility Parameter and Excipient Selection
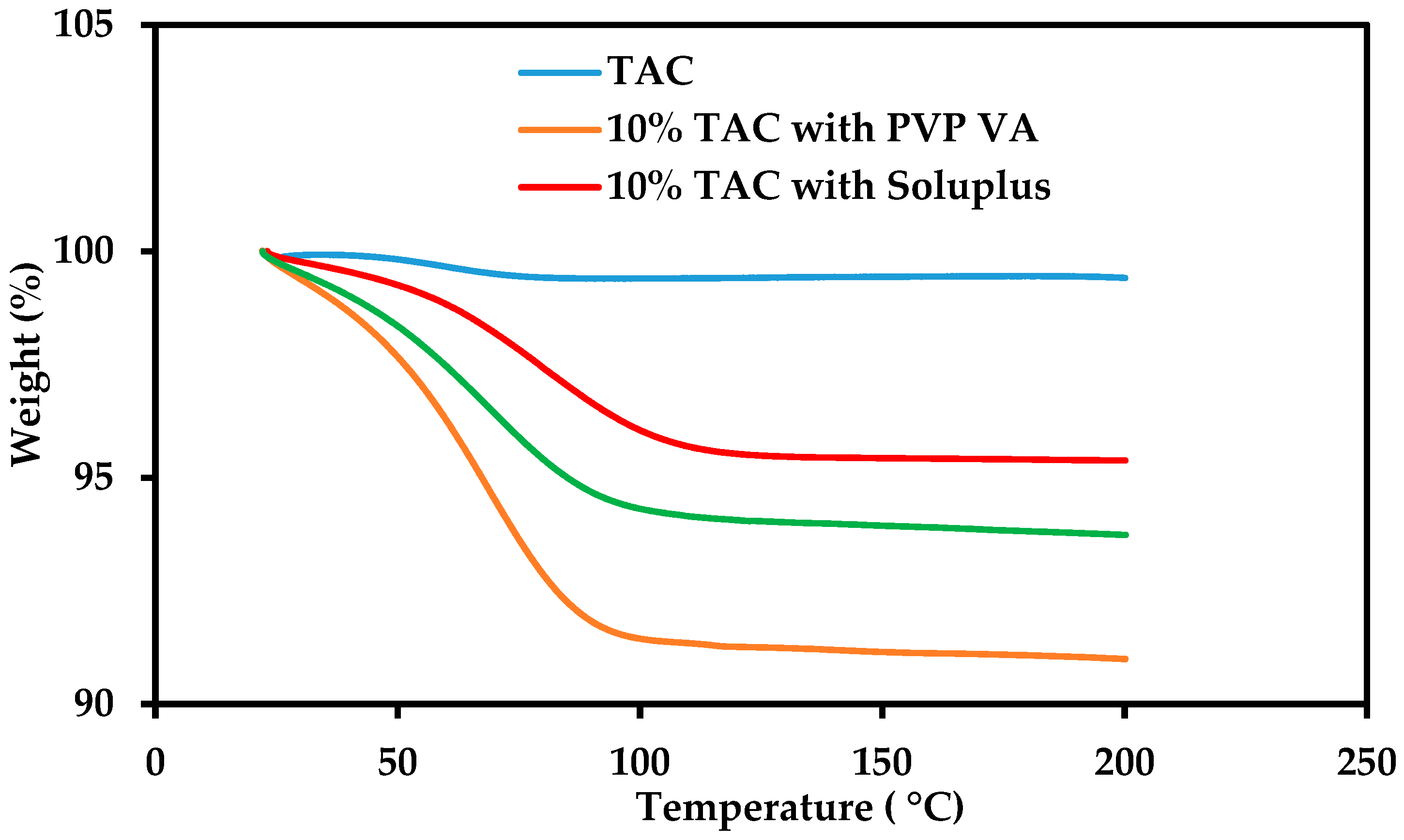
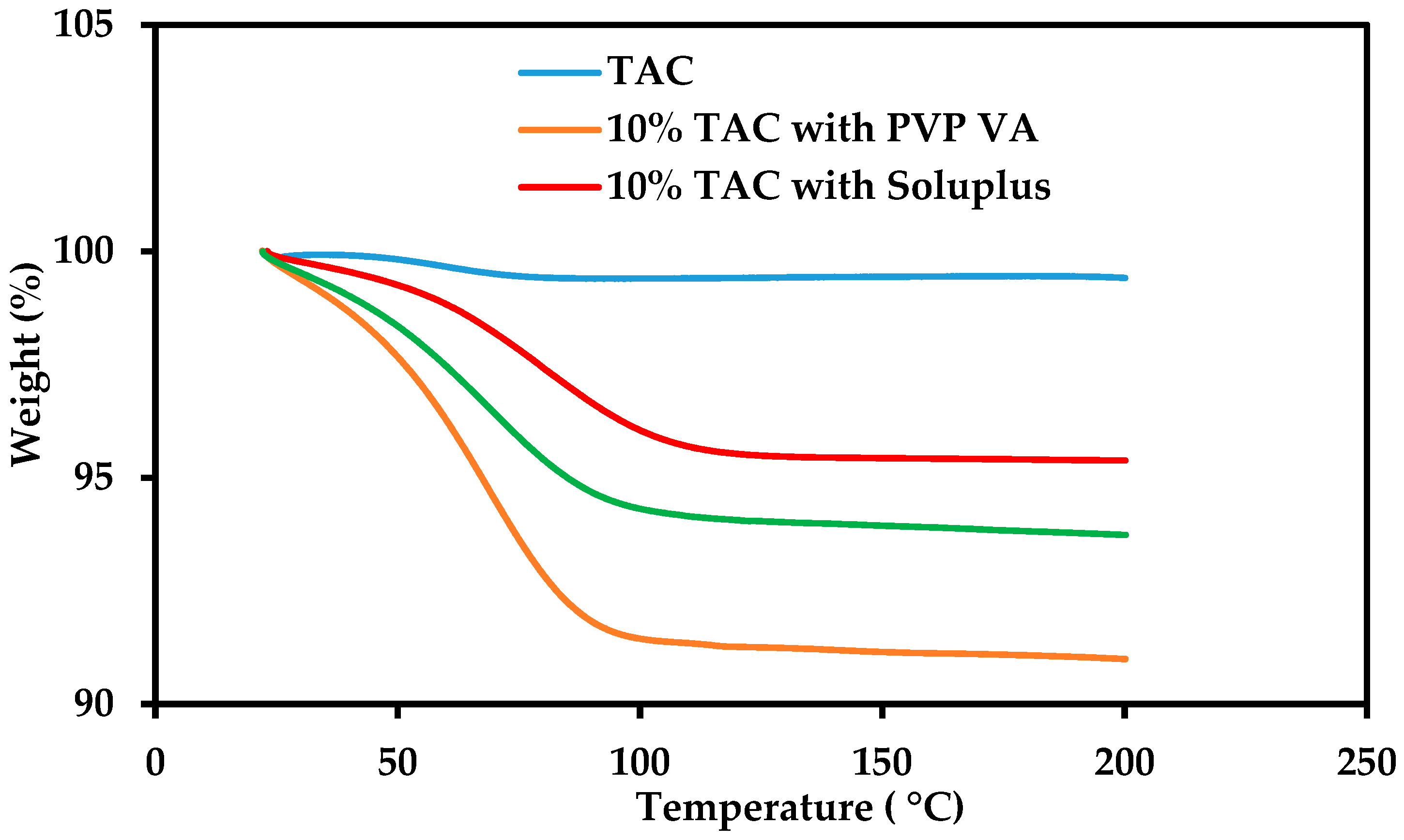
2.3. Thermogravimetric Analysis
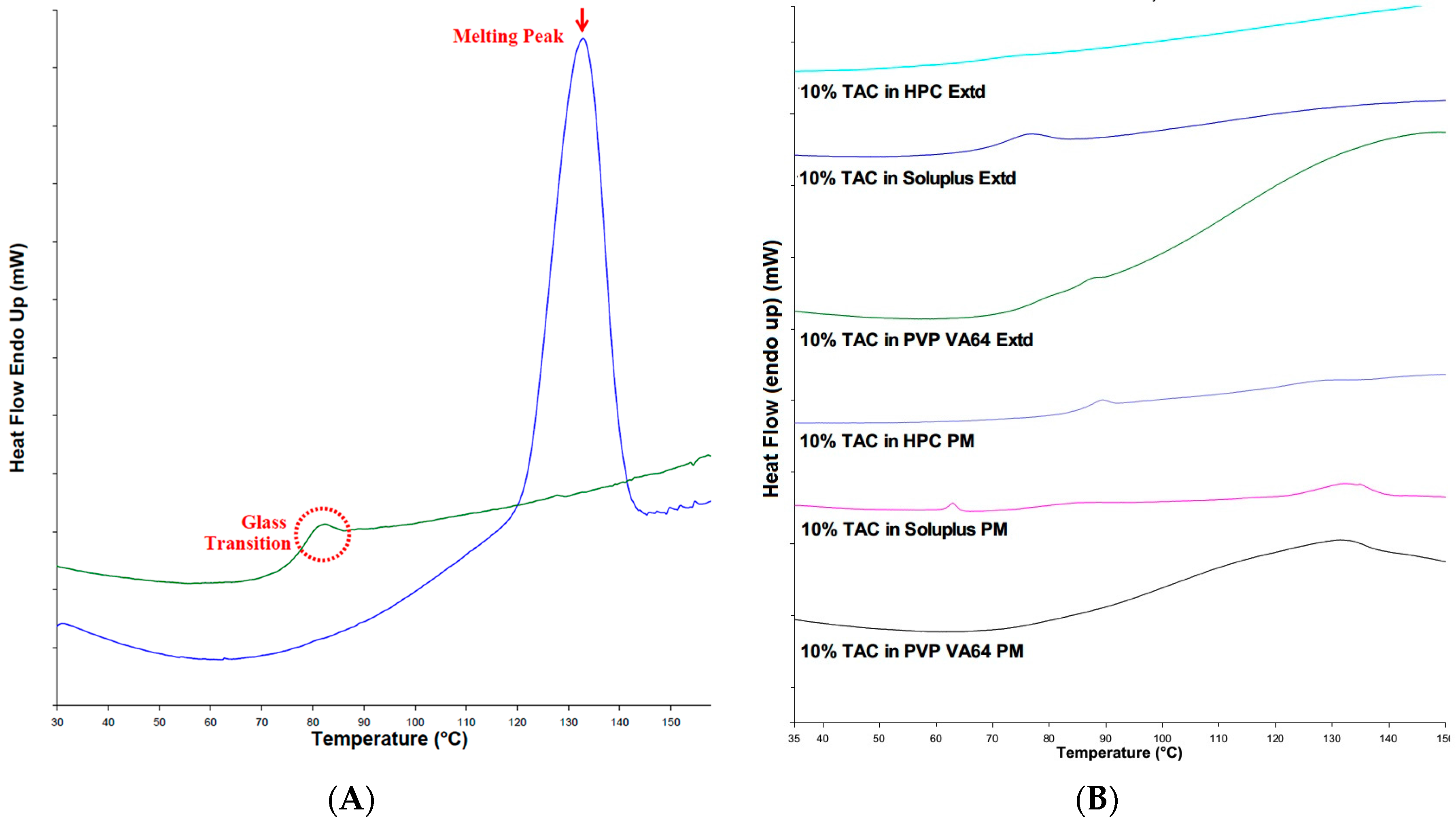
2.4. Differential Scanning Calorimetry
2.5. Preparation of Solid Dispersions by Hot Melt Extrusion (HME)
| Barrel Zone | 1 | 2 | 3 | 4 | 5 | 6 | Rod die |
| Temperature (°C) | not used | not used | 70 | 125 | 135 | 140 | 135 |
2.6. High Performance Liquid Chromatography (HPLC) Analysis
2.7. Microscopy and Imaging
2.8. Powder X-ray Diffraction
2.9. Fourier Transform InfraRed (FTIR) Spectrophotometric Analysis
2.10. Dissolution Testing
2.11. Amorphous Stability Testing
2.12. Orally-Disintegrating Tablet (ODT) Compression and Characterization
3. Results
3.1. Hansen Solubility Parameter and Excipient Selection
3.2. Thermal Stability of Tacrolimus (TAC)
3.3. Hot Melt Extrusion
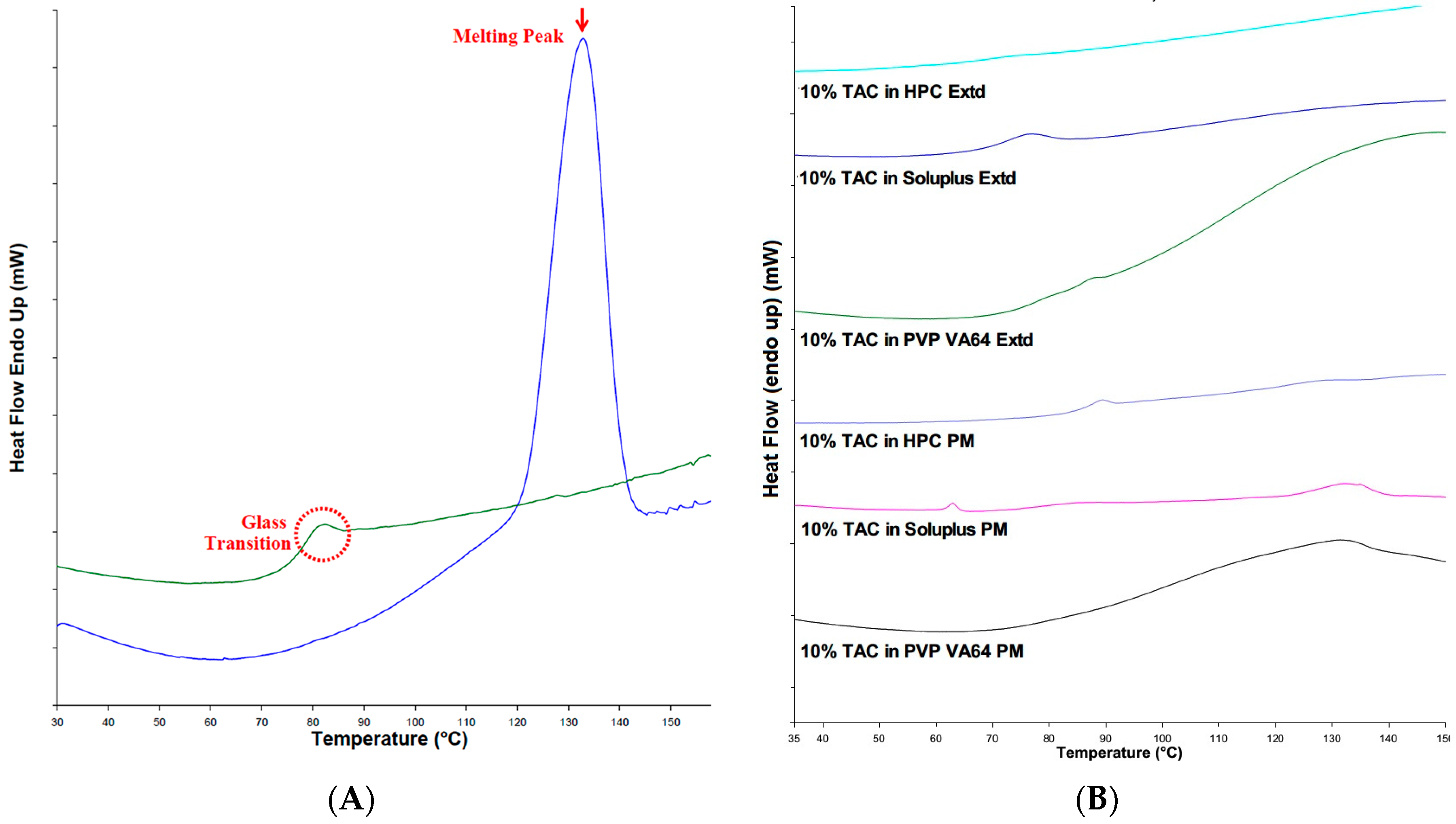
3.4. Differential Scanning Calorimetry (DSC) Analysis
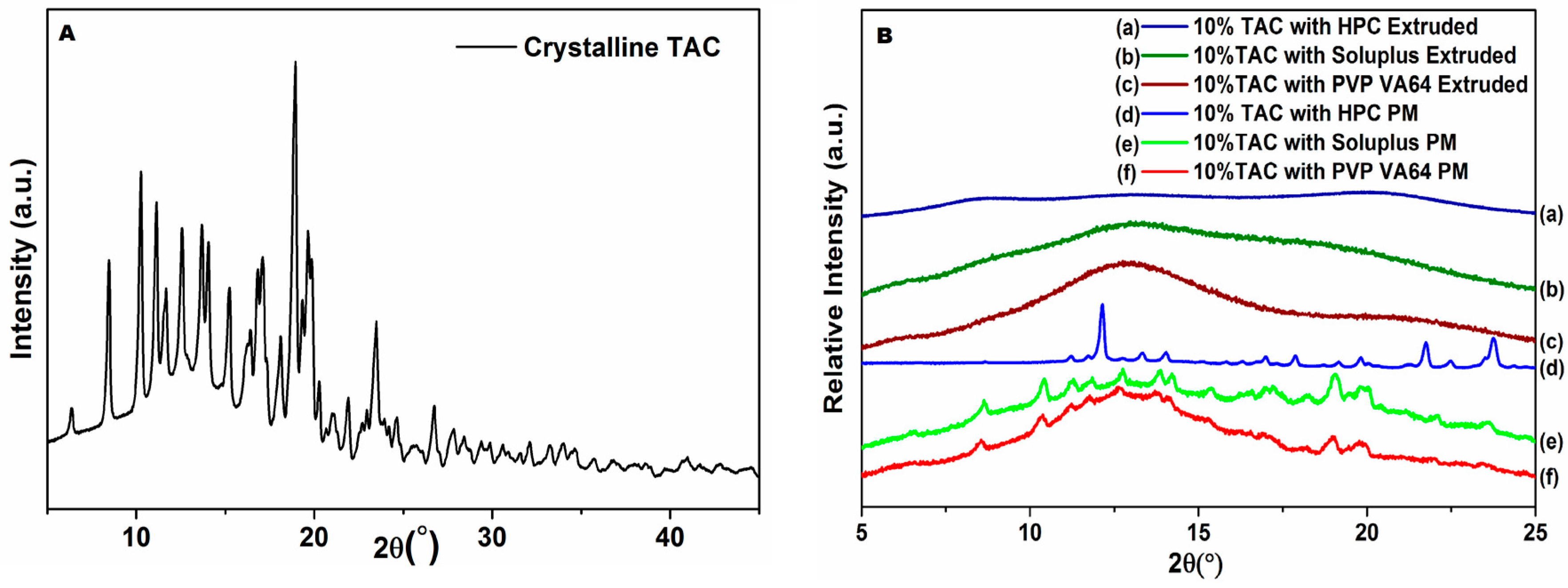
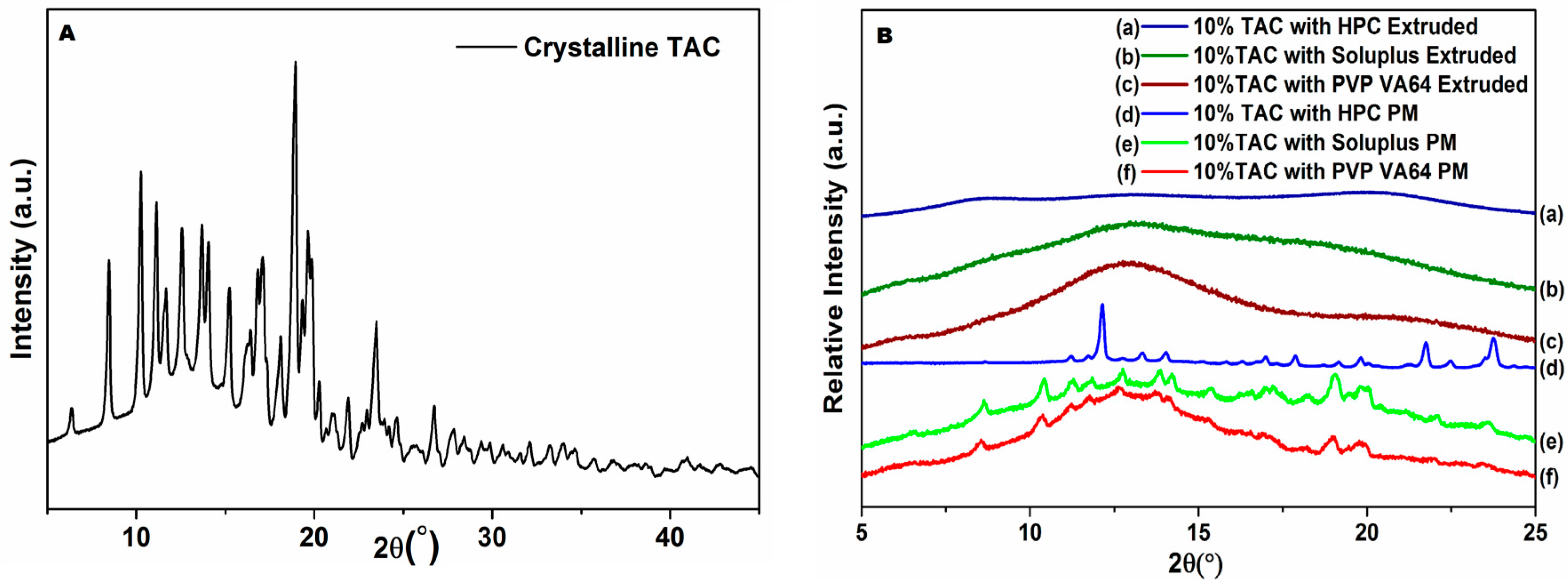
3.5. Powder X-ray Diffraction
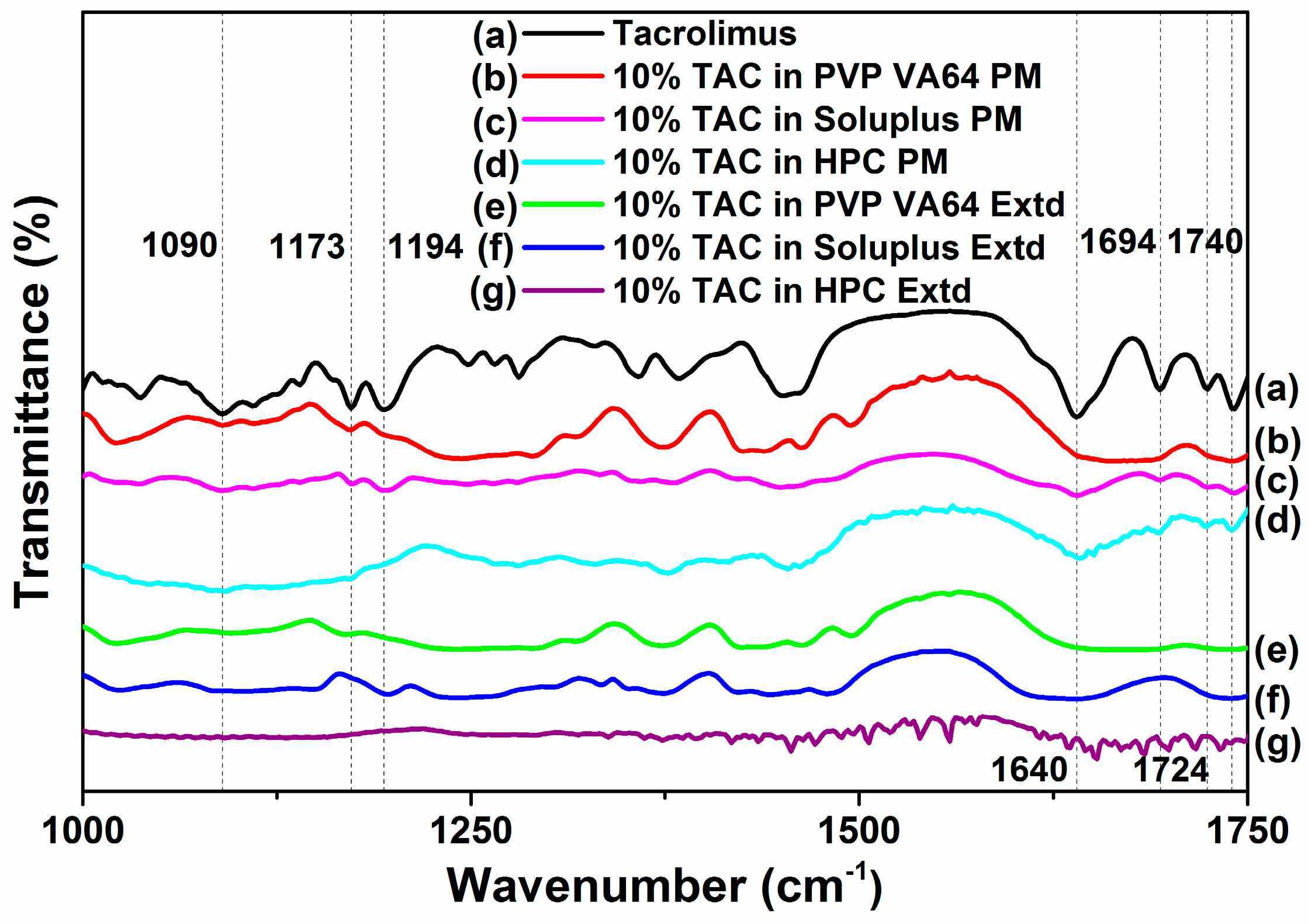
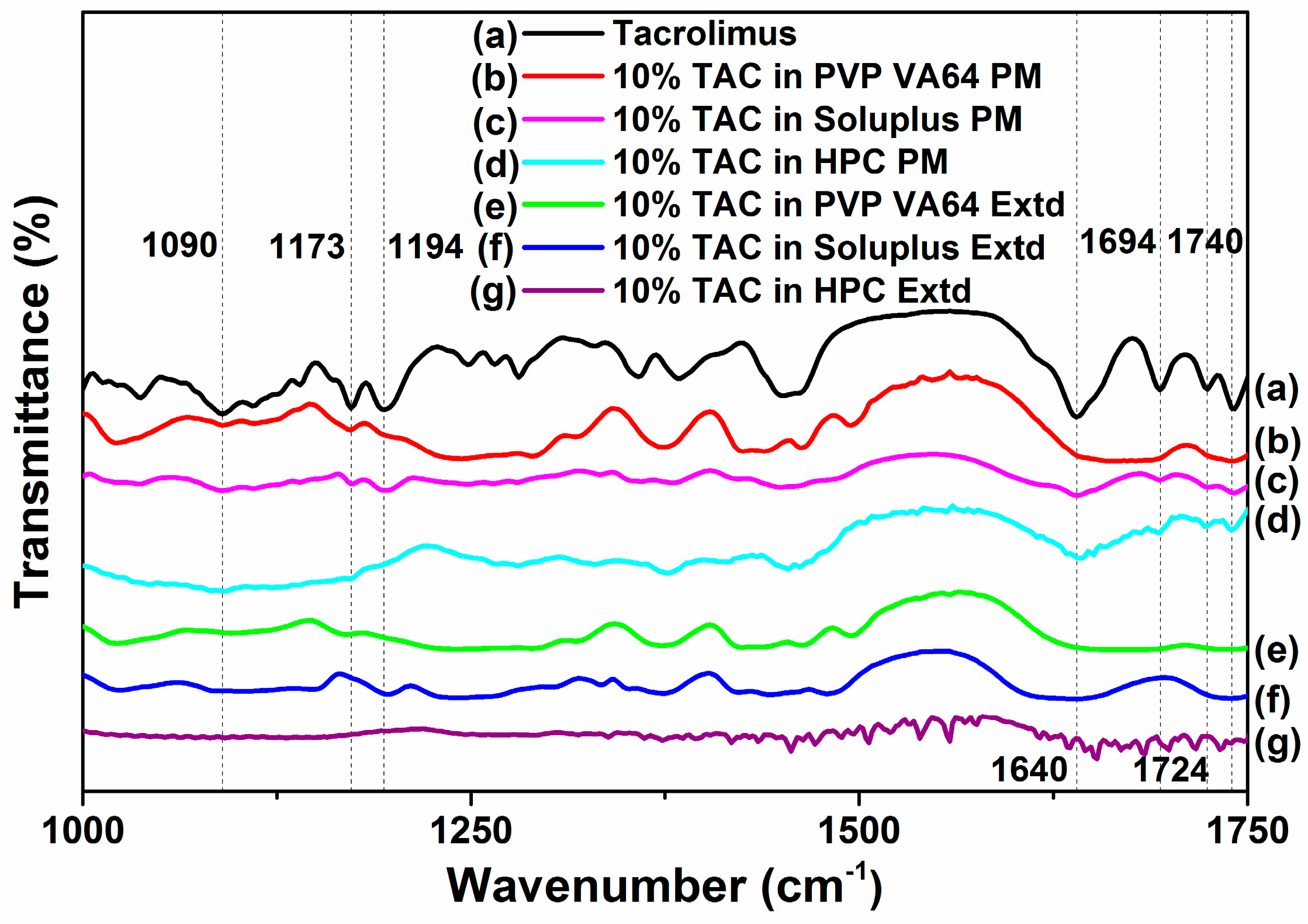
3.6. FTIR
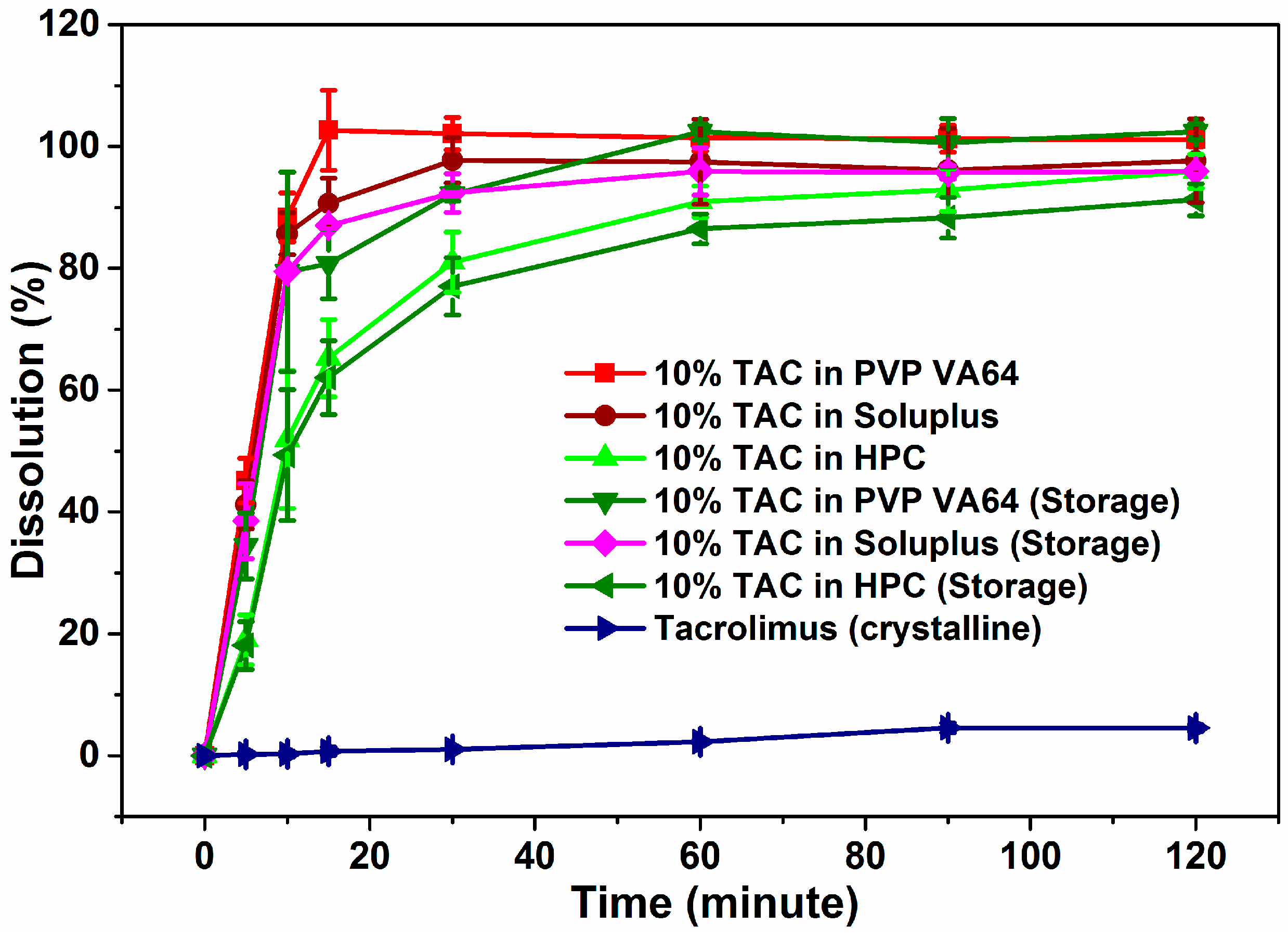
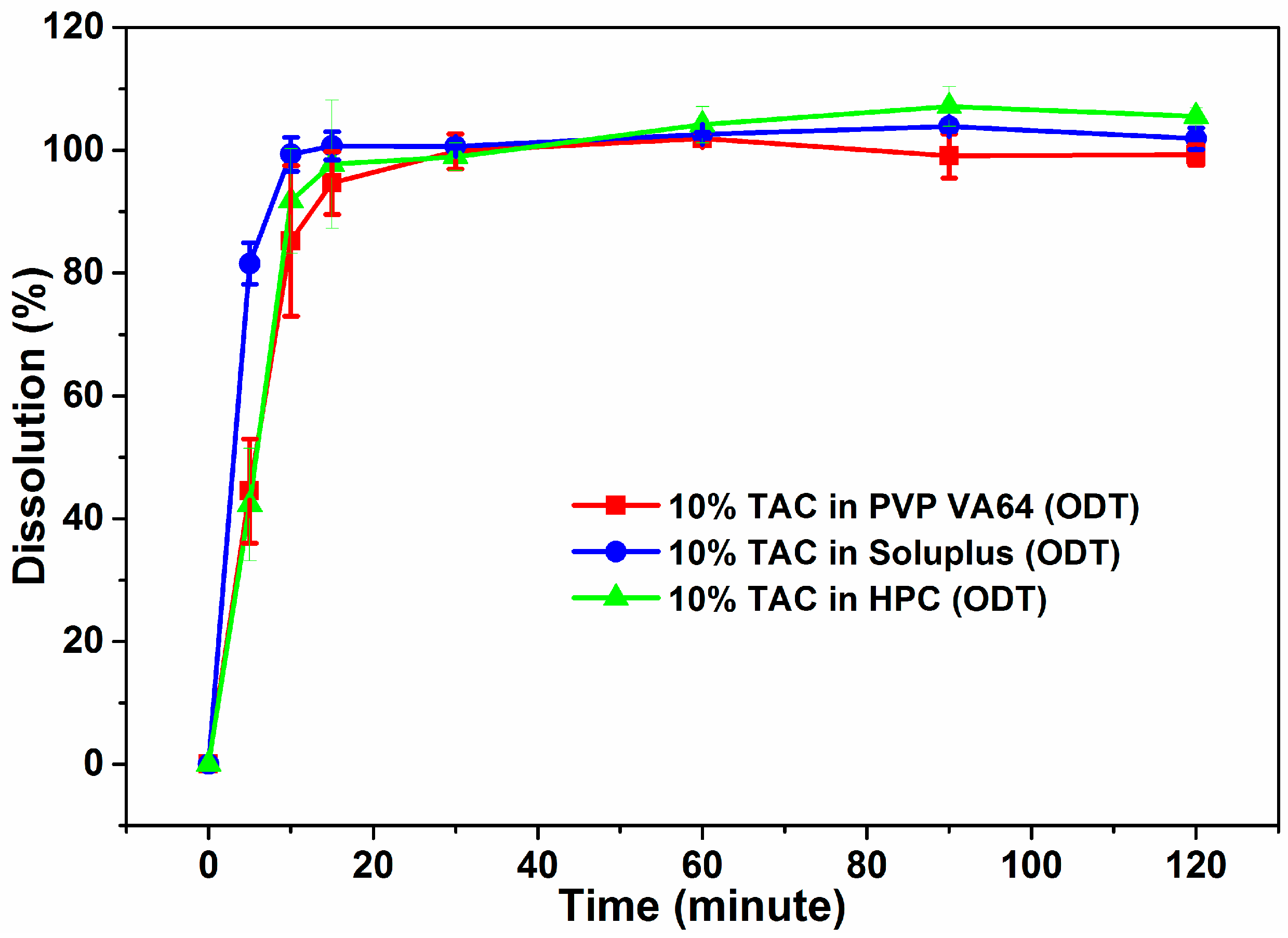
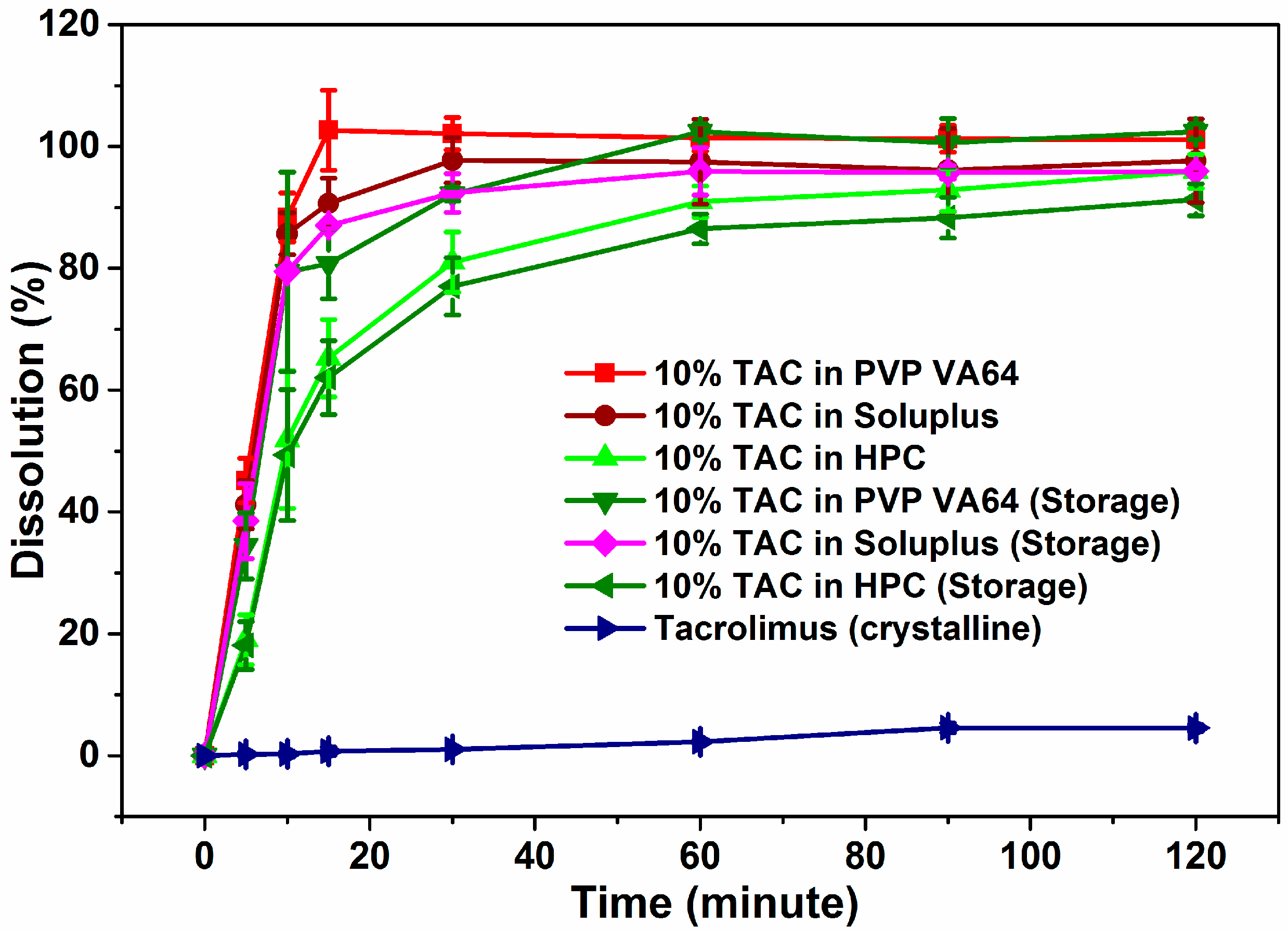
3.7. Dissolution Studies
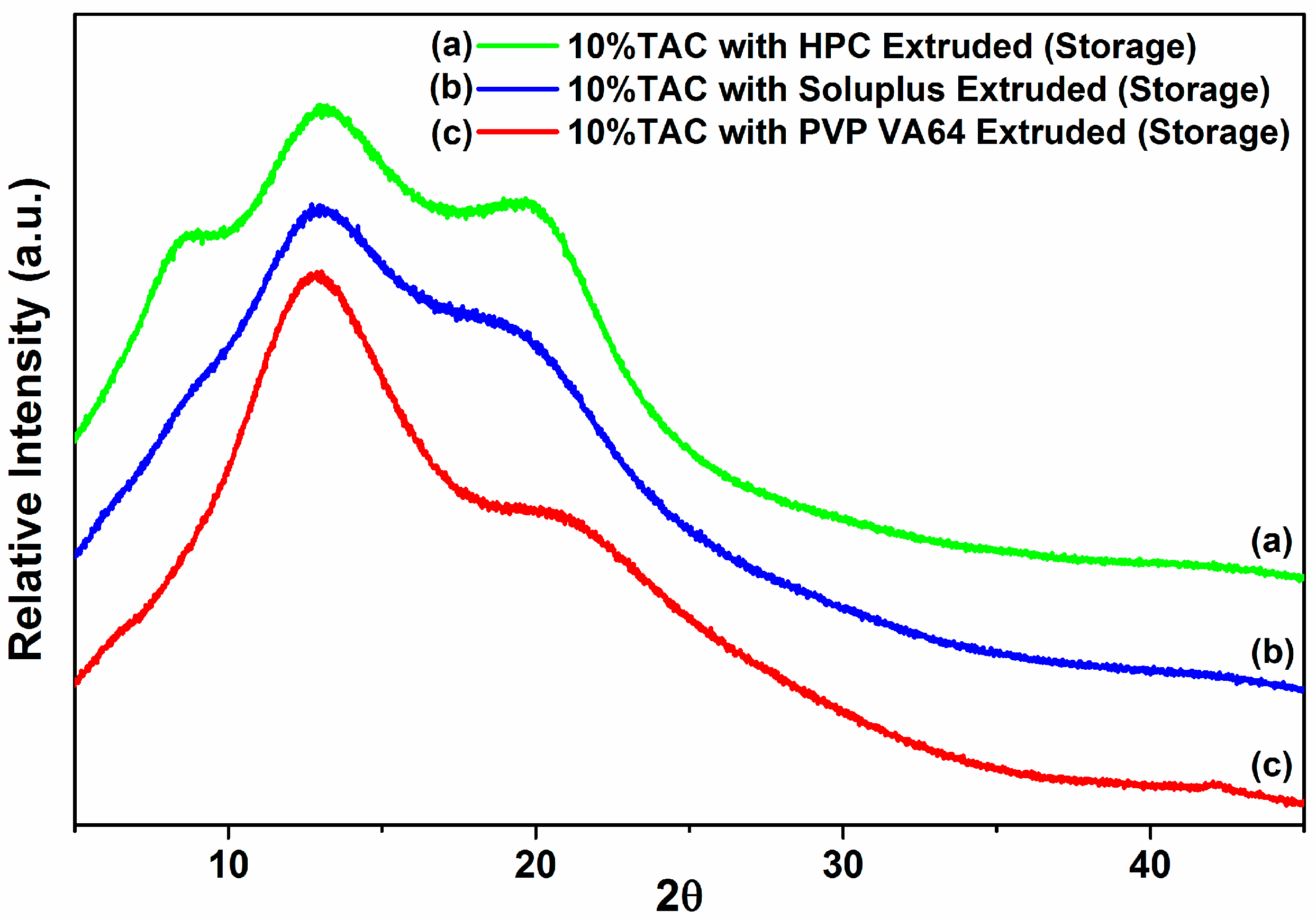
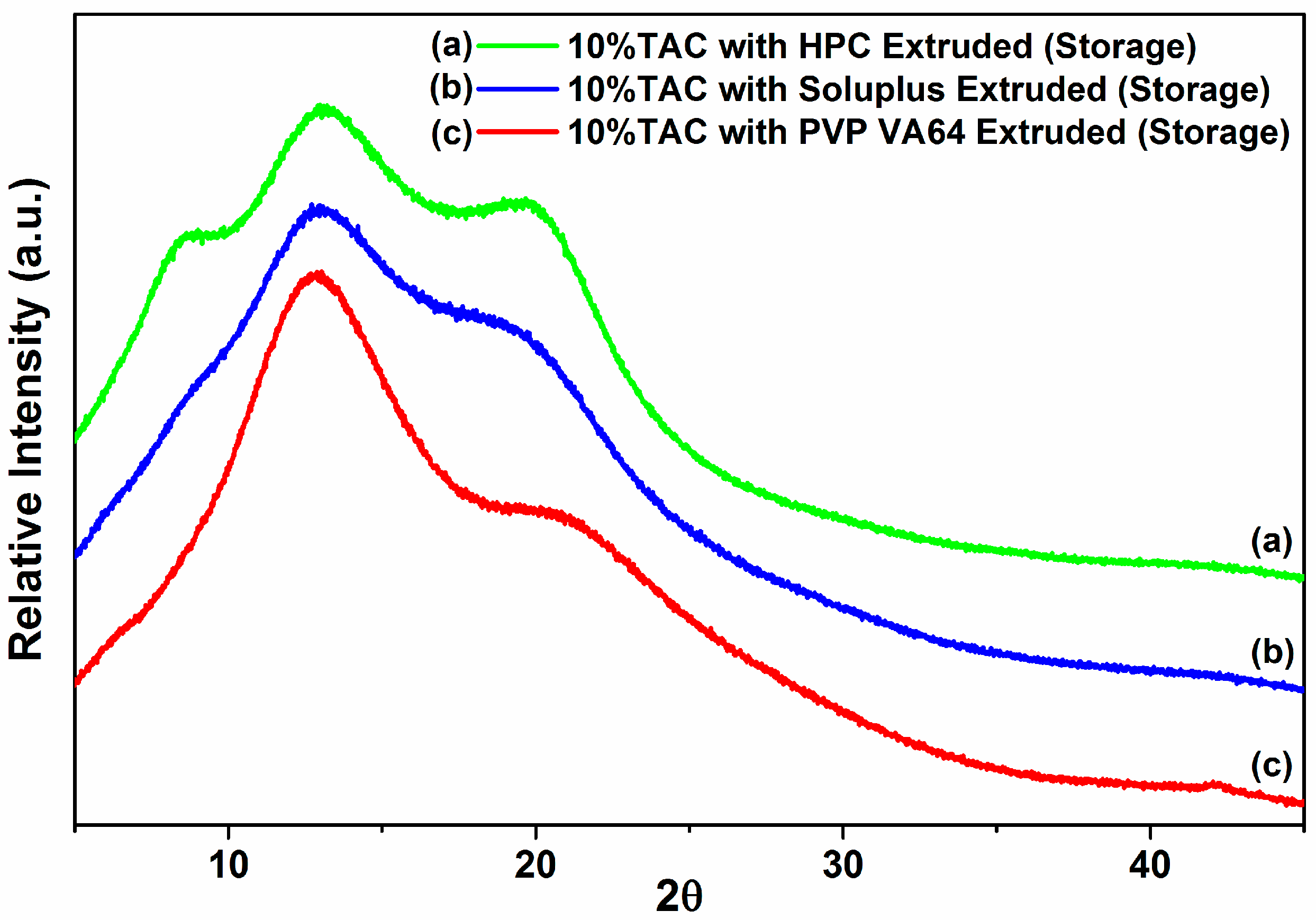
3.8. Stability Studies
3.9. ODT Formulation and Characterization
3.10. Dissolution Testing of TAC ODTs
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halloran, P.F. Immunosuppressive drugs for kidney transplantation. N. Engl. J. Med. 2004, 351, 2715–2729. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hatanaka, H.; Miyata, S.; Inamura, N.; Nishiyama, M.; Yajima, T.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; et al. Fk-506, a novel immunosuppressant isolated from a streptomyces. Ii. Immunosuppressive effect of fk-506 in vitro. J. Antibiot. 1987, 40, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Joe, J.H.; Lee, W.M.; Park, Y.J.; Joe, K.H.; Oh, D.H.; Seo, Y.G.; Woo, J.S.; Yong, C.S.; Choi, H.G. Effect of the solid-dispersion method on the solubility and crystalline property of tacrolimus. Int. J. Pharm. Investig. 2010, 395, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Nakate, T.; Okimoto, K.; Ohike, A.; Tokunaga, Y.; Ibuki, R.; Higaki, K.; Kimura, T. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int. J. Pharm. 2003, 267, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Ohike, A.; Ibuki, R.; Amidon, G.L.; Yamashita, S. Tacrolimus is a class ii low-solubility high-permeability drug: The effect of p-glycoprotein efflux on regional permeability of tacrolimus in rats. J. Pharm. Sci. 2002, 91, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Kagayama, A.; Tanimoto, S.; Fujisaki, J.; Kaibara, A.; Ohara, K.; Iwasaki, K.; Hirano, Y.; Hata, T. Oral absorption of fk506 in rats. Pharm. Res. 1993, 10, 1446–1450. [Google Scholar] [CrossRef] [PubMed]
- Grinyo, J.M.; Petruzzelli, S. Once-daily lcp-tacro meltdose tacrolimus for the prophylaxis of organ rejection in kidney and liver transplantations. Expert Rev. Clin. Immunol. 2014, 10, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Sasa, H.; Shimomura, M.; Inui, K. Effects of intestinal and hepatic metabolism on the bioavailability of tacrolimus in rats. Pharm. Res. 1998, 15, 1609–1613. [Google Scholar] [CrossRef] [PubMed]
- Arima, H.; Yunomae, K.; Hirayama, F.; Uekama, K. Contribution of p-glycoprotein to the enhancing effects of dimethyl-beta-cyclodextrin on oral bioavailability of tacrolimus. J. Pharmacol. Exp. Ther. 2001, 297, 547–555. [Google Scholar] [PubMed]
- Nassar, T.; Rom, A.; Nyska, A.; Benita, S. A novel nanocapsule delivery system to overcome intestinal degradation and drug transport limited absorption of p-glycoprotein substrate drugs. Pharm. Res. 2008, 25, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Haragsim, L. Calcineurin nephrotoxicity. Adv. Chronic Kidney Dis. 2006, 13, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Veroux, P.; Veroux, M.; Puliatti, C.; Valvo, M.; Macarone, M.; Cappello, D. Severe neurotoxicity in tacrolimus-treated living kidney transplantation in two cases. Urol. Int. 2003, 71, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Overhoff, K.A.; McConville, J.T.; Yang, W.; Johnston, K.P.; Peters, J.I.; Williams, R.O., 3rd. Effect of stabilizer on the maximum degree and extent of supersaturation and oral absorption of tacrolimus made by ultra-rapid freezing. Pharm. Res. 2008, 25, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kurimoto, I.; Yoshihara, K.; Umejima, H.; Ito, N.; Watanabe, S.; Sako, K.; Kikuchi, A. Aminoalkyl methacrylate copolymers for improving the solubility of tacrolimus. I: Evaluation of solid dispersion formulations. Int. J. Pharm. 2012, 428, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kurimoto, I.; Yoshihara, K.; Umejima, H.; Ito, N.; Watanabe, S.; Sako, K.; Kikuchi, A. Effect of aminoalkyl methacrylate copolymer e/hcl on in vivo absorption of poorly water-soluble drug. Drug Dev. Ind. Pharm. 2013, 39, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Arima, H.; Yunomae, K.; Miyake, K.; Irie, T.; Hirayama, F.; Uekama, K. Comparative studies of the enhancing effects of cyclodextrins on the solubility and oral bioavailability of tacrolimus in rats. J. Pharm. Sci. 2001, 90, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Nekkanti, V.; Rueda, J.; Wang, Z.; Betageri, G.V. Design, characterization, and in vivo pharmacokinetics of tacrolimus proliposomes. AAPS PharmSciTech 2016, 17, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hayes, D., Jr.; Zwischenberger, J.B.; Kuhn, R.J.; Mansour, H.M. Design and physicochemical characterization of advanced spray-dried tacrolimus multifunctional particles for inhalation. Drug Des. Dev. Ther. 2013, 7, 59–72. [Google Scholar]
- Collin, C.; Boussaud, V.; Lefeuvre, S.; Amrein, C.; Glouzman, A.S.; Havard, L.; Billaud, E.M.; Guillemain, R. Sublingual tacrolimus as an alternative to intravenous route in patients with thoracic transplant: A retrospective study. Transplant. Proc. 2010, 42, 4331–4337. [Google Scholar] [CrossRef] [PubMed]
- Srinarong, P.; Pham, B.T.; Holen, M.; van der Plas, A.; Schellekens, R.C.; Hinrichs, W.L.; Frijlink, H.W. Preparation and physicochemical evaluation of a new tacrolimus tablet formulation for sublingual administration. Drug Dev. Ind. Pharm. 2012, 38, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Breitenbach, J. Melt extrusion: From process to drug delivery technology. Eur. J. Pharm. Biopharm. 2002, 54, 107–117. [Google Scholar] [CrossRef]
- Materials Studio Modeling; 7.0.100; Accelrys Software Inc.: San Diego, CA, USA, 2013.
- Sun, H. Compass: An ab initio force-field optimized for condensed-phase applications-overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation, 2nd ed.; Academic Press: San Deigo, CA, USA, 2002. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Akashi, T.; Nefuji, T.; Yoshida, M.; Hosoda, J. Quantitative determination of tautomeric fk506 by reversed-phase liquid chromatography. J. Pharm. Biomed. Anal. 1996, 14, 339–346. [Google Scholar] [CrossRef]
- Azarmi, S.; Roa, W.; Lobenberg, R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int. J. Pharm. 2007, 328, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Kolter, K.; Karl, M.; Gryczke, A. Hot Melt Extrusion with Basf Pharma Polymers, 2nd ed.; BASF: Ludwigshafen, Germany, 2012. [Google Scholar]
- Mididoddi, P.K.; Repka, M.A. Characterization of hot-melt extruded drug delivery systems for onychomycosis. Eur. J. Pharm. Biopharm. 2007, 66, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Boer, T.M.; Procopio, J.V.; Nascimento, T.G.; Macedo, R.O. Correlation of thermal analysis and pyrolysis coupled to gc-ms in the characterization of tacrolimus. J. Pharm. Biomed. Anal. 2013, 73, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Zidan, A.S.; Rahman, Z.; Sayeed, V.; Raw, A.; Yu, L.; Khan, M.A. Crystallinity evaluation of tacrolimus solid dispersions by chemometric analysis. Int. J. Pharm. 2012, 423, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry Orally Disintegrating Tablets; U.S. Food and drug administration: Silver Spring, MD, USA, 2008.
- McLaughlin, R.; Banbury, S.; Crowley, K. Orally disintegrating tablets: The effect of recent fda guidance on odt technologies and applications. Pharm. Tech. 2009. Available online: http://www.pharmtech.com/orally-disintegrating-tablets-effect-recent-fda-guidance-odt-technologies-and-applications (accessed on 9 February 2018).
- Fitzsimmons, W.E. Tacrolimus. In Immunotherapy in Transplantation; Wiley-Blackwell: Hoboken, NJ, USA, 2010; pp. 224–240. [Google Scholar]
- Wu, C.-Y.; Benet, L.Z. Predicting drug disposition via application of bcs: Transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kadajji, V.G.; Betageri, G.V. Water soluble polymers for pharmaceutical applications. Polymers 2011, 3, 1972–2009. [Google Scholar] [CrossRef]
- Jijun, F.; Lishuang, X.; Xiaoguang, T.; Min, S.; Mingming, Z.; Haibing, H.; Xing, T. The inhibition effect of high storage temperature on the recrystallization rate during dissolution of nimodipine-kollidon va64 solid dispersions (nm-sd) prepared by hot-melt extrusion. J. Pharm. Sci. 2011, 100, 1643–1647. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Polymer | Aqueous Solubility | Molecular Weight | Glass Transition (°C) | Hansen’s Solubility Parameters (MPa1/2) | Interaction Parameter (Δδt) | Solid State |
|---|---|---|---|---|---|---|
| TAC | Insoluble (2.6 µg/mL) | 804.02 g/mol | 78.8 (amorphous form) | 19.1 * | - | Crystalline |
| PVP VA64 | Very soluble | 45–70 kD | 101 | 19.7 [31] | 0.6 | Amorphous |
| Soluplus | Very soluble | 118 kD | 70 | 19.4 [31] | 0.3 | Amorphous |
| HPC | Soluble | 95 kD | 105 | 21.27 [32] | 2.17 | Semi-crystalline |
| Excipient | Formulation 1 | Formulation 2 |
|---|---|---|
| Microcrystalline cellulose | 69.75% | 59.5% |
| Crospovidone | 10% | 10% |
| Mannitol | 10% | 20% |
| Magnesium Stearate | 0.25% | 0.5% |
| Polymer/TAC Solid dispersion | 10% | 10% |
| Formulation | Polymer | Hardness (kP) | Friability (%) | Disintegration Time (s) |
|---|---|---|---|---|
| Formulation 1 | PVP VA64 | 18.6 ± 2.8 | 0% | 83 |
| Soluplus | 11.9 ± 2.5 | 0.07% | 10 | |
| HPC | 20.9 ± 2.7 | 0.06% | 50 | |
| Formulation 2 | PVP VA64 | 23.0 ± 1.8 | 0% | 60 |
| Soluplus | 17.5 ± 0.8 | 0.03% | 18 | |
| HPC | 17.5 ± 1.0 | 0% | 40 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponnammal, P.; Kanaujia, P.; Yani, Y.; Ng, W.K.; Tan, R.B.H. Orally Disintegrating Tablets Containing Melt Extruded Amorphous Solid Dispersion of Tacrolimus for Dissolution Enhancement. Pharmaceutics 2018, 10, 35. https://doi.org/10.3390/pharmaceutics10010035
Ponnammal P, Kanaujia P, Yani Y, Ng WK, Tan RBH. Orally Disintegrating Tablets Containing Melt Extruded Amorphous Solid Dispersion of Tacrolimus for Dissolution Enhancement. Pharmaceutics. 2018; 10(1):35. https://doi.org/10.3390/pharmaceutics10010035
Chicago/Turabian StylePonnammal, Poovizhi, Parijat Kanaujia, Yin Yani, Wai Kiong Ng, and Reginald B. H. Tan. 2018. "Orally Disintegrating Tablets Containing Melt Extruded Amorphous Solid Dispersion of Tacrolimus for Dissolution Enhancement" Pharmaceutics 10, no. 1: 35. https://doi.org/10.3390/pharmaceutics10010035
APA StylePonnammal, P., Kanaujia, P., Yani, Y., Ng, W. K., & Tan, R. B. H. (2018). Orally Disintegrating Tablets Containing Melt Extruded Amorphous Solid Dispersion of Tacrolimus for Dissolution Enhancement. Pharmaceutics, 10(1), 35. https://doi.org/10.3390/pharmaceutics10010035