Citrate- and Succinate-Modified Carbonate Apatite Nanoparticles with Loaded Doxorubicin Exhibit Potent Anticancer Activity against Breast Cancer Cells

Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Synthesis of CA, CMCA and SMCA NPs
2.2.1. DOX-Loaded CA NPs
2.2.2. DOX-Loaded CMCA NPs
2.2.3. Succinate-Modified CA (SMCA) NPs
2.2.4. DOX-Loaded SMCA NPs
2.3. Optical Images of Free DOX, DOX-Loaded CA and DOX-Loaded CMCA and SMCA Particles
2.4. Estimation of Drug Binding Affinity towards NPs
2.5. Particle Size and Zeta Potential Measurement
2.6. Cell Culture and Seeding
2.7. Cell Treatment with Free DOX and DOX-Apatite Complexes
2.8. MTT Assay in MCF-7 and 4T1 Cell Lines
2.9. Data Analysis
2.10. Cellular Uptake of Free DOX and DOX-Loaded NPs in MCF-7 Cell Lines
2.11. Estimation of Bound Citrate in CMCA NPs
2.12. pH Sensitivity of CA, CMCA and SMCA NPs
2.13. Release Profile of DOX
2.14. Characterization of NPs by Fourier Transform-Infrared Spectroscopy (FTIR)
2.15. Characterization of NPs by Field Emission Scanning Electron Microscope (FE-SEM)
2.16. Characterization of NPs by Dynamic Light Scattering (DLS)
2.17. Statistical Analysis
3. Results
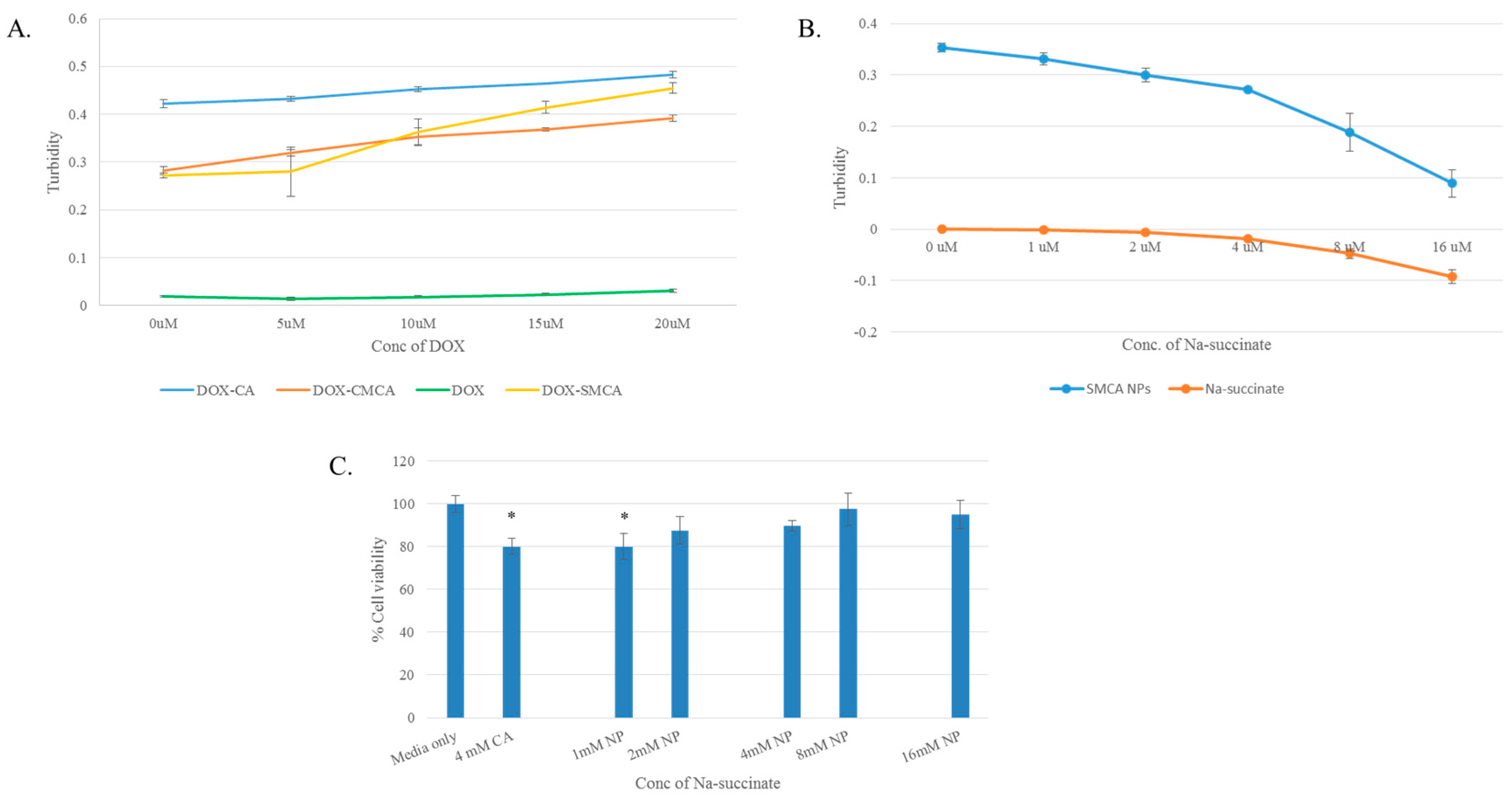
3.1. Synthesis and Characterization of DOX-Loaded CA, CMCA and SMCA NPs
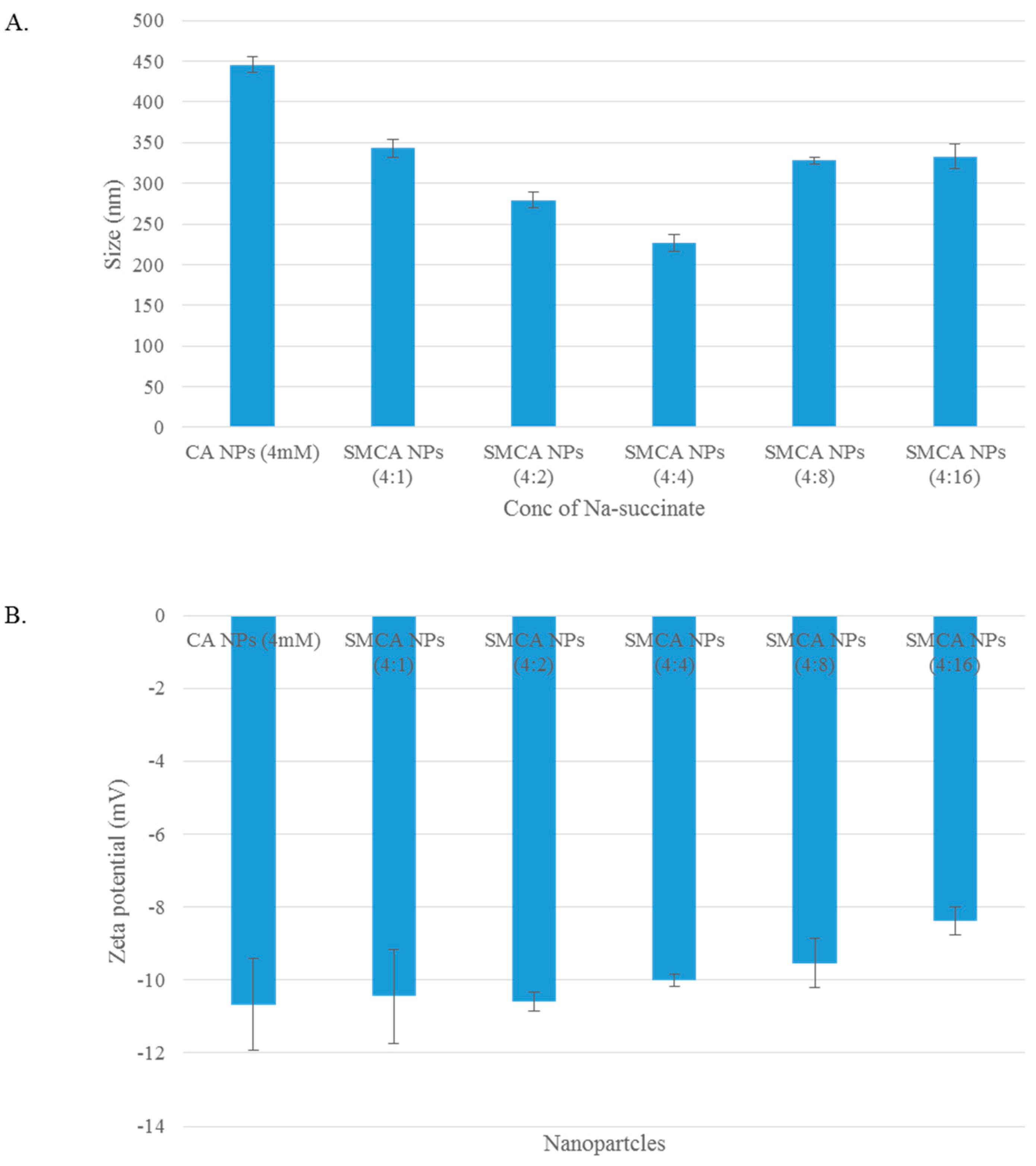
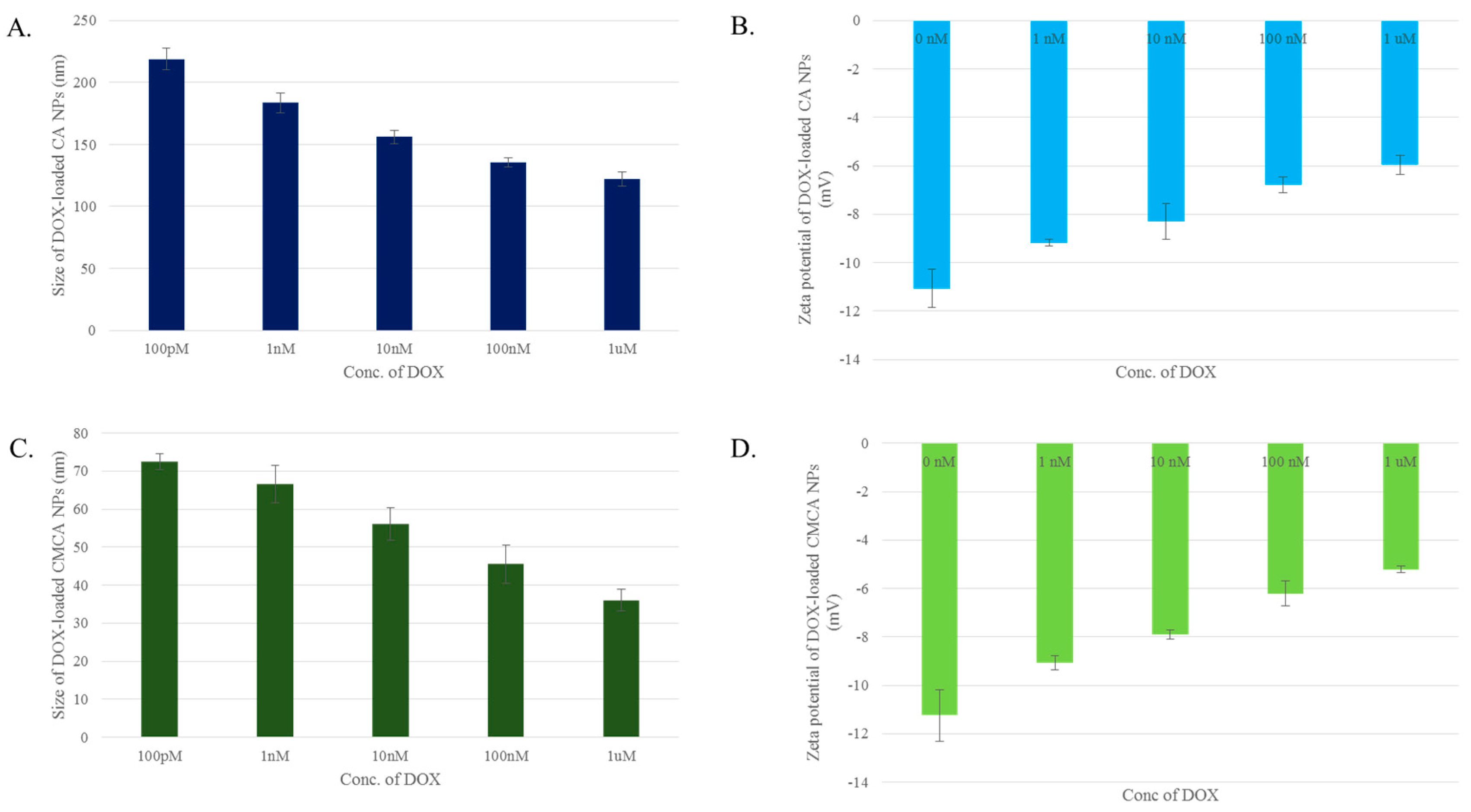
3.2. Particle Size and Surface Charge
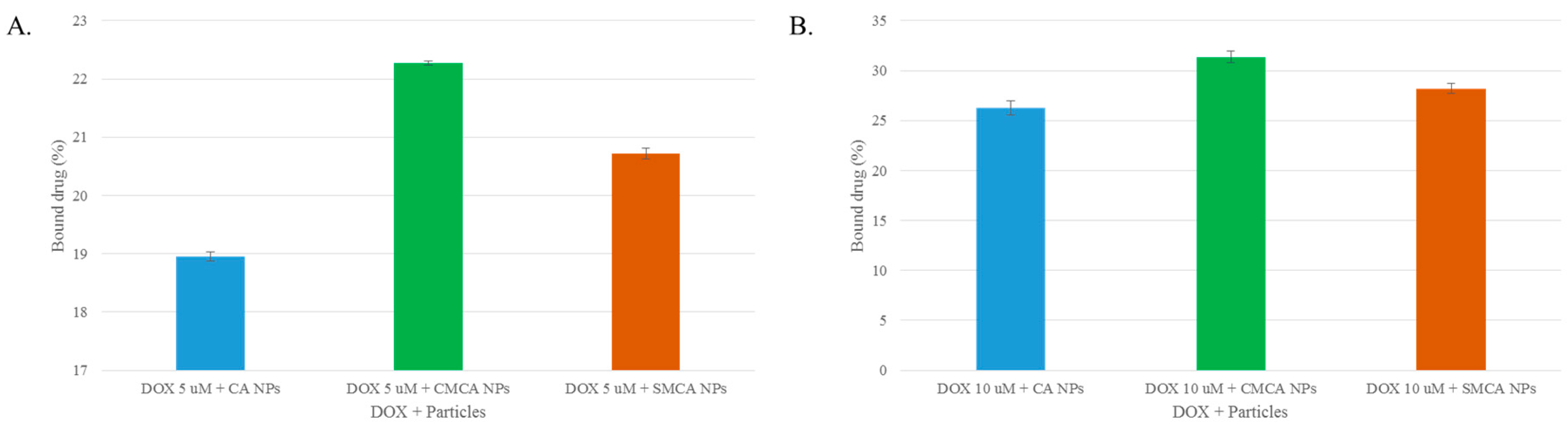
3.3. Estimation of DOX Adsorption onto Apatite-Based NPs
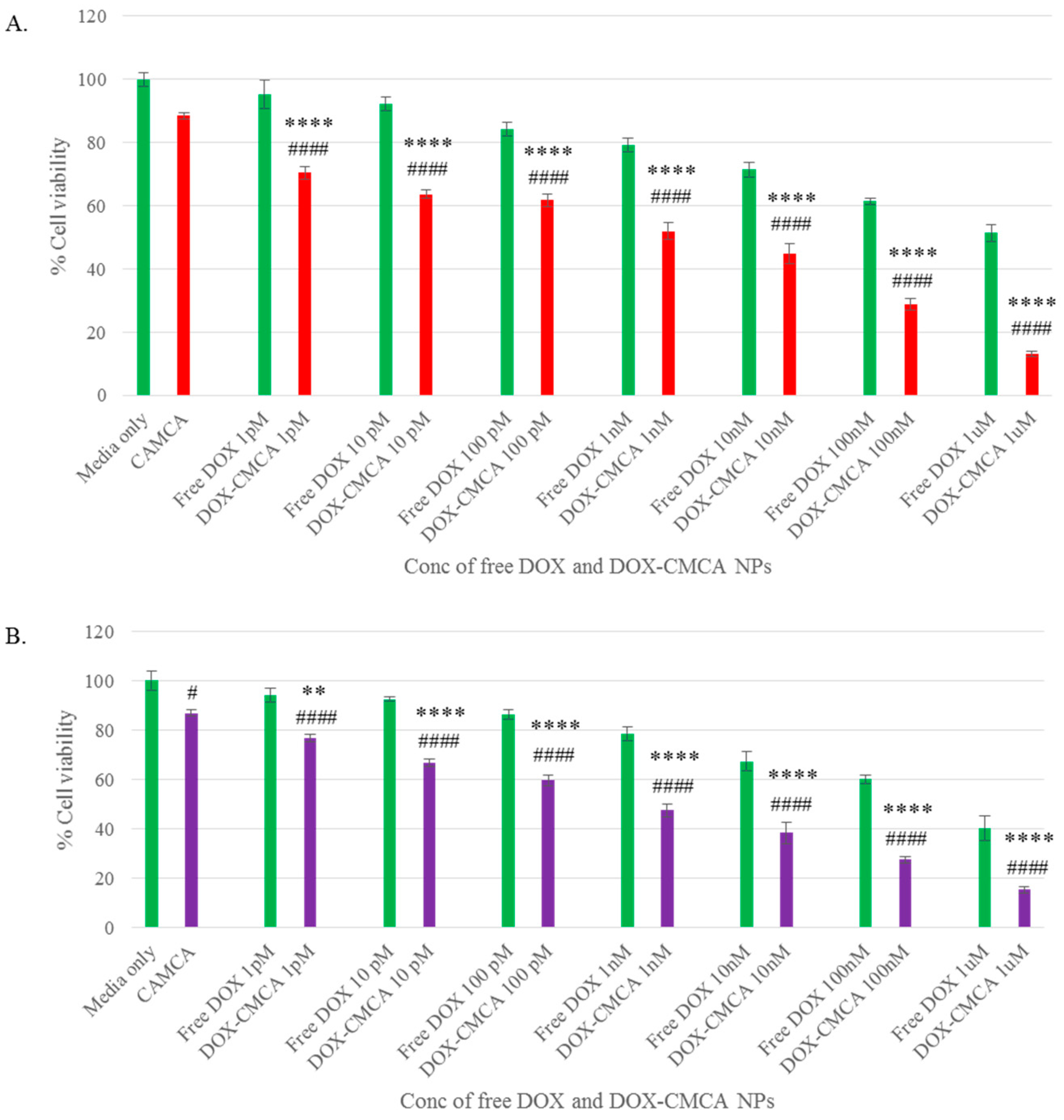
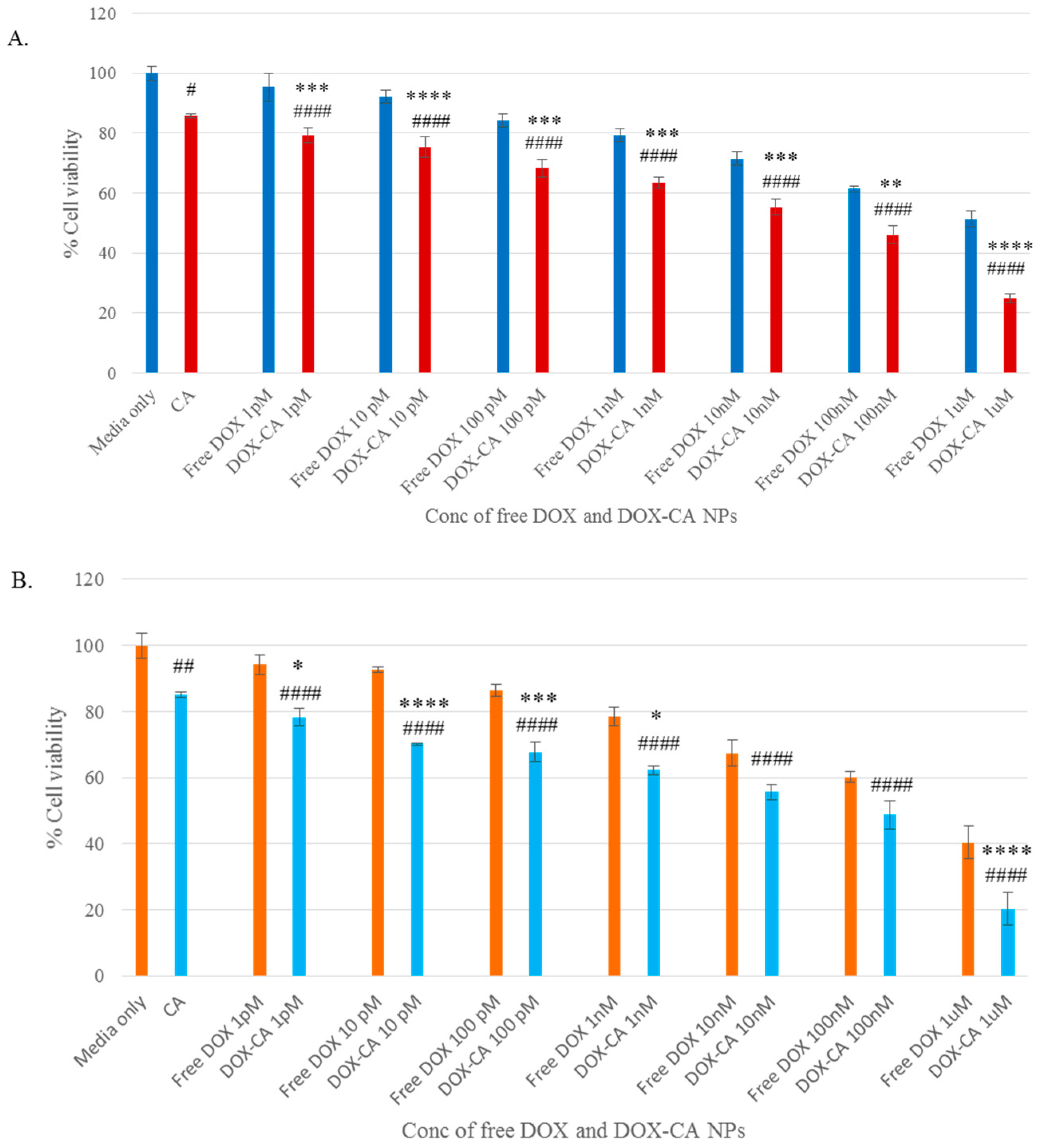
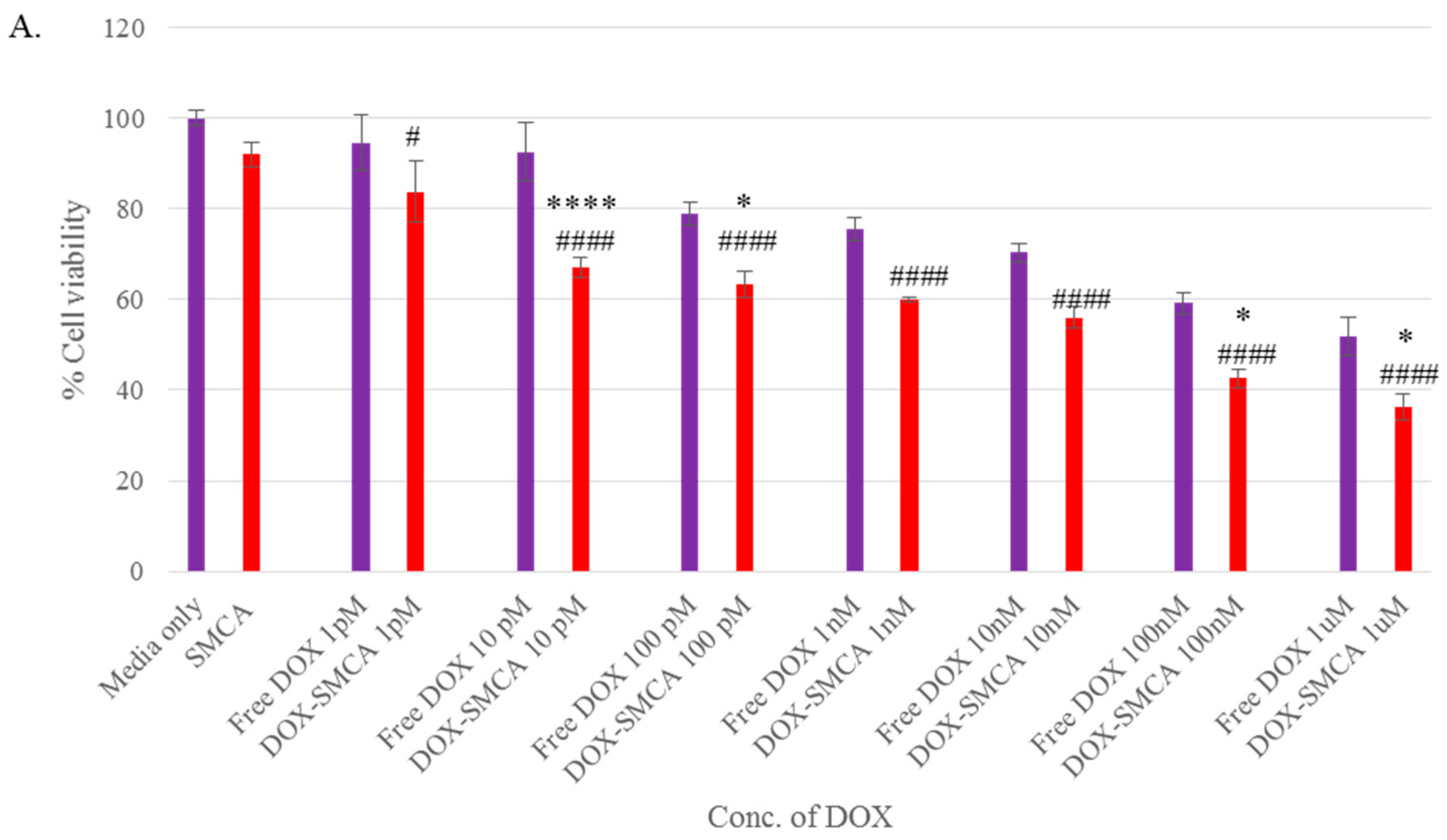
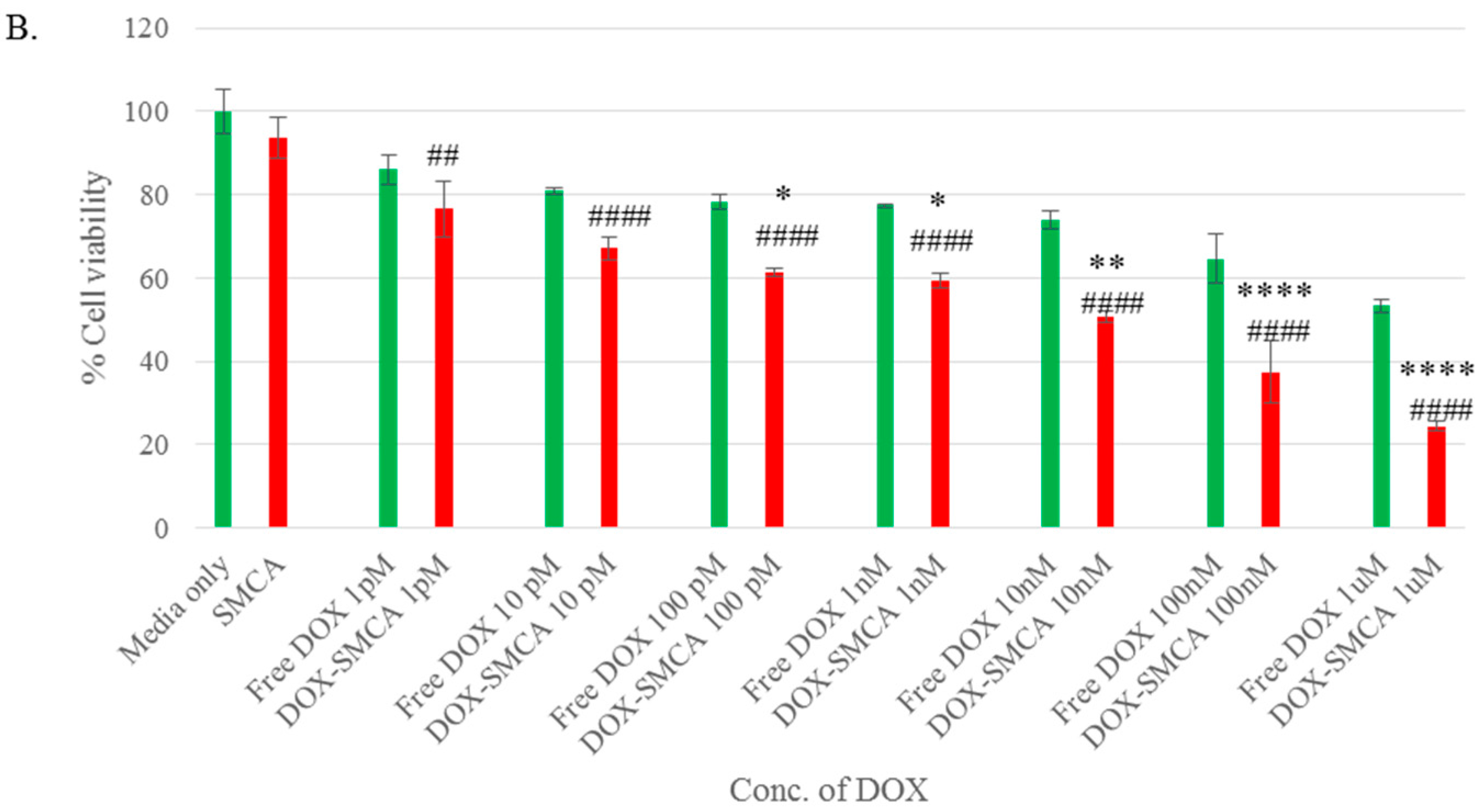
3.4. Cytotoxicity of Free DOX and DOX-Loaded CA, CMCA and SMCA NPs
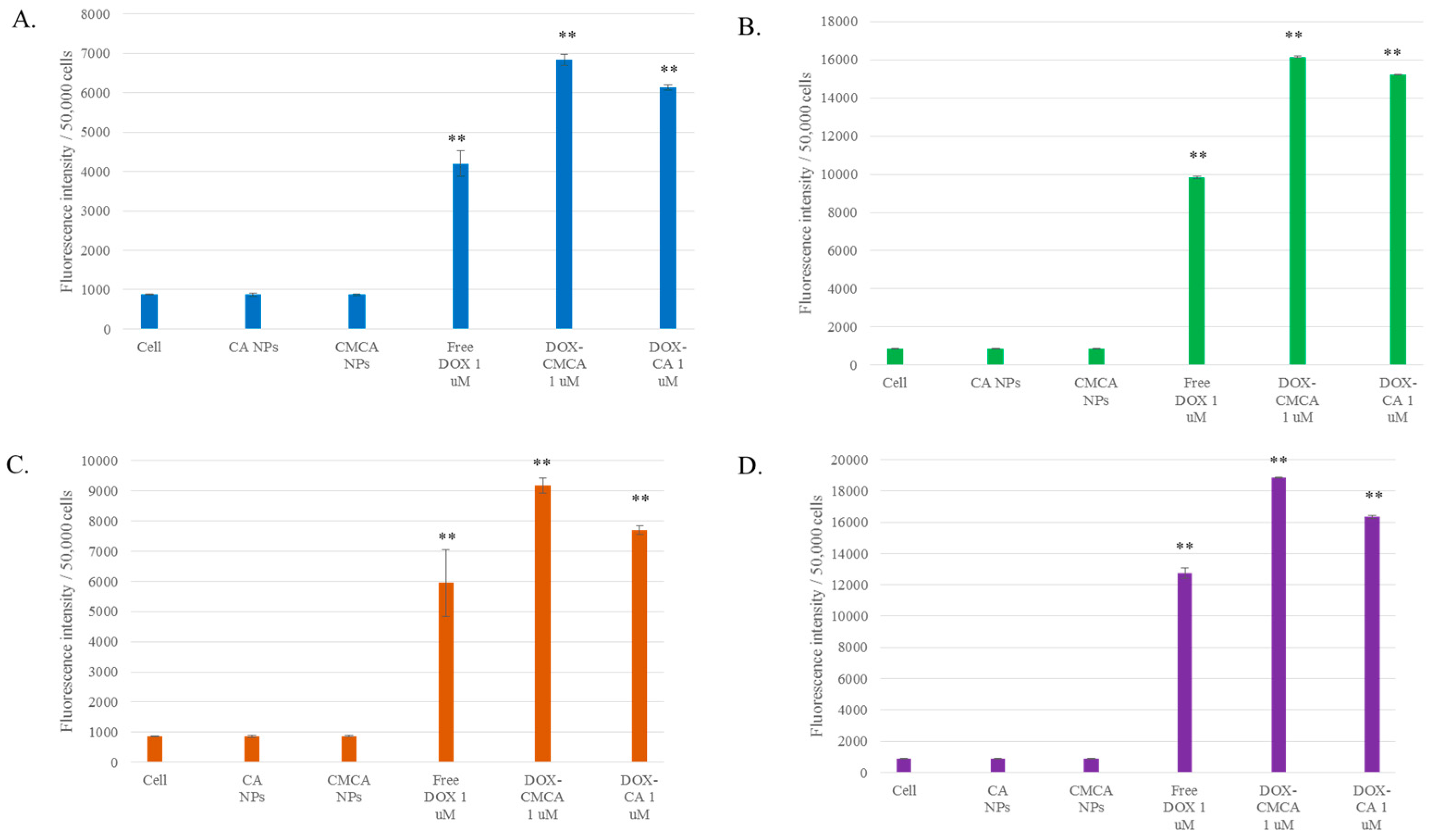
3.5. Cellular Uptake Study by Quantitative Fluorescence Intensity Measurement of DOX
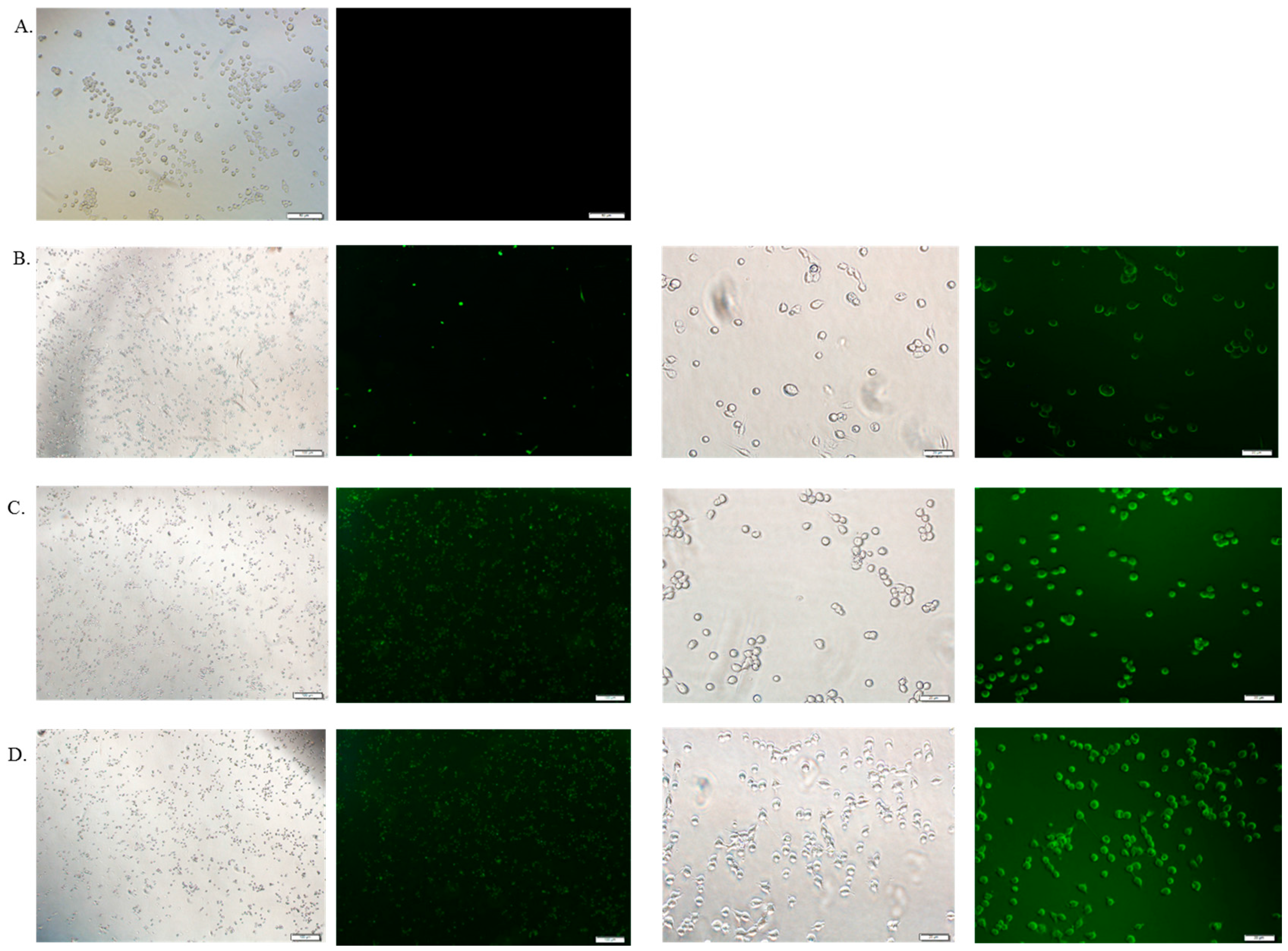
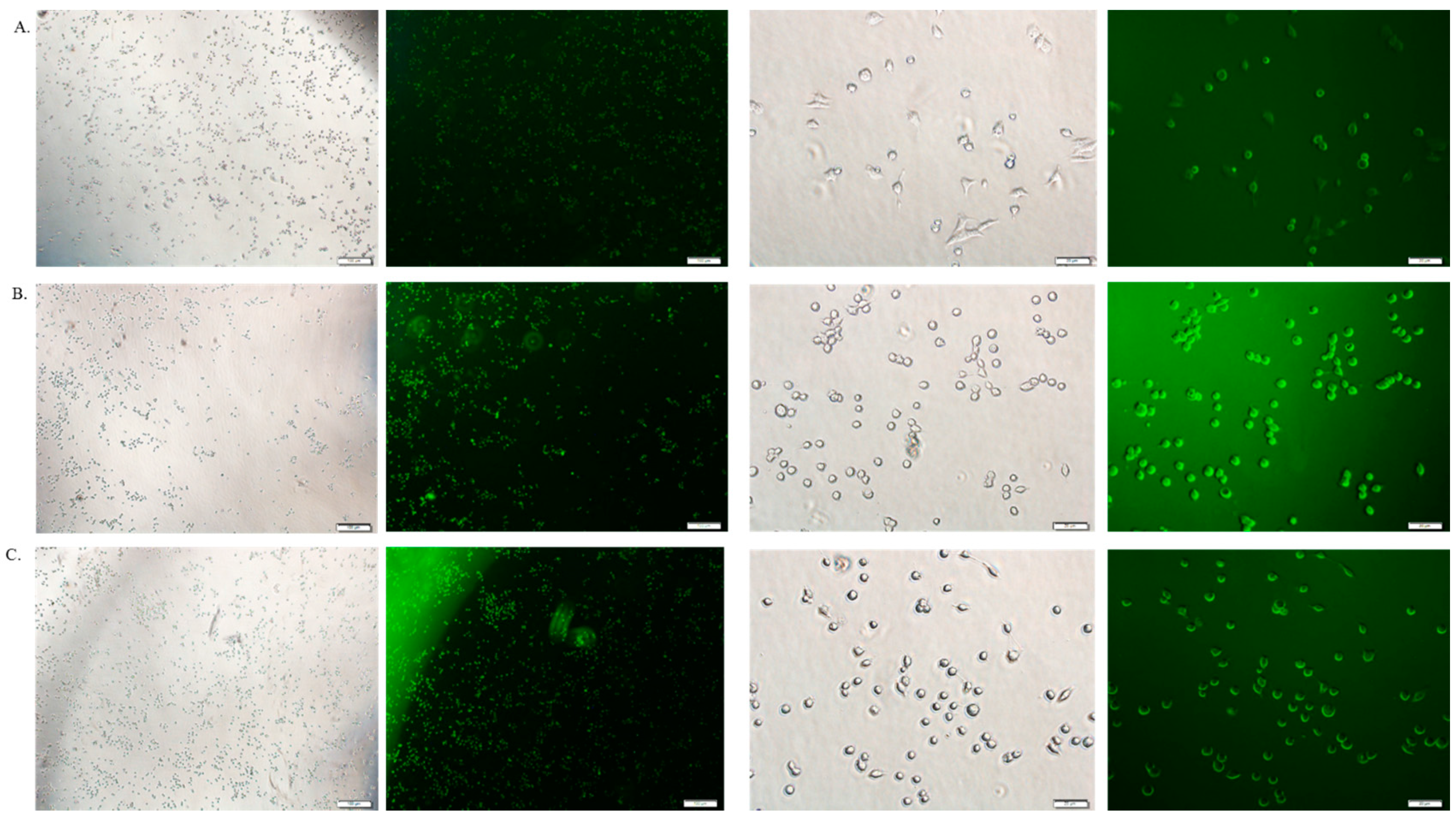
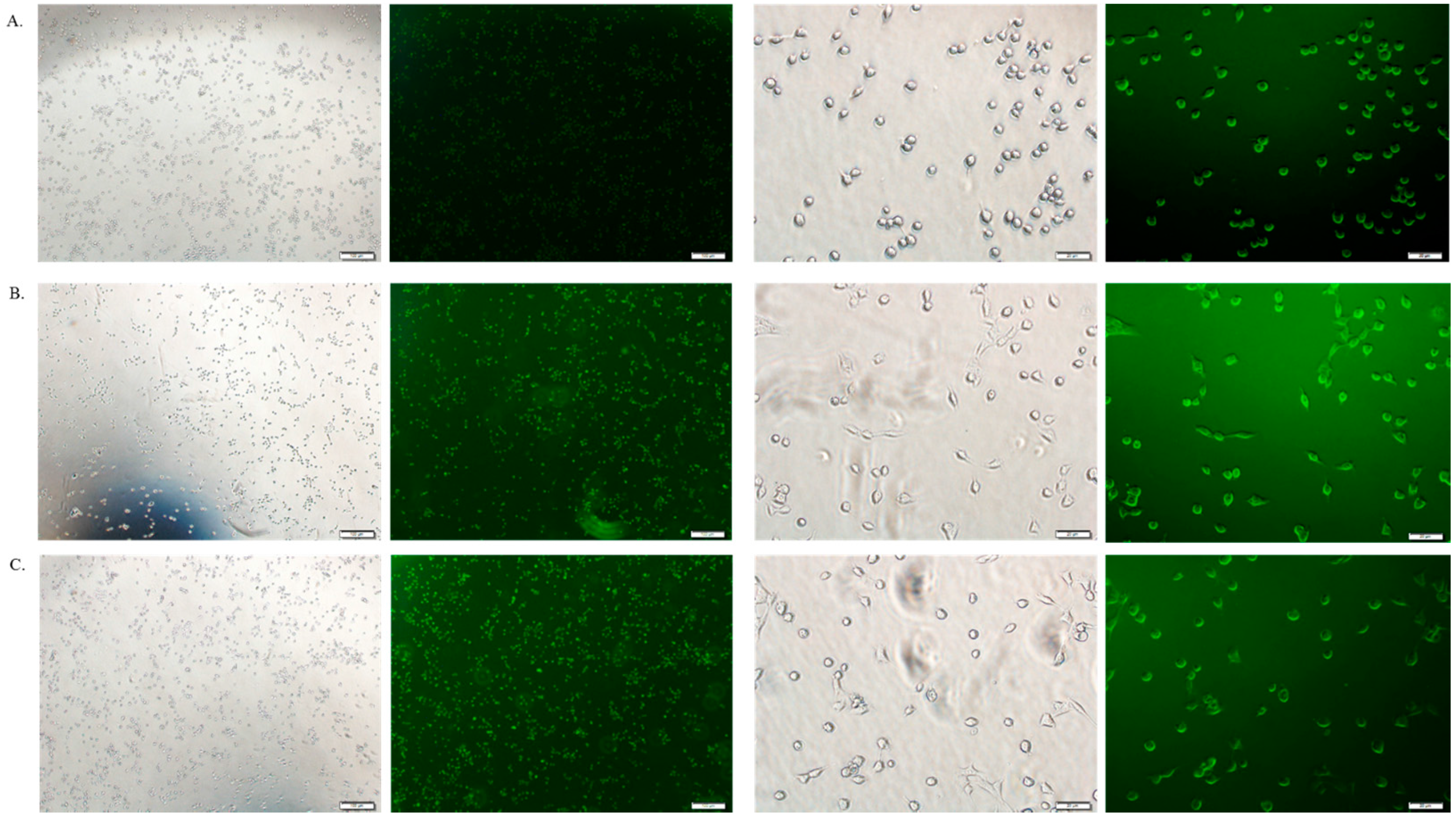
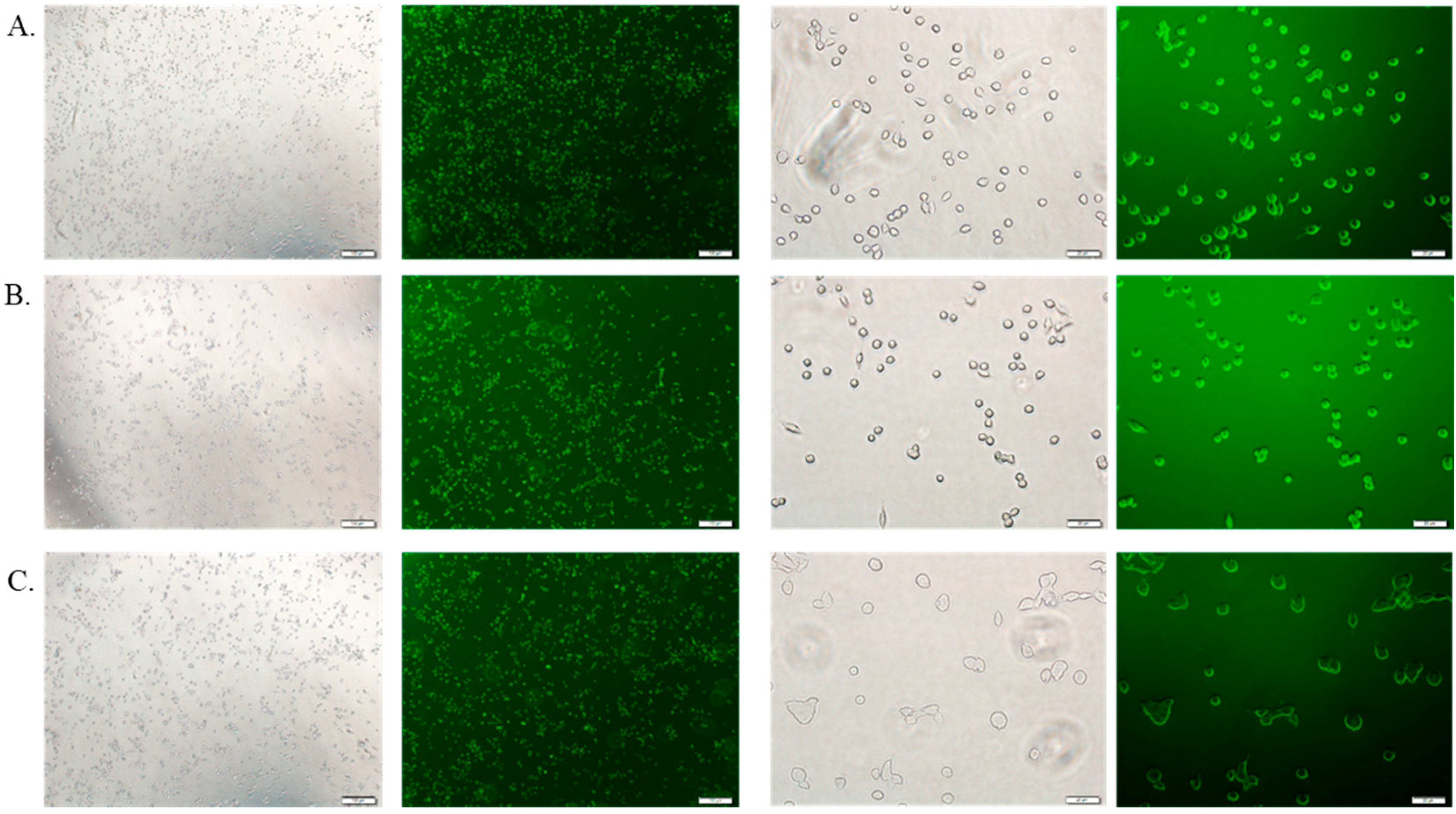
3.6. Cellular Uptake Study by Fluorescence Microscopy
3.7. Estimation of Unbound Citrate in CMCA NPs
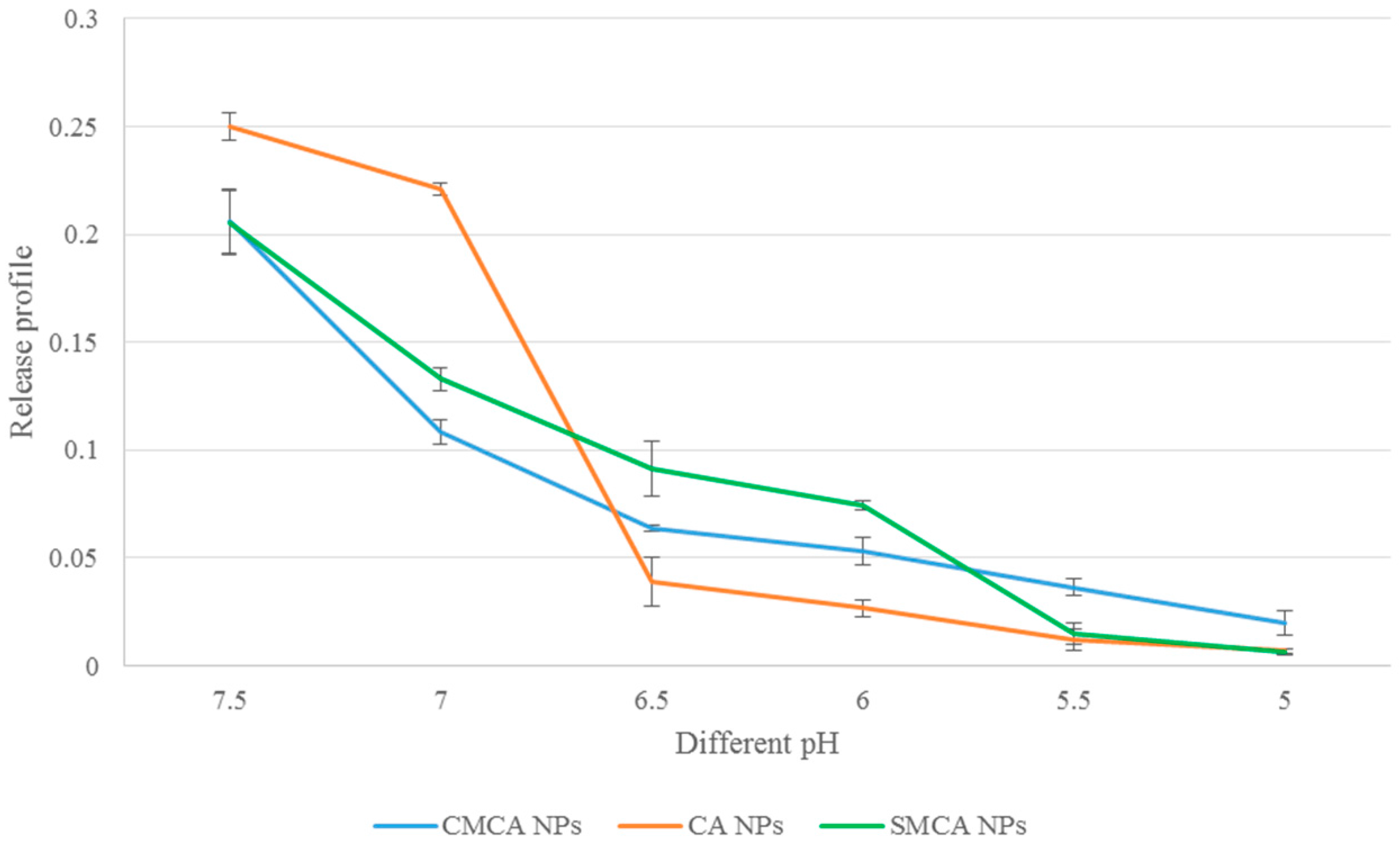
3.8. Confirmation of pH-Sensitivity of CA, CMCA and SMCA NPs
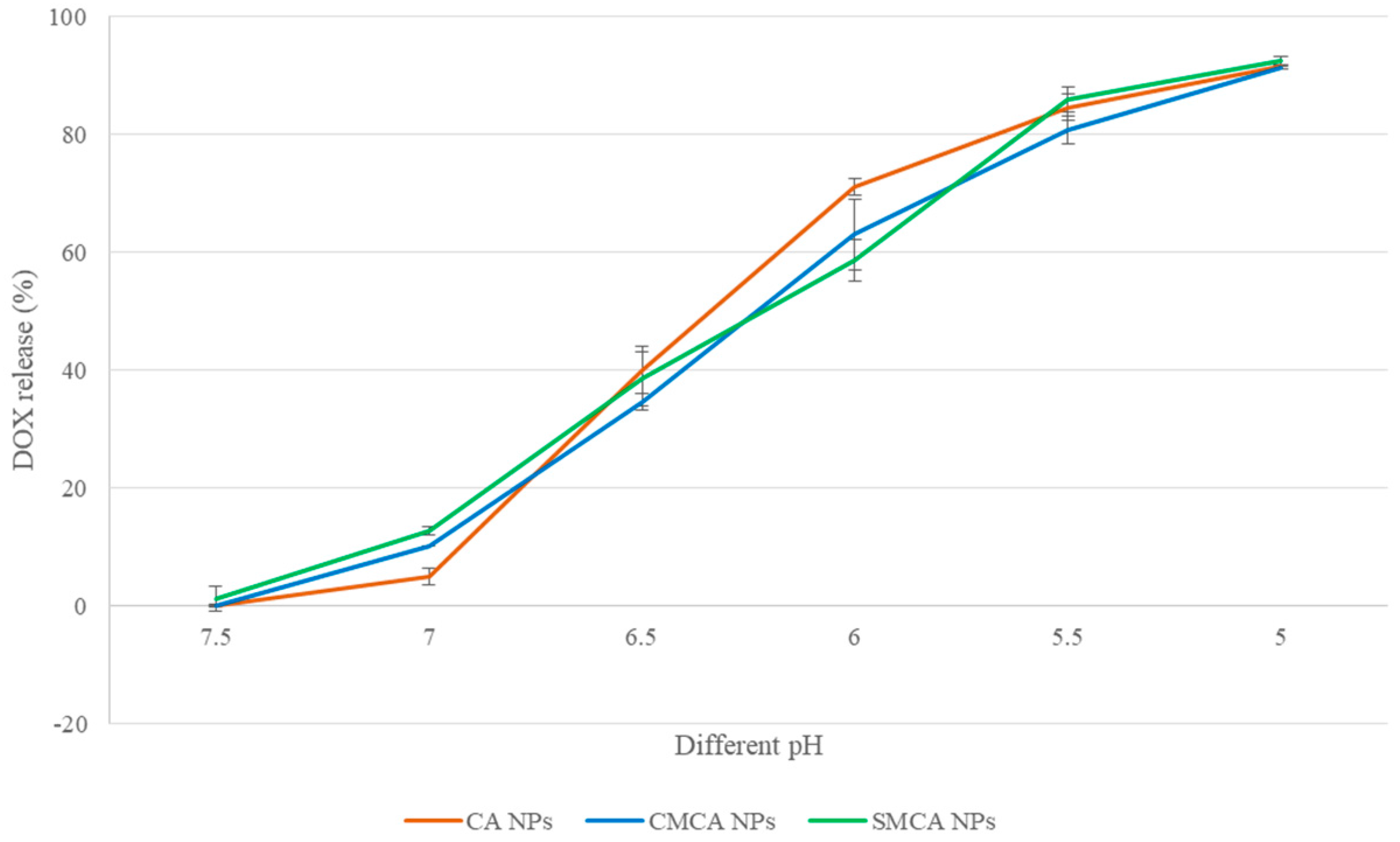
3.9. pH-Dependent DOX Release from NPs
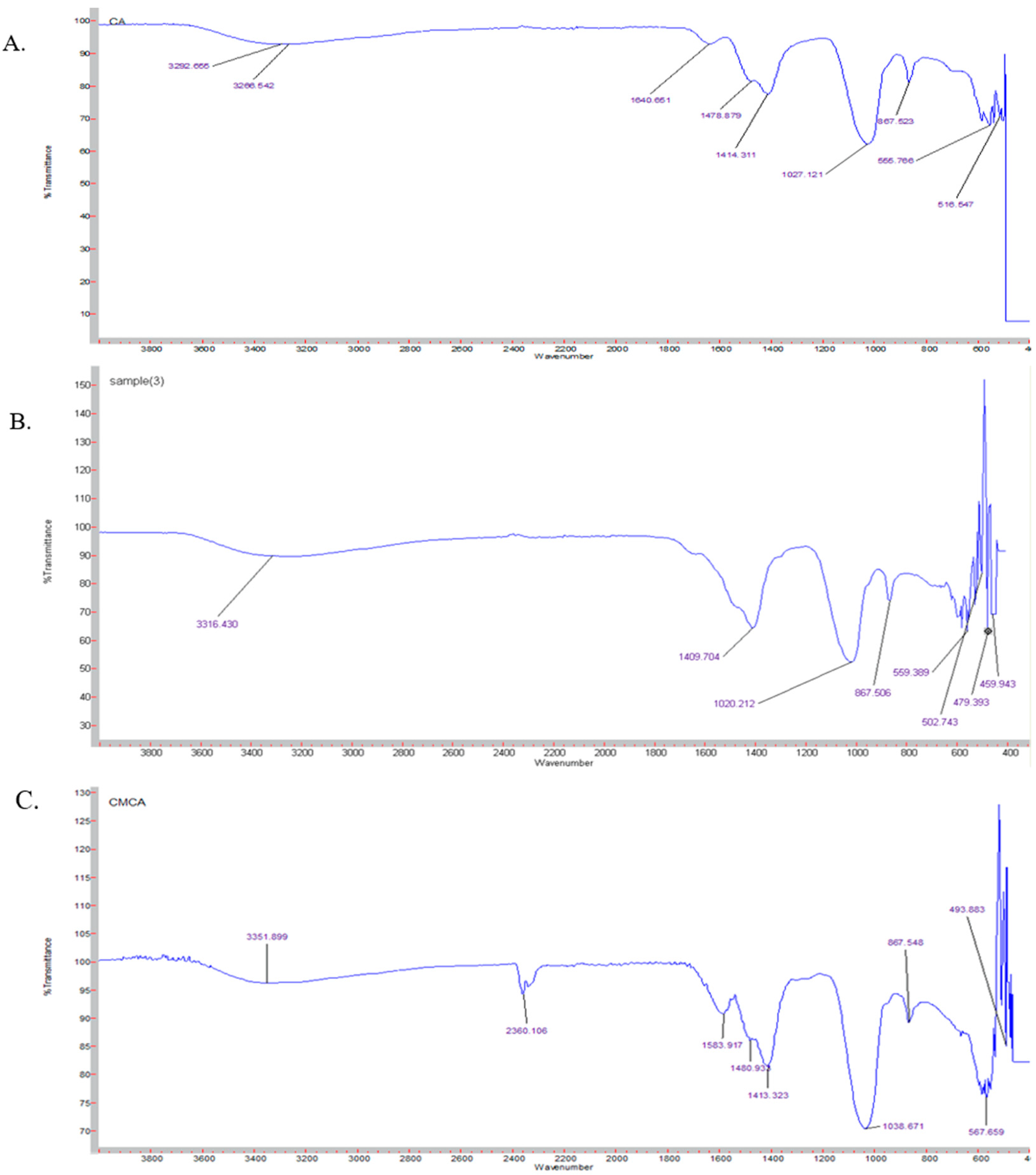
3.10. Characterization of SMCA NPs by FTIR
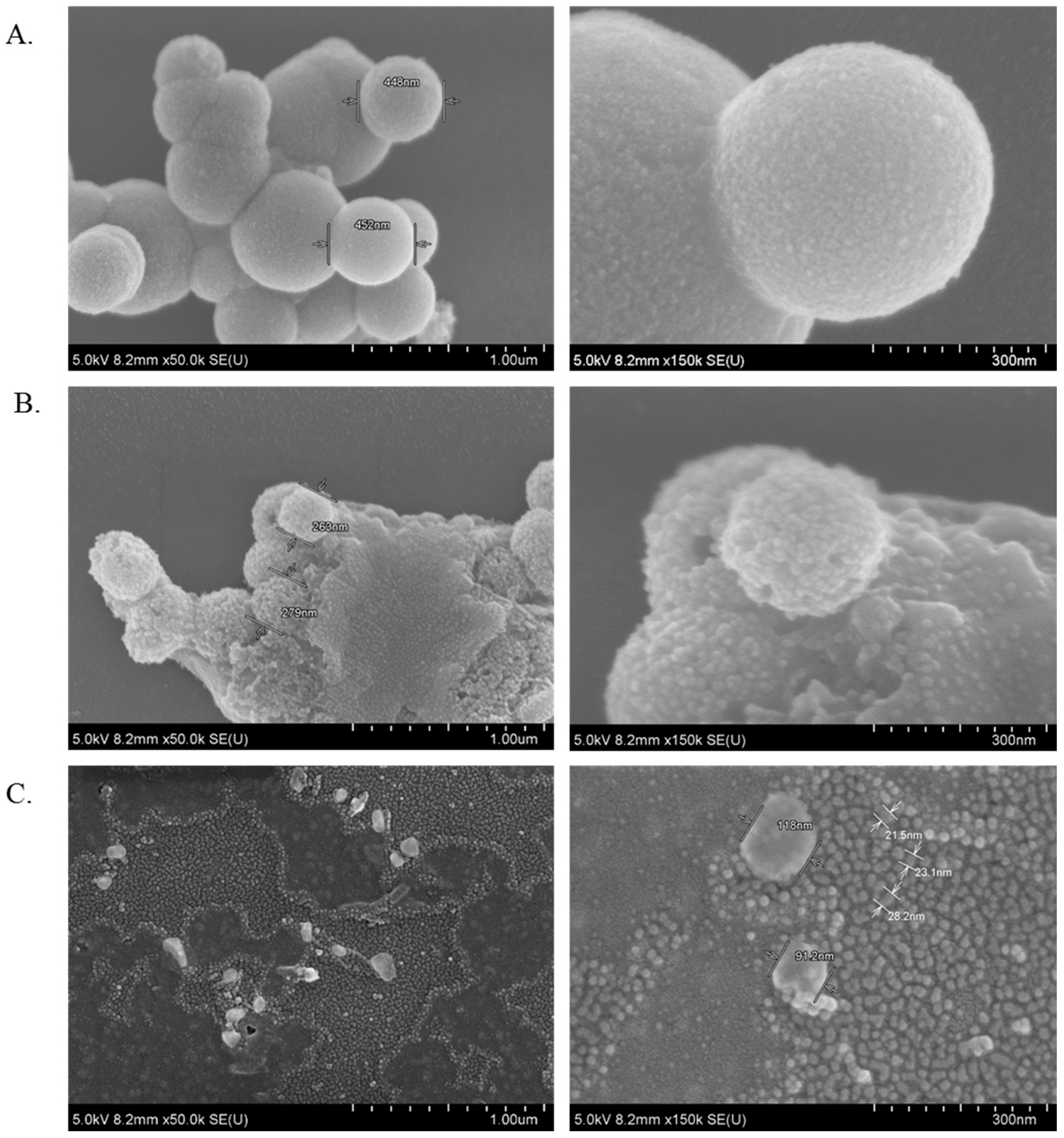
3.11. Characterization of CA, CMCA and SMCA NPs by FE-SEM
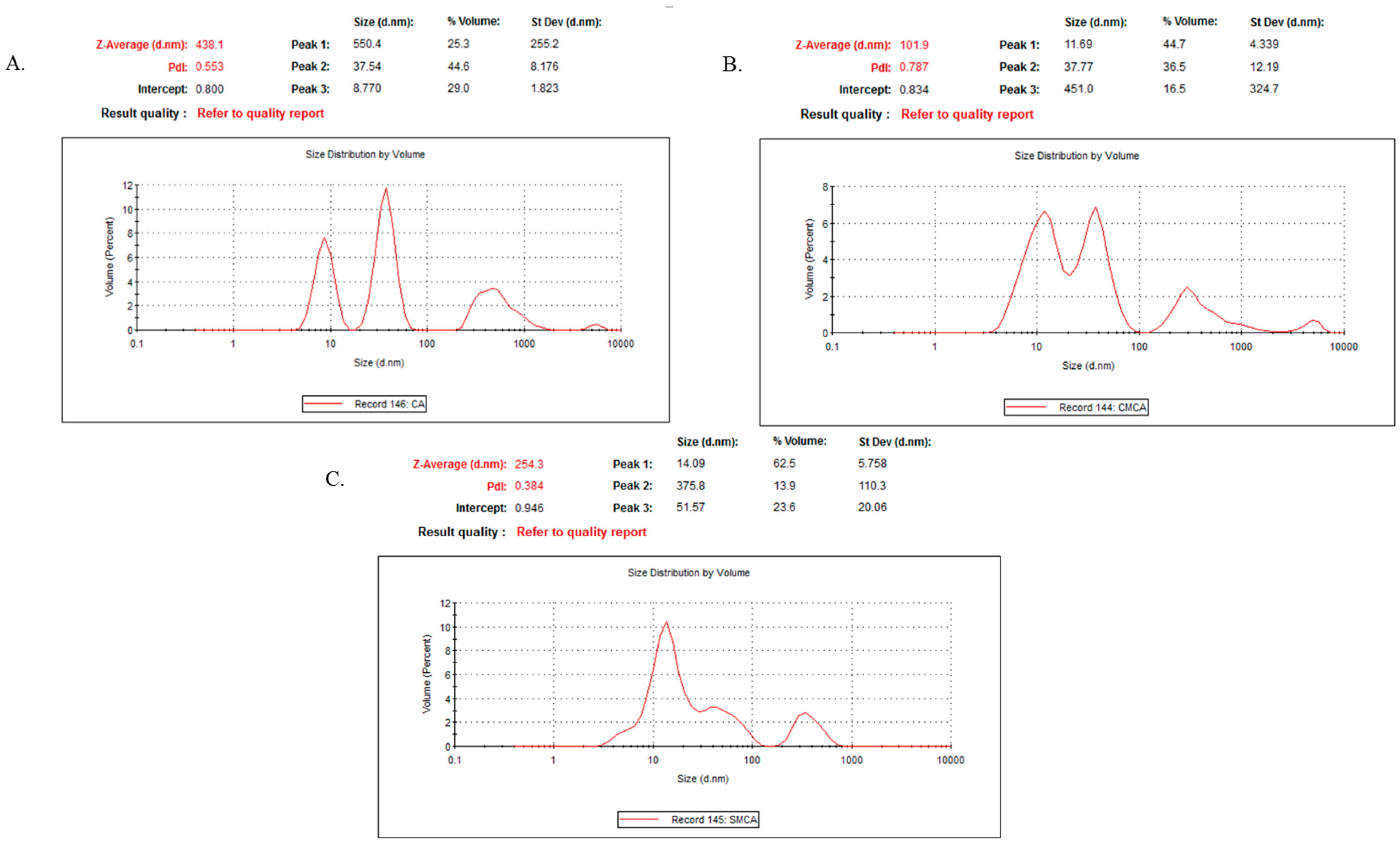
3.12. Characterization of CA, CMCA and SMCA NPs by DLS
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Liu, Y.; Ye, G.; Yan, D.; Zhang, L.; Fan, F.; Feng, J. Role of nab-paclitaxel in metastatic breast cancer: A meta-analysis of randomized clinical trials. Oncotarget 2017, 8, 72950–72958. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Kim, G.J.; Nie, S.; Shin, D.M. Nanotechnology in cancer therapeutics: Bioconjugated nanoparticles for drug delivery. Mol. Cancer Ther. 2006, 5, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Perez-Herrero, E.; Fernandez-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.A.; Randolph, J.K.; Yalowich, J.C.; Ritke, M.K.; Gewirtz, D.A. Interference by doxorubicin with DNA unwinding in mcf-7 breast tumor cells. Mol. Pharmacol. 1994, 45, 649–656. [Google Scholar]
- Momparler, R.L.; Karon, M.; Siegel, S.E.; Avila, F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976, 36, 2891–2895. [Google Scholar] [PubMed]
- Mohan, P.; Rapoport, N. Doxorubicin as a molecular nanotheranostic agent: Effect of doxorubicin encapsulation in micelles or nanoemulsions on the ultrasound-mediated intracellular delivery and nuclear trafficking. Mol. Pharm. 2010, 7, 1959–1973. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Nijsen, J.F.; Hennink, W.E. Polymeric micelles in anticancer therapy: Targeting, imaging and triggered release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef] [PubMed]
- Coelho, S.C.; Pereira, M.C.; Juzeniene, A.; Juzenas, P.; Coelho, M.A. Supramolecular nanoscale assemblies for cancer diagnosis and therapy. J. Control. Release 2015, 213, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Devalapally, H.; Chakilam, A.; Amiji, M.M. Role of nanotechnology in pharmaceutical product development. J. Pharm. Sci. 2007, 96, 2547–2565. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the epr effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Yezhelyev, M.; Yacoub, R.; O’Regan, R. Inorganic nanoparticles for predictive oncology of breast cancer. Nanomedicine (Lond.) 2009, 4, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Mozar, F.S.; Chowdhury, E.H. Surface-modification of carbonate apatite nanoparticles enhances delivery and cytotoxicity of gemcitabine and anastrozole in breast cancer cells. Pharmaceutics 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug. Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Vail, D.M.; Amantea, M.A.; Colbern, G.T.; Martin, F.J.; Hilger, R.A.; Working, P.K. Pegylated liposomal doxorubicin: Proof of principle using preclinical animal models and pharmacokinetic studies. Semin. Oncol. 2004, 31, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H. Diversity of bioactive nanoparticles from biological, chemical, and physical perspectives. In Nanotherapeutics: From Laboratory to Clinic; CRC Press: Boca Raton, FL, USA, 2016; pp. 29–41. [Google Scholar]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 12320–12364. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H. pH-sensitive nano-crystals of carbonate apatite for smart and cell-specific transgene delivery. Expert Opin. Drug Deliv. 2007, 4, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H.; Maruyama, A.; Kano, A.; Nagaoka, M.; Kotaka, M.; Hirose, S.; Kunou, M.; Akaike, T. pH-sensing nano-crystals of carbonate apatite: Effects on intracellular delivery and release of DNA for efficient expression into mammalian cells. Gene 2006, 376, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Tiash, S.; Othman, I.; Rosli, R.; Chowdhury, E.H. Methotrexate- and cyclophosphamide-embedded pure and strontiumsubstituted carbonate apatite nanoparticles for augmentation of chemotherapeutic activities in breast cancer cells. Curr. Drug Deliv. 2014, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H.; Akaike, T. A bio-recognition device developed onto nano-crystals of carbonate apatite for cell-targeted gene delivery. Biotech. Bioeng. 2005, 90, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Kingston, J.K.; Bayly, W.M.; Sellon, D.C.; Meyers, K.M.; Wardrop, K.J. Effects of sodium citrate, low molecular weight heparin, and prostaglandin E1 on aggregation, fibrinogen binding, and enumeration of equine platelets. Am. J. Vet. Res. 2001, 62, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Whitcroft, K.L.; Merkonidis, C.; Cuevas, M.; Haehner, A.; Philpott, C.; Hummel, T. Intranasal sodium citrate solution improves olfaction in post-viral hyposmia. Rhinology 2016, 54, 368–374. [Google Scholar] [PubMed]
- Hoffman, J.F. Annual Review of Physiology; Hoffman, J.F., De Weer, P., Eds.; Annual Reviews Inc.: Palo Alto, CA, USA, 1999; Volume 61, ISBN 9780824303518. [Google Scholar]
- Chowdhury, E.H.; Akaike, T. High performance DNA nano-carriers of carbonate apatite: Multiple factors in regulation of particle synthesis and transfection efficiency. Int. J. Nanomed. 2007, 2, 101–106. [Google Scholar] [CrossRef]
- Lalatsa, A.; Schatzlein, A.G.; Mazza, M.; Le, T.B.; Uchegbu, I.F. Amphiphilic poly(L-amino acids)—New materials for drug delivery. J. Control. Release 2012, 161, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Subbenaik, S.C. Physical and chemical nature of nanoparticles. In Plant Nanotechnology; Springer: Cham, Switzerland, 2016; pp. 15–27. ISBN 9783319421520. [Google Scholar]
- Kaasalainen, M.; Aseyev, V.; Von Haartman, E.; Karaman, D.S.; Makila, E.; Tenhu, H.; Rosenholm, J.; Salonen, J. Size, stability, and porosity of mesoporous nanoparticles characterized with light scattering. Nanoscale Res. Lett. 2017, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.H.; Lee, S.; Hwang, Y.S. Characterization of silver nanoparticles under environmentally relevant conditions using asymmetrical flow field-flow fractionation (AF4). PLoS ONE 2015, 10, e0143149. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H.; Kunou, M.; Nagaoka, M.; Kundu, A.K.; Hoshiba, T.; Akaike, T. High-efficiency gene delivery for expression in mammalian cells by nanoprecipitates of Ca-Mg phosphate. Gene 2004, 341, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Sui, J.; He, M.; Xu, Z.; Sun, Y.; Liang, J.; Fan, Y.; Zhang, X. Reduction-degradable polymeric micelles decorated with parg for improving anticancer drug delivery efficacy. ACS Appl. Mater. Interfaces 2016, 8, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the epr effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Serra, A.C.; Coelho, J.F. Bioabsorbable polymers in cancer therapy: Latest developments. EPMA J. 2015, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Huang, L. Stealth nanoparticles: High density but sheddable peg is a key for tumor targeting. J. Control. Release 2010, 145, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The epr effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Cheng, S.H.; Huang, I.P.; Souris, J.S.; Yang, C.S.; Mou, C.Y.; Lo, L.W. Intracellular pH-responsive mesoporous silica nanoparticles for the controlled release of anticancer chemotherapeutics. Angew. Chem. Int. Ed. Engl. 2010, 49, 8214–8219. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.; Huck, W.T.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xue, M.; Xia, T.; Zhao, Y.L.; Tamanoi, F.; Stoddart, J.F.; Zink, J.I.; Nel, A.E. Autonomous in vitro anticancer drug release from mesoporous silica nanoparticles by pH-sensitive nanovalves. J. Am. Chem. Soc. 2010, 132, 12690–12697. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shi, J.; Shen, W.; Dong, X.; Feng, J.; Ruan, M.; Li, Y. Stimuli-responsive controlled drug release from a hollow mesoporous silica sphere/polyelectrolyte multilayer core-shell structure. Angew. Chem. Int. Ed. Engl. 2005, 44, 5083–5087. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H. Ph-responsive magnesium- and carbonate-substituted apatite nano-crystals for efficient and cell-targeted delivery of transgenes. OJGen Open J. Gene. 2013, 03, 38–44. [Google Scholar] [CrossRef]
- Hossain, S.; Stanislaus, A.; Chua, M.J.; Tada, S.; Tagawa, Y.; Chowdhury, E.H.; Akaike, T. Carbonate apatite-facilitated intracellularly delivered sirna for efficient knockdown of functional genes. J. Control. Release 2010, 147, 101–108. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Takeshima, S.N.; Tada, S.; Akaike, T.; Ito, Y.; Aida, Y. pH-sensitive carbonate apatite nanoparticles as DNA vaccine carriers enhance humoral and cellular immunity. Vaccine 2014, 32, 6199–6205. [Google Scholar] [CrossRef] [PubMed]
- Dzamukova, M.R.; Naumenko, E.A.; Lvov, Y.M.; Fakhrullin, R.F. Enzyme-activated intracellular drug delivery with tubule clay nanoformulation. Sci. Rep. 2015, 5, 10560. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, Y.; Chen, M.; Hu, Y.; Jiang, W.; Zhou, L.; Li, S.; Xu, M.; Zhao, Q.; Wan, R. Enhanced antitumor efficacy of doxorubicin-encapsulated halloysite nanotubes. Int. J. Nanomed. 2018, 13, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, J.; Gao, H.; Shen, Y.; Jiang, L.; Zhou, C.; Li, Y.; He, R.; Liu, M. Folate-conjugated halloysite nanotubes, an efficient drug carrier, deliver doxorubicin for targeted therapy of breast cancer. ACS Appl. Nano Mater. 2018, 1, 595–608. [Google Scholar] [CrossRef]
- Yuan, H.; Bao, X.; Du, Y.Z.; You, J.; Hu, F.Q. Preparation and evaluation of SiO2-deposited stearic acid-g-chitosan nanoparticles for doxorubicin delivery. Int. J. Nanomed. 2012, 7, 5119–5128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, Y.; Chen, C.K.; Law, W.C.; Weinheimer, E.; Sengupta, S.; Prasad, P.N.; Cheng, C. Polylactide-graft-doxorubicin nanoparticles with precisely controlled drug loading for pH-triggered drug delivery. Biomacromolecules 2014, 15, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Prabaharan, M.; Grailer, J.J.; Pilla, S.; Steeber, D.A.; Gong, S. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials 2009, 30, 5757–5766. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Bickerton, S.; Zhuang, J.; Thayumanavan, S. Ligand-decorated nanogels: Fast one-pot synthesis and cellular targeting. Biomacromolecules 2012, 13, 1515–1522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qi, J.; Yao, P.; He, F.; Yu, C.; Huang, C. Nanoparticles with dextran/chitosan shell and bsa/chitosan core—Doxorubicin loading and delivery. Int. J. Pharm. 2010, 393, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Haque, S.A.; Yang, S.T.; Luo, P.G.; Gu, L.; Kitaygorodskiy, A.; Li, H.; Lacher, S.; Sun, Y.P. Aqueous compatible fullerene-doxorubicin conjugates. J. Phys. Chem. C. Nanomater. Interfaces 2009, 113, 17768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Akbarzadeh, A.; Zarghami, N.; Mikaeili, H.; Asgari, D.; Goganian, A.M.; Khiabani, H.K.; Samiei, M.; Davaran, S. Synthesis, characterization, and in vitro evaluation of novel polymer-coated magnetic nanoparticles for controlled delivery of doxorubicin. Nanotechnol. Sci. Appl. 2012, 5, 13–25. [Google Scholar] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of DOX | MCF-7 | 4T1 |
|---|---|---|
| 1 pM | 13.35 ± 1.92 | 4.14 ± 1.4 |
| 10 pM | 16.90 ± 1.34 | 12.42 ± 1.62 |
| 100 pM | 10.85 ± 1.87 | 13.45 ± 2.27 |
| 1 nM | 15.66 ± 2.69 | 17.72 ± 2.58 |
| 10 nM | 15.12 ± 3.25 | 15.78 ± 4.32 |
| 100 nM | 20.99 ± 1.93 | 19.40 ±1.34 |
| 1 µM | 25.62 ± 0.82 | 11.77 ± 1.25 |
| Concentration of DOX | MCF-7 | 4T1 |
|---|---|---|
| 1 pM | 1.78 ± 2.52 | 0.91 ± 2.58 |
| 10 pM | 2.67 ± 3.5 | 7.37 ± 0.22 |
| 100 pM | 1.60 ± 2.97 | 3.62 ± 3.01 |
| 1 nM | 1.42 ± 1.92 | 1.16 ± 1.24 |
| 10 nM | 1.94 ± 2.7 | 3.23 ±2.20 |
| 100 nM | 1.11 ± 2.93 | 3.62 ± 4.25 |
| 1 µM | 11.03 ± 1.34 | 5.04 ± 5.1 |
| Concentration of DOX | MCF-7 | 4T1 |
|---|---|---|
| 1 pM | 2.89 ± 6.68 | 2.99 ± 6.76 |
| 10 pM | 17.33 ± 2.24 | 7.49 ± 2.79 |
| 100 pM | 7.64 ± 2.93 | 10.64 ± 1.04 |
| 1 nM | 7.52 ± 0.54 | 11.64 ± 1.94 |
| 10 nM | 6.25 ± 2.56 | 16.93 ± 1.41 |
| 100 nM | 8.55 ± 2.11 | 20.88 ± 7.48 |
| 1 µM | 7.60 ± 2.92 | 22.53 ± 1.29 |
| Formulation | % Cellular Uptake 1 h of Treatment | % Cellular Uptake 4 h of Treatment |
|---|---|---|
| DOX | 22.4 ± 1.73 | 31.71 ± 5.92 |
| DOX-CA | 32.69 ± 0.38 | 41.09 ± 0.79 |
| DOX-CMCA | 36.45 ± 0.72 | 48.93 ± 1.32 |
| Formulation | % Cellular Uptake 1 h of Treatment | % Cellular Uptake 4 h of Treatment |
|---|---|---|
| DOX | 24.75 ± 0.14 | 32.07 ± 0.82 |
| DOX-CA | 38.26 ± 0.07 | 41.14 ± 0.26 |
| DOX-CMCA | 40.64 ± 0.12 | 47.41 ± 0.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehbuba Hossain, S.; Chowdhury, E.H. Citrate- and Succinate-Modified Carbonate Apatite Nanoparticles with Loaded Doxorubicin Exhibit Potent Anticancer Activity against Breast Cancer Cells. Pharmaceutics 2018, 10, 32. https://doi.org/10.3390/pharmaceutics10010032
Mehbuba Hossain S, Chowdhury EH. Citrate- and Succinate-Modified Carbonate Apatite Nanoparticles with Loaded Doxorubicin Exhibit Potent Anticancer Activity against Breast Cancer Cells. Pharmaceutics. 2018; 10(1):32. https://doi.org/10.3390/pharmaceutics10010032
Chicago/Turabian StyleMehbuba Hossain, Sultana, and Ezharul Hoque Chowdhury. 2018. "Citrate- and Succinate-Modified Carbonate Apatite Nanoparticles with Loaded Doxorubicin Exhibit Potent Anticancer Activity against Breast Cancer Cells" Pharmaceutics 10, no. 1: 32. https://doi.org/10.3390/pharmaceutics10010032
APA StyleMehbuba Hossain, S., & Chowdhury, E. H. (2018). Citrate- and Succinate-Modified Carbonate Apatite Nanoparticles with Loaded Doxorubicin Exhibit Potent Anticancer Activity against Breast Cancer Cells. Pharmaceutics, 10(1), 32. https://doi.org/10.3390/pharmaceutics10010032