Solvation and Aggregation of Meta-Aminobenzoic Acid in Water: Density Functional Theory and Molecular Dynamics Study


Abstract
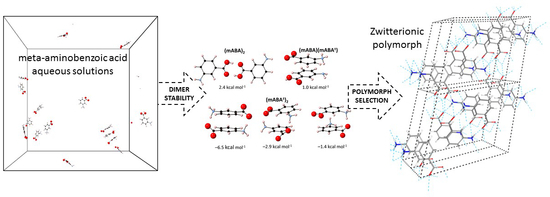
1. Introduction
2. Computational Methods
2.1. Density Functional Theory Calculations
2.2. Molecular Dynamics Simulations
3. Results
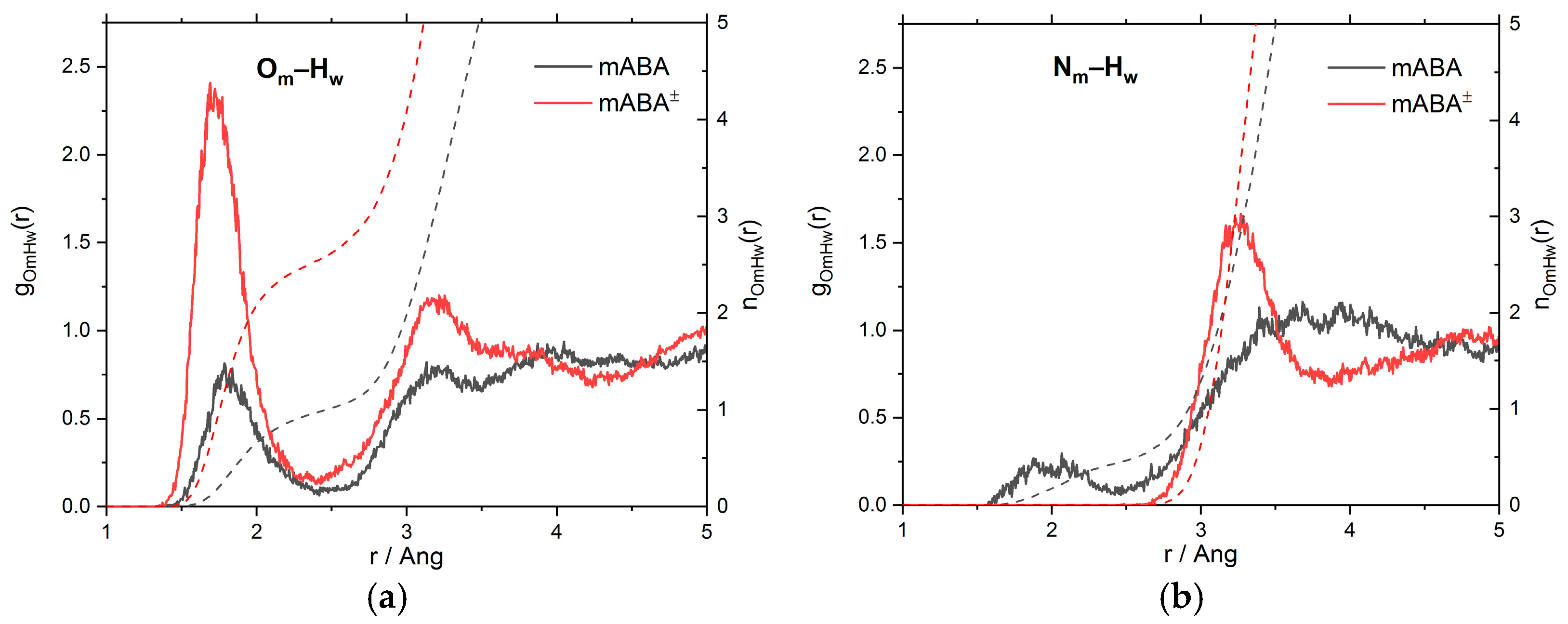
3.1. Intermolecular Properties and Hydration Structure
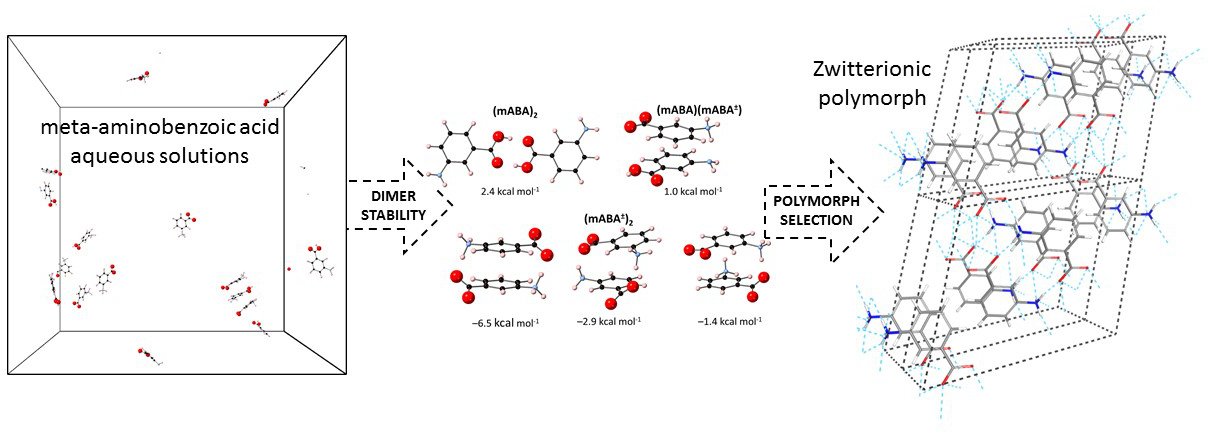
3.2. Dimerization of Meta-Aminobenzoic Acid
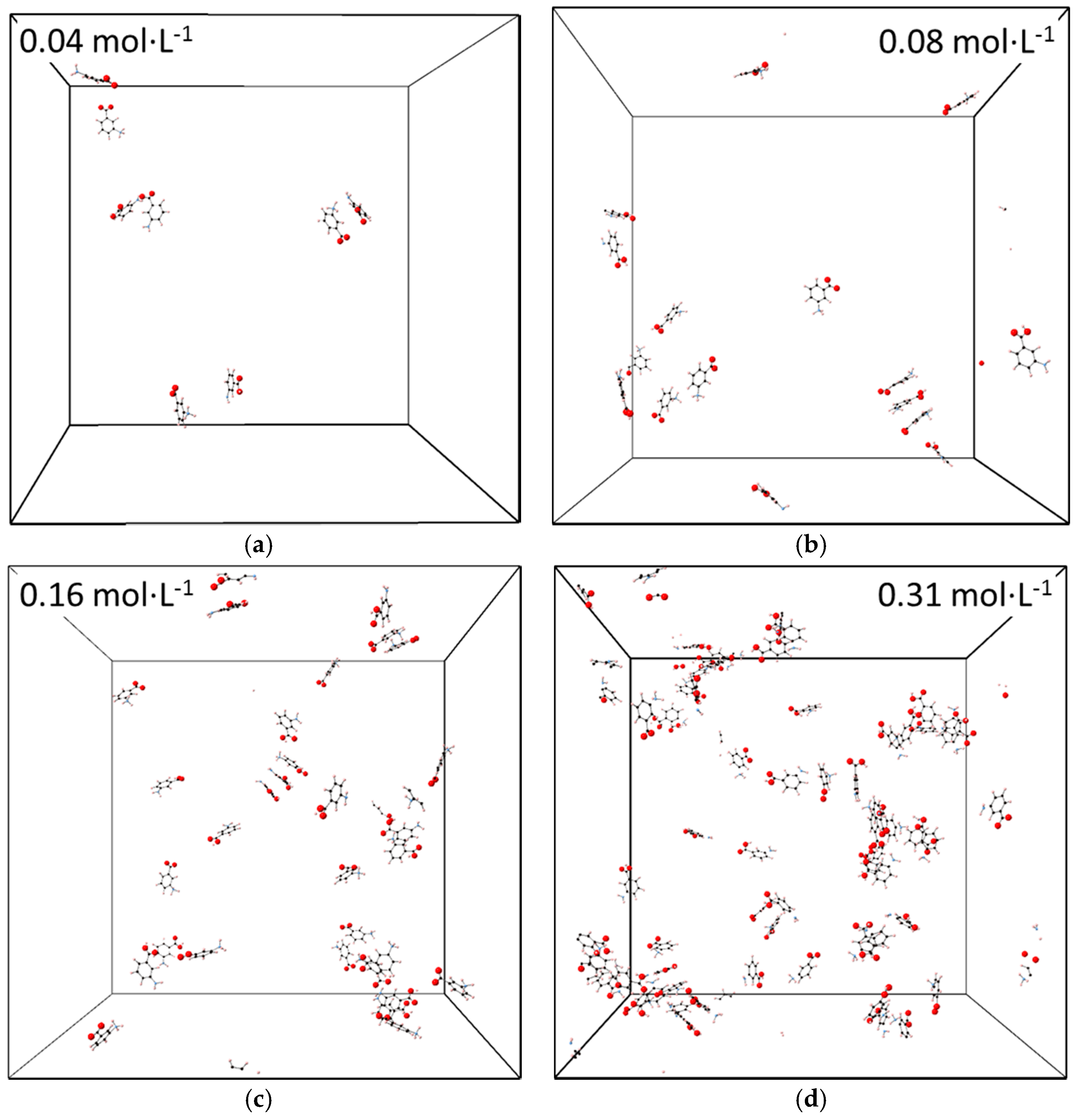
3.3. Molecular Aggregation in Mixed mABA–mABA± Aqueous Solutions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Palafox, M.A.; Gill, M.; Núñez, J.L. Meta-aminobenzoic acid: Structures and spectral characteristics. Spectrosc. Lett. 1996, 29, 609–629. [Google Scholar] [CrossRef]
- Williams, P.A.; Hughes, C.E.; Lim, G.K.; Kariuki, B.M.; Harris, K.D. Discovery of a new system exhibiting abundant polymorphism: M-aminobenzoic acid. Cryst. Growth Des. 2012, 12, 3104–3113. [Google Scholar] [CrossRef]
- Théorêt, A. Structure moléculaire et zwitterion des acides aminés—I. Spectres infrarouges des acides o, m et p-aminobenzoïques dans différentes formes cristallines. Spectrochim. Acta Part A Mol. Spectrosc. 1971, 27, 11–18. [Google Scholar] [CrossRef]
- Svärd, M.; Nordström, F.L.; Jasnobulka, T.; Rasmuson, Å.C. Thermodynamics and nucleation kinetics of m-aminobenzoic acid polymorphs. Cryst. Growth Des. 2010, 10, 195–204. [Google Scholar] [CrossRef]
- Kumar, S.S.; Nangia, A. A solubility comparison of neutral and zwitterionic polymorphs. Cryst. Growth Des. 2014, 14, 1865–1881. [Google Scholar] [CrossRef]
- Lahav, M.; Leiserowitz, L. The effect of solvent on crystal growth and morphology. Chem. Eng. Sci. 2001, 56, 2245–2253. [Google Scholar] [CrossRef]
- Ter Horst, J.H.; Geertman, R.M.; van Rosmalen, G.M. The effect of solvent on crystal morphology. J. Cryst. Growth 2001, 230, 277–284. [Google Scholar] [CrossRef]
- Gaines, E.; Maisuria, K.; Di Tommaso, D. The role of solvent in the self-assembly of m-aminobenzoic acid: A density functional theory and molecular dynamics study. CrystEngComm 2016, 18, 2937–2948. [Google Scholar] [CrossRef]
- Blagden, N.; Davey, R.J. Polymorph selection: Challenges for the future? Cryst. Growth Des. 2003, 3, 873–885. [Google Scholar] [CrossRef]
- Musumeci, D.; Hunter, C.A.; McCabe, J.F. Solvent effects on acridine polymorphism. Cryst. Growth Des. 2010, 10, 1661–1664. [Google Scholar] [CrossRef]
- Kitamura, M.; Umeda, E.; Miki, K. Mechanism of solvent effect in polymorphic crystallization of BPT. Ind. Eng. Chem. Res. 2012, 51, 12814–12820. [Google Scholar] [CrossRef]
- Hughes, C.E.; Williams, P.A.; Harris, K.D.M. “CLASSIC NMR”: An In-Situ NMR Strategy for Mapping the Time-Evolution of Crystallization Processes by Combined Liquid-State and Solid-State Measurements. Angew. Chem. Int. Ed. 2014, 53, 8939–8943. [Google Scholar] [CrossRef] [PubMed]
- Kumler, W.D. Acidic and basic dissociation constants and structure. J. Org. Chem. 1955, 20, 700–706. [Google Scholar] [CrossRef]
- Cohn, E.J.; Edsall, J.T. Proteins, Amino Acids and Peptides; Reinhold Publishing Corporation: New York, NY, USA, 1943. [Google Scholar]
- Bjerrun, N.Z. Die Konstitution der Ampholyte, besonders der Aminosäuren, und ihre Dissoziationskonstanten. Phys. Chem. 1923, 104, 147–173. [Google Scholar]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian Software; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acta 2008, 120, 215–241. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J.Phys.Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Klamt, A.; Coote, M.L. Comment on the correct use of continuum solvent models. J. Phys. Chem. A 2010, 114, 13442–13444. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.F.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. The solvation, partitioning, hydrogen bonding, and dimerization of nucleotide bases: A multifaceted challenge for quantum chemistry. Phys. Chem. Chem. Phys. 2011, 13, 10908–10922. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Besley, N.A. Structural optimization of molecular clusters with density functional theory combined with basin hopping. J. Chem. Phys. 2012, 137, 134106. [Google Scholar] [CrossRef] [PubMed]
- Montero, L.A.; Esteva, A.M.; Molina, J.; Zapardiel, A.; Herna, L.; Màrquez, H.; Acosta, A. A theoretical approach to analytical properties of 2, 4-diamino-5-phenylthiazole in water solution. Tautomerism and dependence on pH. J. Am. Chem. Soc. 1998, 120, 12023–12033. [Google Scholar] [CrossRef]
- Sanchez-Garcia, E.; Studentkowski, M.; Montero Luis, A.; Sander, W. Noncovalent Complexes between Dimethyl Ether and Formic Acid—An Ab Initio and Matrix Isolation Study. ChemPhysChem 2005, 6, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Salvalaglio, M.; Perego, C.; Giberti, F.; Mazzotti, M.; Parrinello, M. Molecular-dynamics simulations of urea nucleation from aqueous solution. Proc. Natl. Acad. Sci. USA 2014, 112, E6–E14. [Google Scholar] [CrossRef] [PubMed]
- Salvalaglio, M.; Giberti, F.; Parrinello, M. 1,3,5-Tris (4-bromophenyl) benzene prenucleation clusters from metadynamics. Acta Crystallogr. Sect. C Struct. Chem. 2014, 70, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Toroz, D.; Hammond, R.B.; Roberts, K.J.; Harris, S.; Ridley, T. Molecular dynamics simulations of organic crystal dissolution: The lifetime and stability of the polymorphic forms of para-amino benzoic acid in aqueous environment. J. Cryst. Growth 2014, 401, 38–43. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Gaigeot, M.-P.; Sprik, M. Ab initio molecular dynamics study of uracil in aqueous solution. J. Phys. Chem. B 2004, 108, 7458–7467. [Google Scholar] [CrossRef]
- Tang, E.; Di Tommaso, D.; de Leeuw, N.H. Hydrogen transfer and hydration properties of HnPO4 3 − n (n = 0–3) in water studied by first principles molecular dynamics simulations. J. Chem. Phys. 2009, 130, 234502. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.J.; Dent, G.; Mughal, R.K.; Parveen, S. Concerning the relationship between structural and growth synthons in crystal nucleation: Solution and crystal chemistry of carboxylic acids as revealed through IR spectroscopy. Cryst. Growth Des. 2006, 6, 1788–1796. [Google Scholar] [CrossRef]
- Du, W.; Cruz-Cabeza, A.J.; Woutersen, S.; Davey, R.J.; Yin, Q. Can the study of self-assembly in solution lead to a good model for the nucleation pathway? The case of tolfenamic acid. Chem. Sci. 2015, 6, 3515–3524. [Google Scholar] [CrossRef]
- Khamar, D.; Zeglinski, J.; Mealey, D.; Rasmuson, Å.C. Investigating the Role of Solvent–Solute Interaction in Crystal Nucleation of Salicylic Acid from Organic Solvents. J. Am. Chem. Soc. 2014, 136, 11664–11673. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mABA | mABA± | |
|---|---|---|
| 1.79 | 1.72 | |
| 0.81 | 2.38 | |
| 2.50 | 2.52 | |
| 0.09 | 0.17 | |
| 9.00 | 14.00 | |
| 1.0 | 2.6 | |
| 1.88 | - | |
| 0.27 | - | |
| 2.46 | - | |
| 0.06 | - | |
| 4.50 | - | |
| 0.5 | 0 |
| mABA | mABA± | |
|---|---|---|
| 1.51 | - | |
| 3.06 | - | |
| 2.31 | - | |
| 0.01 | - | |
| 306.00 | - | |
| 1.0 | - | |
| - | 1.77 | |
| - | 2.15 | |
| - | 2.23 | |
| - | 0.03 | |
| - | 71.7 | |
| 0 | 1.0 |
| Reaction | |||
|---|---|---|---|
| 2 mABA → (mABA)2 | −18.3 | –6.6 | –0.1 1 |
| 2.4 2 | |||
| mABA + mABA± → (mABA)(mABA±) | 1.3 2 | ||
| 2 mABA± → (mABA±)2 | – | – | –5.8 2 |
| A* | B* | C* | A | B | C | |
|---|---|---|---|---|---|---|
| A* | 0.2 | 0.6 | 8.7 | 5.3 | 3.4 | 3.5 |
| B* | 9.1 | 2.7 | 2.6 | 10.0 | 7.6 | |
| C* | 0.1 | 3.6 | 2.3 | 3.6 | ||
| A | 4.3 | 4.2 | 5.2 | |||
| B | 6.1 | 10.6 | ||||
| C | 6.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaines, E.; Di Tommaso, D. Solvation and Aggregation of Meta-Aminobenzoic Acid in Water: Density Functional Theory and Molecular Dynamics Study. Pharmaceutics 2018, 10, 12. https://doi.org/10.3390/pharmaceutics10010012
Gaines E, Di Tommaso D. Solvation and Aggregation of Meta-Aminobenzoic Acid in Water: Density Functional Theory and Molecular Dynamics Study. Pharmaceutics. 2018; 10(1):12. https://doi.org/10.3390/pharmaceutics10010012
Chicago/Turabian StyleGaines, Etienne, and Devis Di Tommaso. 2018. "Solvation and Aggregation of Meta-Aminobenzoic Acid in Water: Density Functional Theory and Molecular Dynamics Study" Pharmaceutics 10, no. 1: 12. https://doi.org/10.3390/pharmaceutics10010012
APA StyleGaines, E., & Di Tommaso, D. (2018). Solvation and Aggregation of Meta-Aminobenzoic Acid in Water: Density Functional Theory and Molecular Dynamics Study. Pharmaceutics, 10(1), 12. https://doi.org/10.3390/pharmaceutics10010012