
Characterization of vB_SauM-fRuSau02, a Twort-Like Bacteriophage Isolated from a Therapeutic Phage Cocktail


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phages and Media
2.2. Phage Purification
2.3. Electron Microscopy
2.4. Infection Growth Curves
2.5. DNA Isolation and Phage Genome Sequencing
2.6. Determination of Physical Ends of the Phage Genome
2.7. In Silico Analysis of Phage Genome
2.8. Proteome Analysis
2.9. Host Range Screening
2.10. Efficiency of Plating and Adsorption Assay
2.11. Staphylococcal Enterotoxin Measurement
3. Results
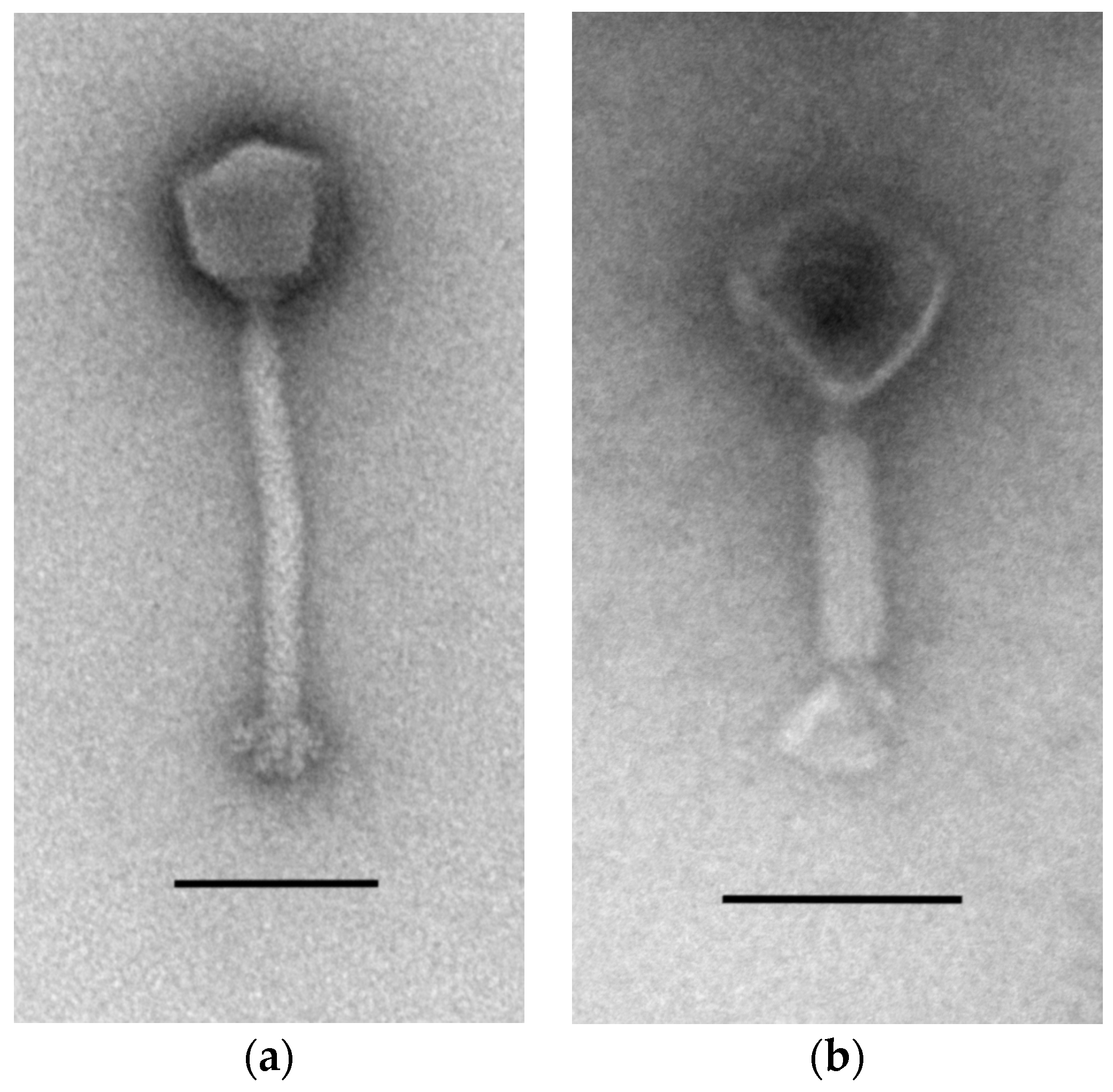
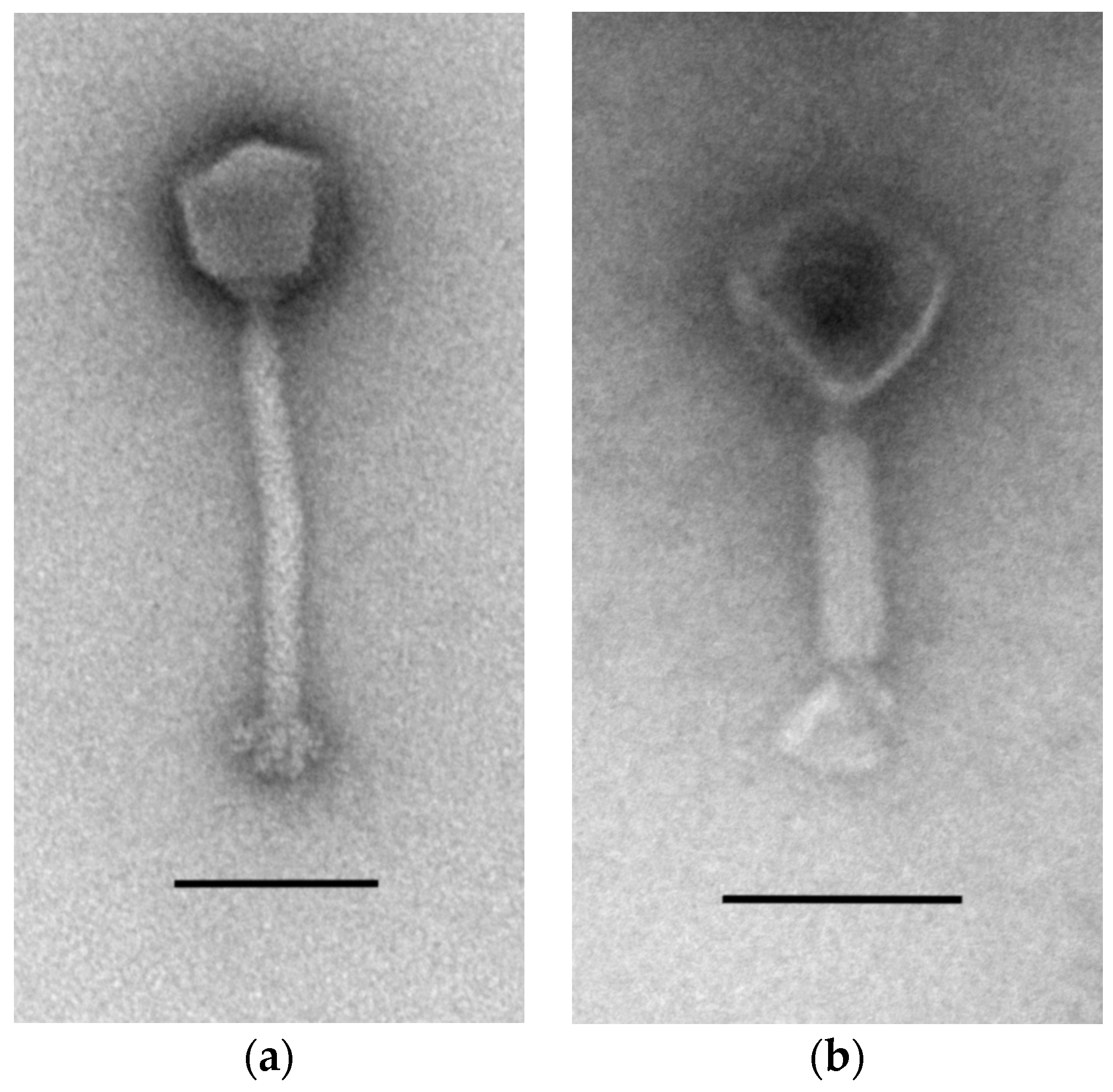
3.1. Isolation and Morphology
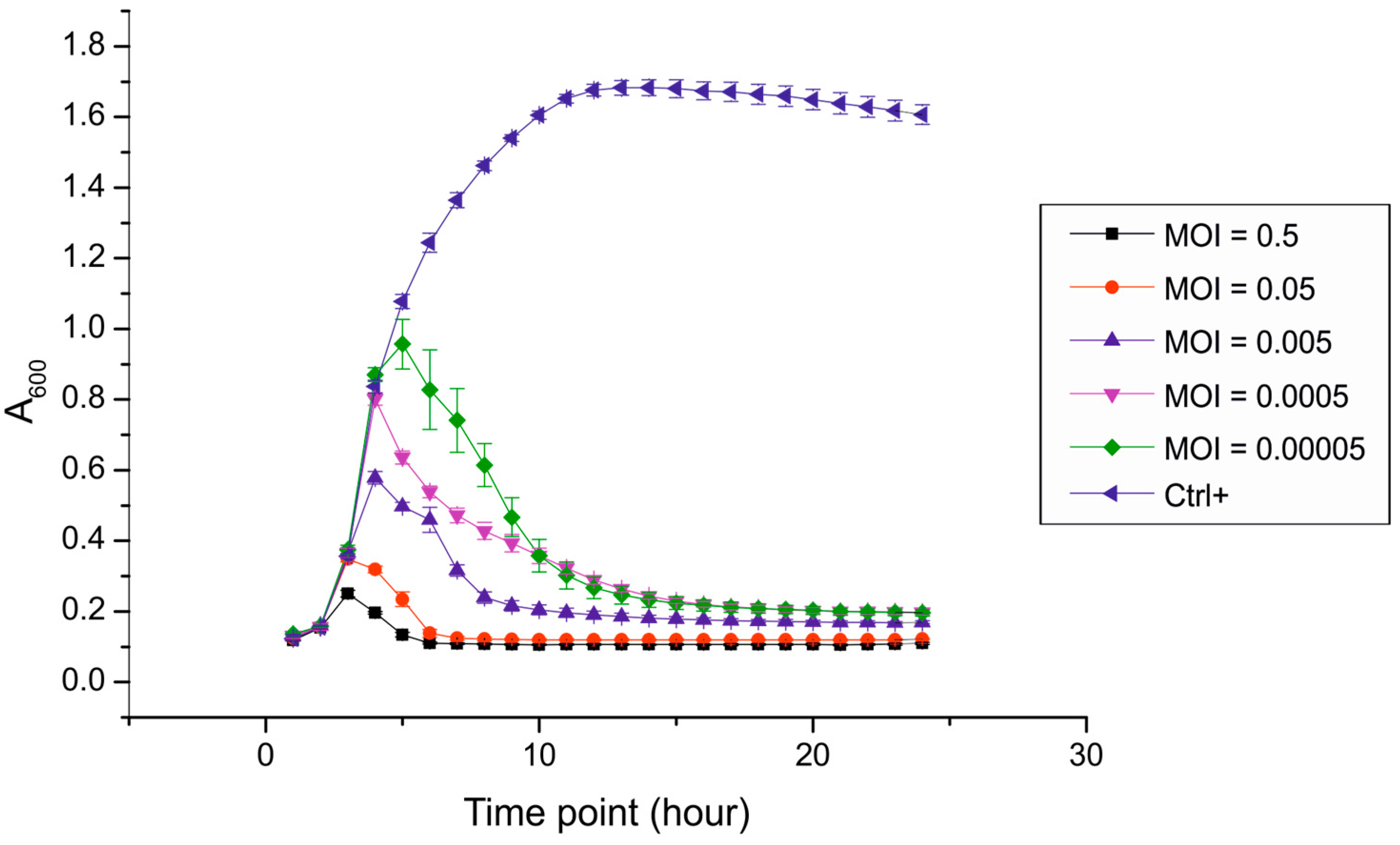
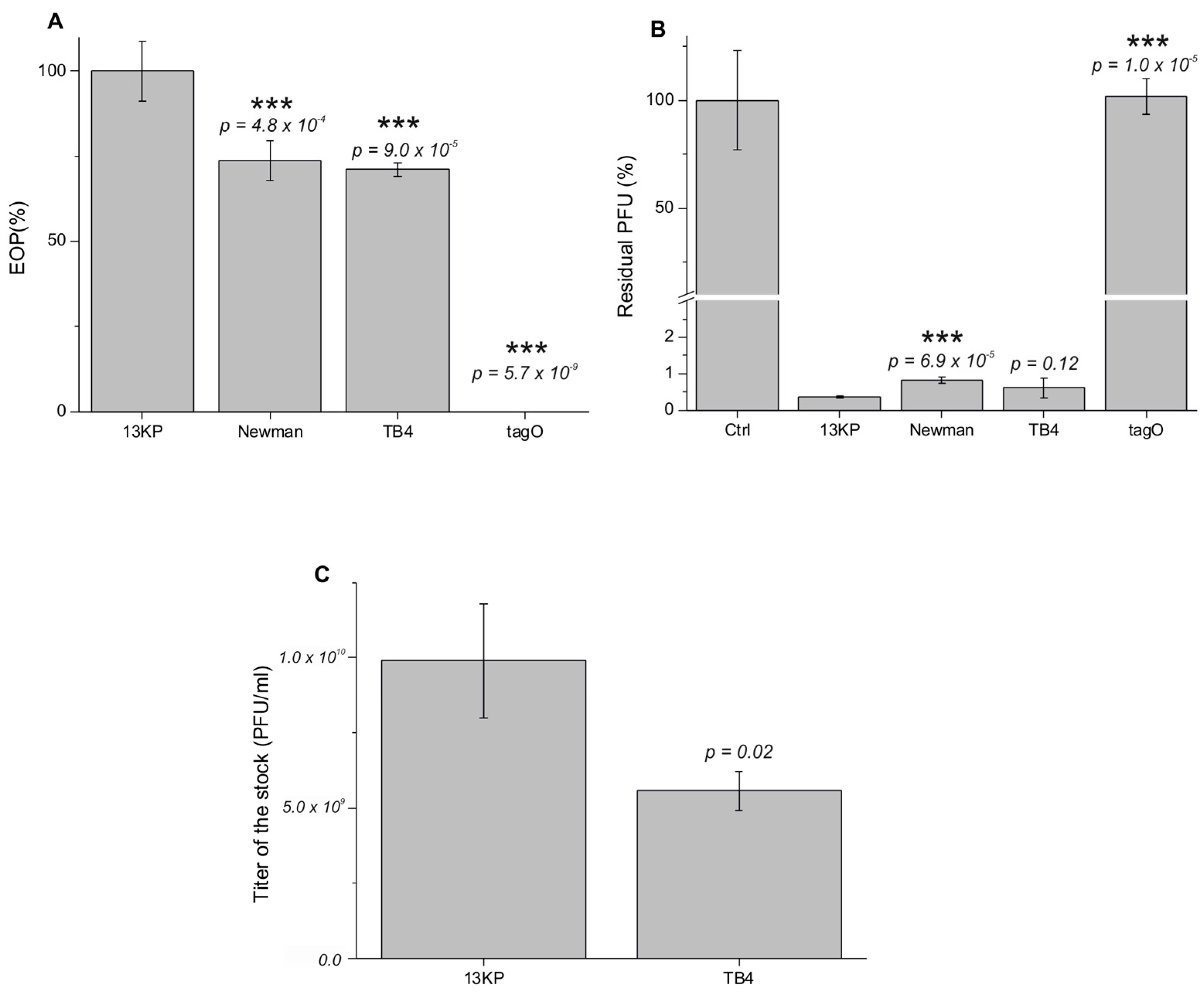
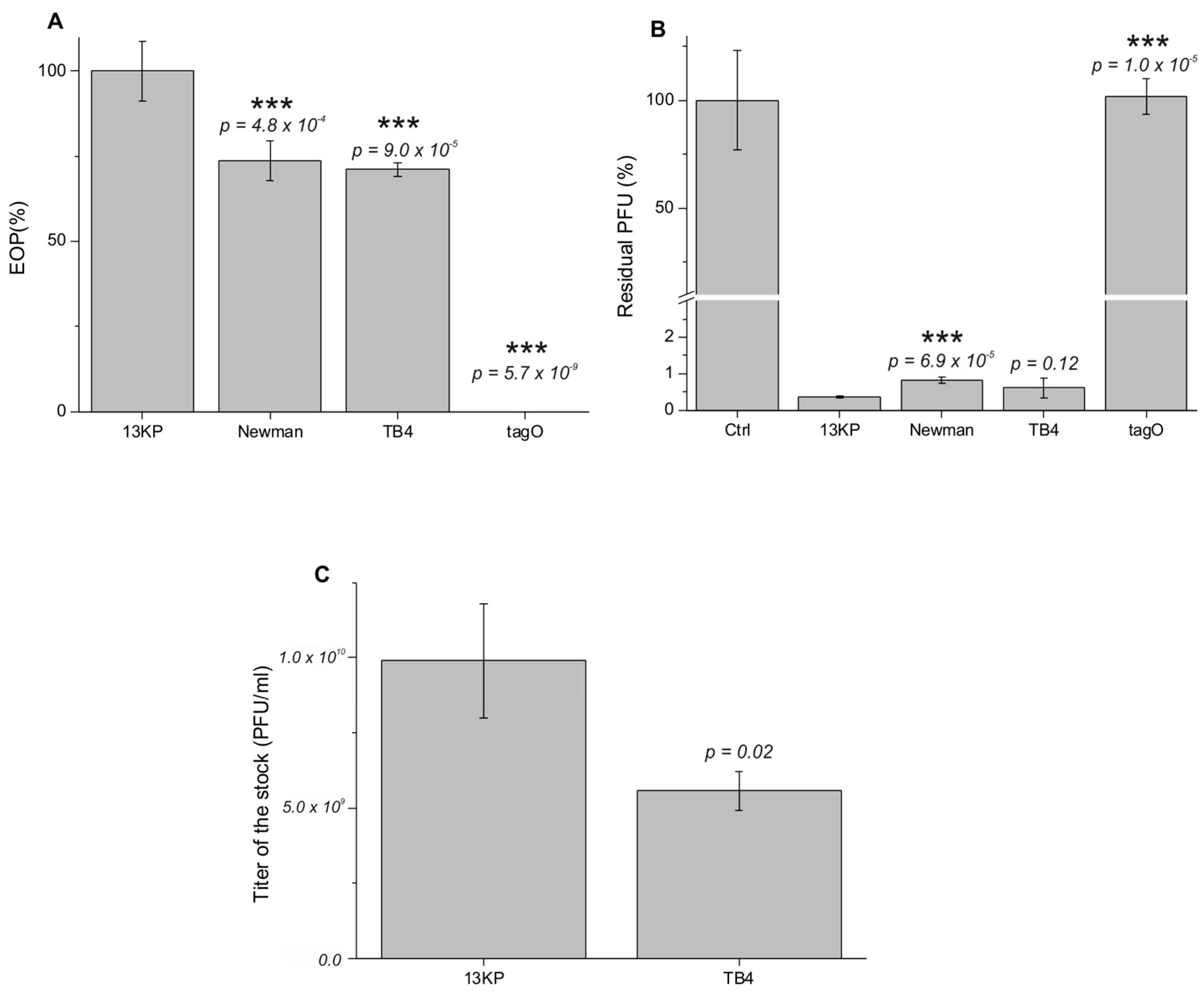
3.2. The Efficiency of Infection
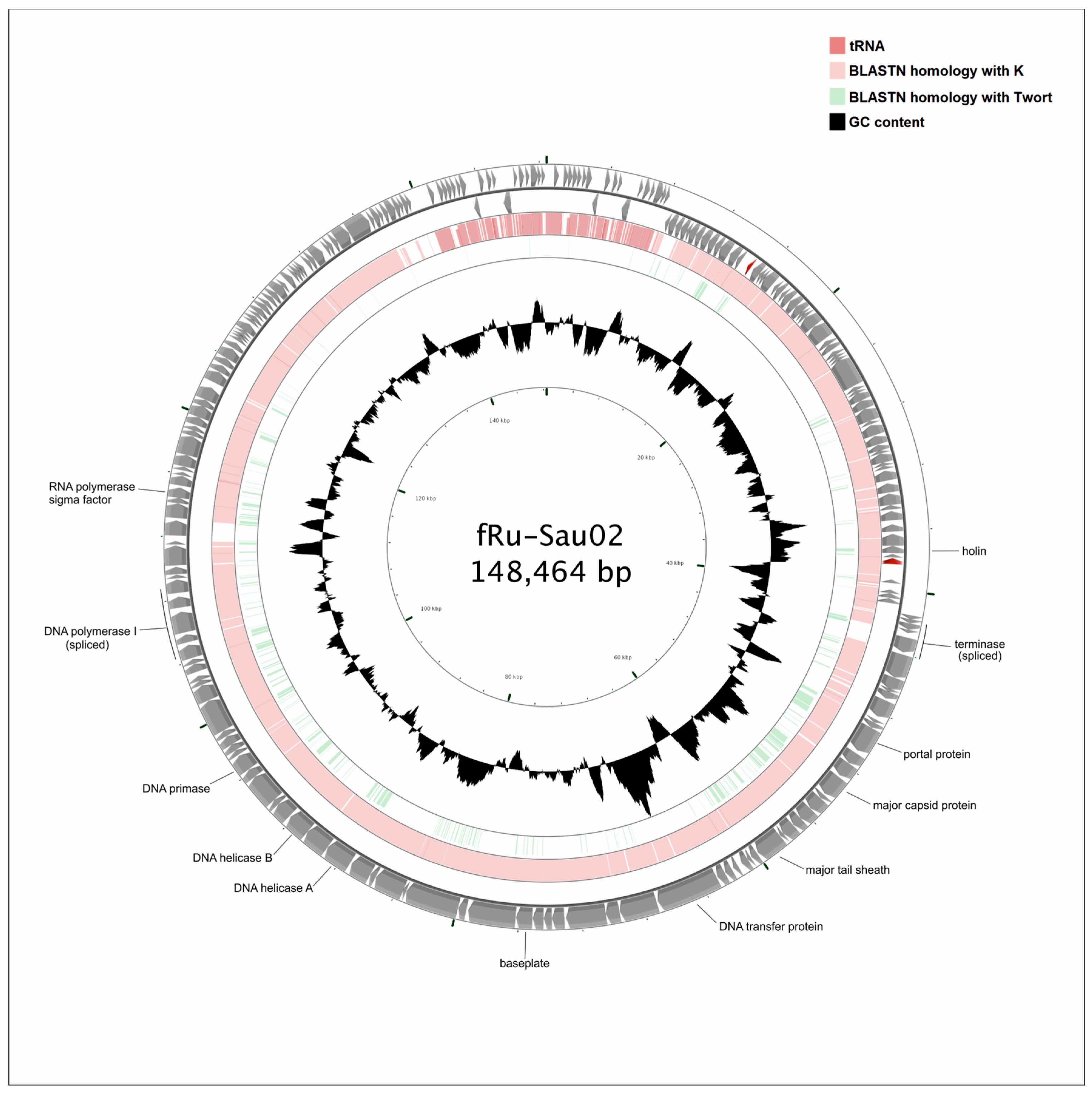
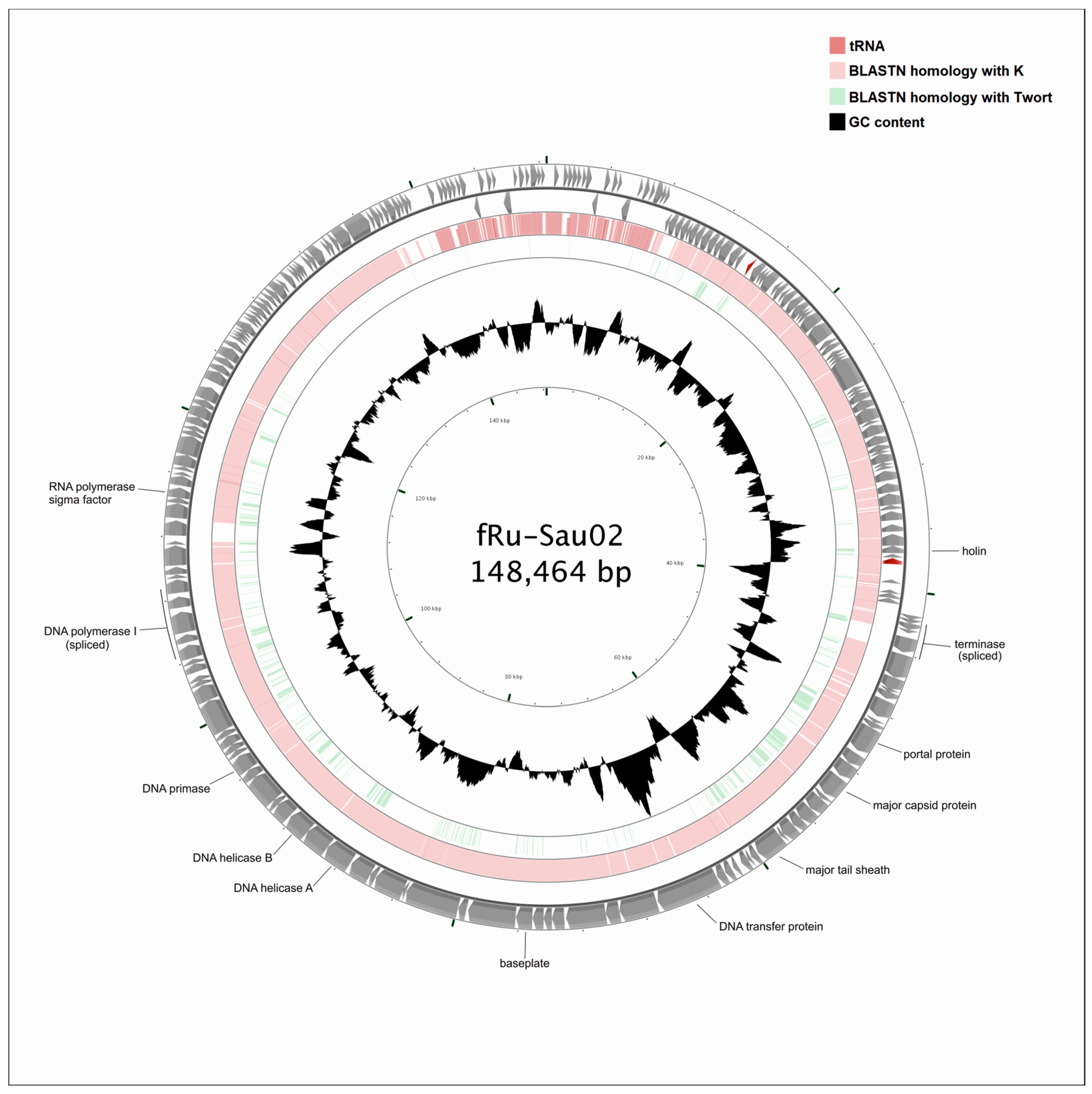
3.3. General Genome Analysis
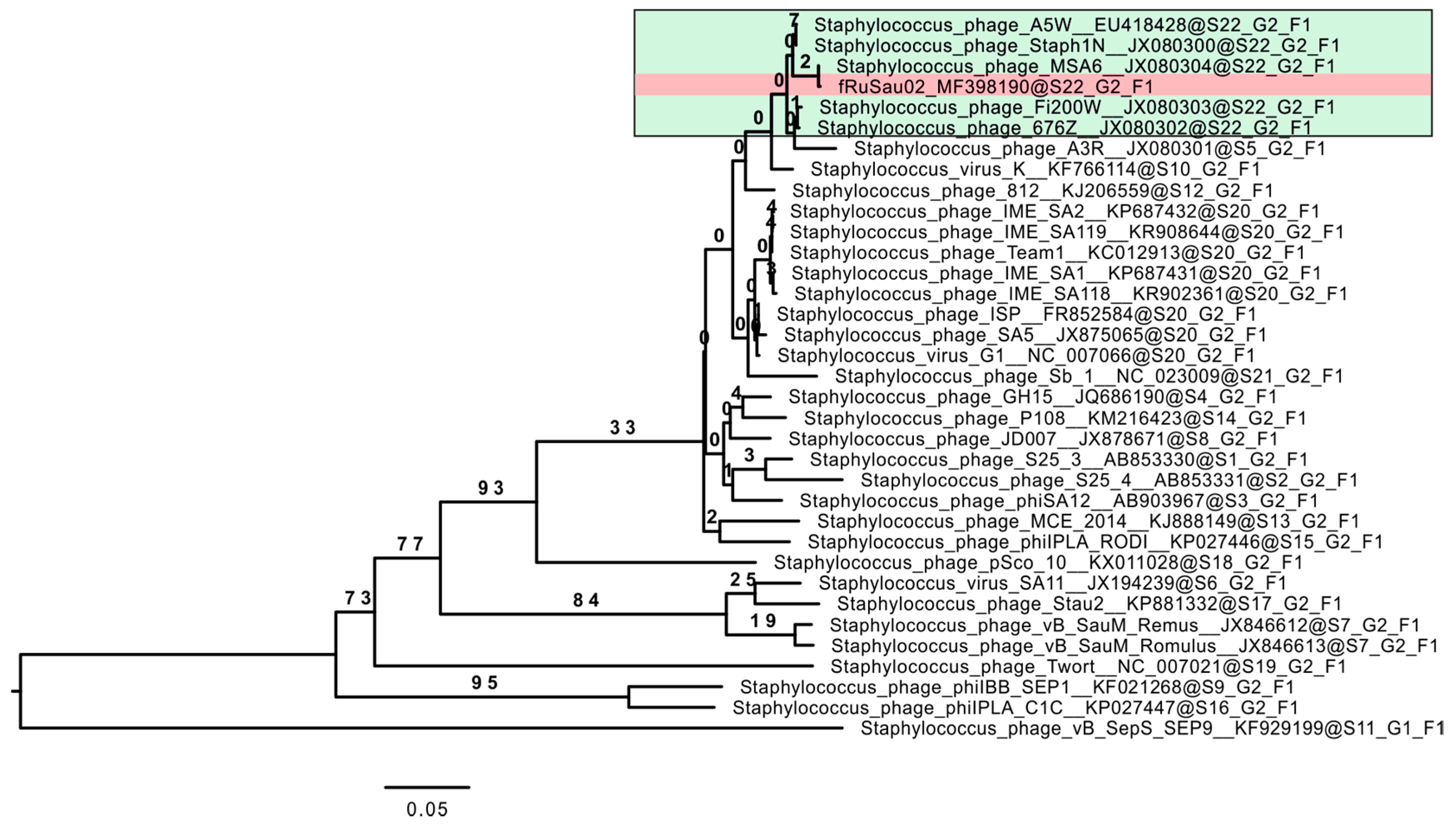
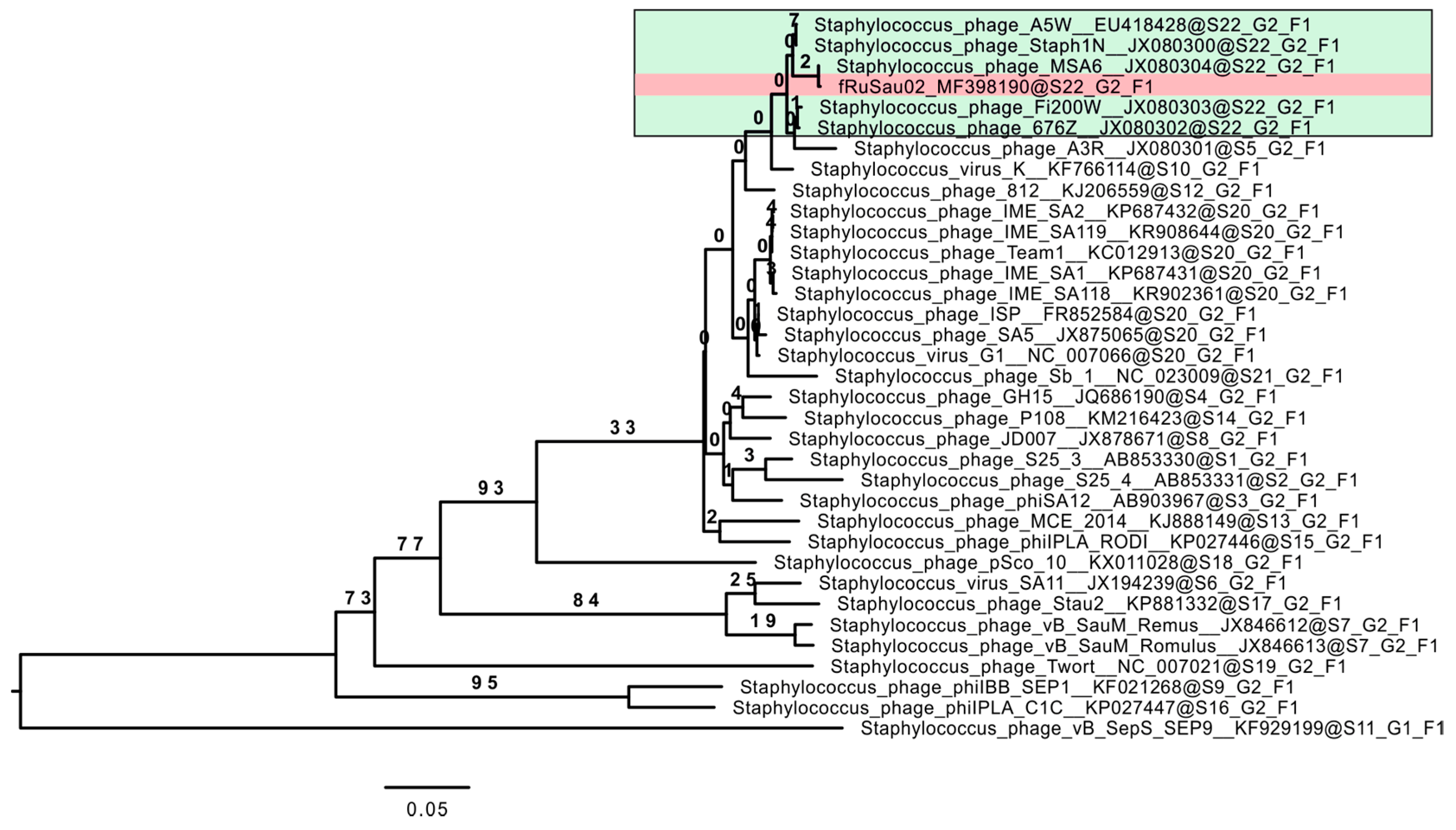
3.4. Comparative Genome Analysis
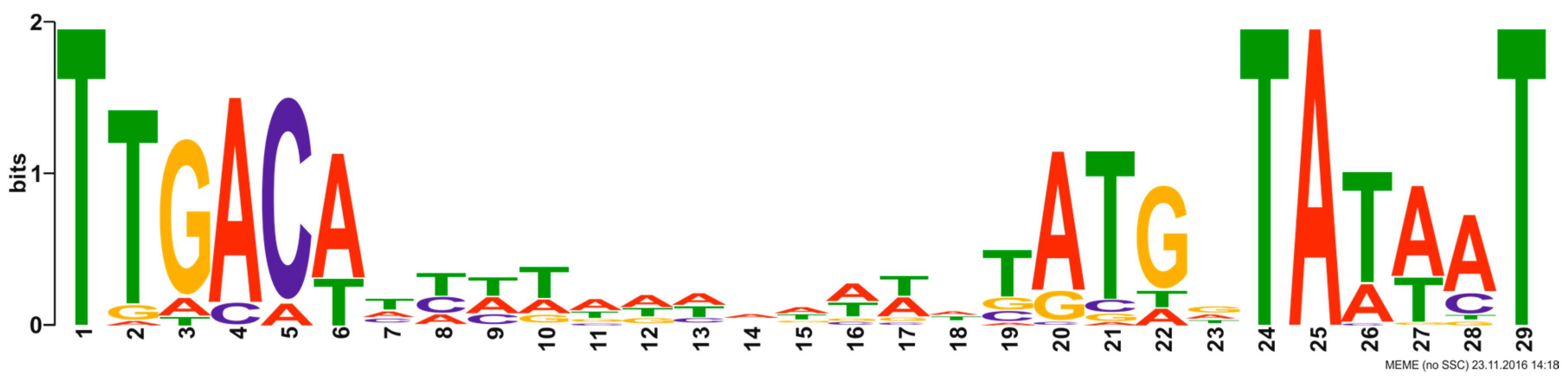
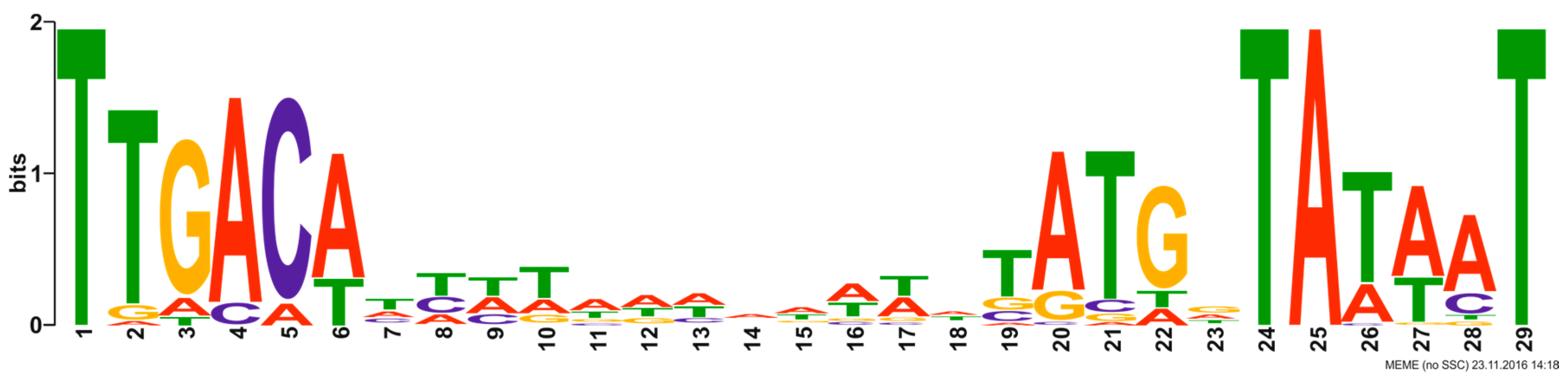
3.5. Genes Interrupted by Self-Splicing Elements
3.6. Proteomic Analysis of the Phage Structural Proteins
3.7. fRuSau02 Host Range
3.8. fRuSau02 Receptor
3.9. The Choice of Optimal Host Strain for Therapeutic Phage Production
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gordon, R.J.; Lowy, F.D. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008, 46, S350–S359. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Dantes, R.; Mu, Y.; Belflower, R.; Aragon, D.; Dumyati, G.; Harrison, L.H.; Lessa, F.C.; Lynfield, R.; Nadle, J.; Petit, S.; et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, united states, 2011. JAMA Intern. Med. 2013, 173, 1970–1978. [Google Scholar] [PubMed]
- Bal, A.M.; Coombs, G.W.; Holden, M.T.G.; Lindsay, J.A.; Nimmo, G.R.; Tattevin, P.; Skov, R.L. Genomic insights into the emergence and spread of international clones of healthcare-, community- and livestock-associated meticillin-resistant Staphylococcus aureus: Blurring of the traditional definitions. J. Glob. Antimicrob. Resist. 2016, 6, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Enright, M.C.; Robinson, D.A.; Randle, G.; Feil, E.J.; Grundmann, H.; Spratt, B.G. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. USA 2002, 99, 7687–7692. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.M.; Gorman, S.P.; Donnelly, R.F.; Gilmore, B.F. Recent advances in bacteriophage therapy: How delivery routes, formulation, concentration and timing influence the success of phage therapy. J. Pharm. Pharmacol. 2011, 63, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M.; Strauch, E. Phage therapy: Facts and fiction. Int. J. Med. Microbiol. 2006, 296, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; De Roock, S.; Opal, S.M. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Pirnay, J.P.; Blasdel, B.G.; Bretaudeau, L.; Buckling, A.; Chanishvili, N.; Clark, J.R.; Corte-Real, S.; Debarbieux, L.; Dublanchet, A.; De Vos, D.; et al. Quality and safety requirements for sustainable phage therapy products. Pharm. Res. 2015, 32, 2173–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deghorain, M.; Van Melderen, L. The staphylococci phages family: An overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, Z.; Gorski, A.; Dabrowska, K. Facing antibiotic resistance: Staphylococcus aureus phages as a medical tool. Viruses 2014, 6, 2551–2570. [Google Scholar] [CrossRef] [PubMed]
- Lobocka, M.; Hejnowicz, M.S.; Dabrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dabrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of staphylococcal Twort-like phages—Potential therapeutics of the post-antibiotic era. Adv. Virus Res. 2012, 83, 143–216. [Google Scholar] [PubMed]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microb. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Vandersteegen, K.; Kropinski, A.M.; Nash, J.H.; Noben, J.P.; Hermans, K.; Lavigne, R. Romulus and Remus, two phage isolates representing a distinct clade within the Twortlikevirus genus, display suitable properties for phage therapy applications. J. Virol. 2013, 87, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- O'Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of staphylococcal phage K: A new lineage of myoviridae infecting gram-positive bacteria with a low G+C content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Kwiatek, M.; Parasion, S.; Mizak, L.; Gryko, R.; Bartoszcze, M.; Kocik, J. Characterization of a bacteriophage, isolated from a cow with mastitis, that is lytic against Staphylococcus aureus strains. Arch. Virol. 2012, 157, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Liu, X.; Lu, R.; Li, Y.; Song, J.; Lei, L.; Sun, C.; Feng, X.; Du, C.; Yu, H.; et al. Complete genome sequence of Staphylococcus aureus bacteriophage GH15. J. Virol. 2012, 86, 8914–8915. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, I.; Osada, K.; Azam, A.H.; Asakawa, H.; Miyanaga, K.; Tanji, Y. The presence of two receptor-binding proteins contributes to the wide host range of staphylococcal Twort-like phages. Appl. Environ. Microbiol. 2016, 82, 5763–5774. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning, a Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- FIMM Sequencing Unit. Available online: https://www.fimm.fi/en/services/technology-centre/sequencing/ (accessed on 13 September 2017).
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- RAST (Rapid Annotation Using Subsystem Technology). Available online: http://rast.nmpdr.org/ (accessed on 13 September 2017).
- De Jong, A.; Pietersma, H.; Cordes, M.; Kuipers, O.P.; Kok, J. PePPER: A webserver for prediction of prokaryote promoter elements and regulons. BMC Genom. 2012, 13, 299. [Google Scholar] [CrossRef] [PubMed]
- Gautheret, D.; Lambert, A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001, 313, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Macke, T.J.; Ecker, D.J.; Gutell, R.R.; Gautheret, D.; Case, D.A.; Sampath, R. RNAmotif, an RNA secondary structure definition and search algorithm. Nucl. Acids Res. 2001, 29, 4724–4735. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.Y.; Li, W.W.; Noble, W.S. MEME suite: Tools for motif discovery and searching. Nucl. Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using GCView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed]
- EMBOSS Stretcher. Available online: http://www.ebi.ac.uk/Tools/psa/emboss_stretcher (accessed on 13 September 2017).
- Meier-Kolthoff, J.P.; Göker, M. Victor: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 1–9. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. Fastme 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 13 September 2017).
- Göker, M.; Garcia-Blazquez, G.; Voglmayr, H.; Telleria, M.T.; Martin, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom.Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E. Phage host range and efficiency of plating. Methods Mol. Biol. 2009, 501, 141–149. [Google Scholar] [PubMed]
- Leon-Velarde, C.G.; Happonen, L.; Pajunen, M.; Leskinen, K.; Kropinski, A.M.; Mattinen, L.; Rajtor, M.; Zur, J.; Smith, D.; Chen, S.; et al. Yersinia enterocolitica-specific infection by bacteriophages TG1 and varphiR1-RT is dependent on temperature-regulated expression of the phage host receptor OmpF. Appl. Environ. Microbiol. 2016, 82, 5340–5353. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Bacteriophage observations and evolution. Res. Microbiol. 2003, 154, 245–251. [Google Scholar] [CrossRef]
- Maniloff, J.; Ackermann, H.W. Taxonomy of bacterial viruses: Establishment of tailed virus genera and the order Caudovirales. Arch. Virol. 1998, 143, 2051–2063. [Google Scholar] [CrossRef] [PubMed]
- Dehbi, M.; Moeck, G.; Arhin, F.F.; Bauda, P.; Bergeron, D.; Kwan, T.; Liu, J.; McCarty, J.; Dubow, M.; Pelletier, J. Inhibition of transcription in Staphylococcus aureus by a primary sigma factor-binding polypeptide from phage G1. J. Bacteriol. 2009, 191, 3763–3771. [Google Scholar] [CrossRef] [PubMed]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.J.; Bilocq, F.; De Vos, D.; Pirnay, J.P.; Noben, J.P.; Merabishvili, M.; Lipinska, U.; Hermans, K.; et al. Microbiological and molecular assessment of bacteriophage ISP for the control of Staphylococcus aureus. PLoS ONE 2011, 6, e24418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landthaler, M.; Begley, U.; Lau, N.C.; Shub, D.A. Two self-splicing group i introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucl. Acids Res. 2002, 30, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Landthaler, M.; Shub, D.A. Unexpected abundance of self-splicing introns in the genome of bacteriophage Twort: Introns in multiple genes, a single gene with three introns, and exon skipping by group I ribozymes. Proc. Natl. Acad. Sci. USA 1999, 96, 7005–7010. [Google Scholar] [CrossRef] [PubMed]
- Casey, E.; Mahony, J.; Neve, H.; Noben, J.P.; Dal Bello, F.; van Sinderen, D. Novel phage group infecting Lactobacillus delbrueckii subsp. Lactis, as revealed by genomic and proteomic analysis of bacteriophage Ldl1. Appl. Environ. Microbiol. 2015, 81, 1319–1326. [Google Scholar] [PubMed]
- Casey, E.; Mahony, J.; O’Connell-Motherway, M.; Bottacini, F.; Cornelissen, A.; Neve, H.; Heller, K.J.; Noben, J.P.; Dal Bello, F.; van Sinderen, D. Molecular characterization of three Lactobacillus delbrueckii subsp. Bulgaricus phages. Appl. Environ. Microbiol. 2014, 80, 5623–5635. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Baba, T.; Hiramatsu, K.; Schneewind, O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006, 62, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Soldo, B.; Lazarevic, V.; Karamata, D. TagO is involved in the synthesis of all anionic cell-wall polymers in Bacillus subtilis 168. Microbiology 2002, 148, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Betley, M.J.; Mekalanos, J.J. Staphylococcal enterotoxin a is encoded by phage. Science 1985, 229, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Goerke, C.; Wirtz, C.; Fluckiger, U.; Wolz, C. Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 2006, 61, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Hetrick, D.L.; Bielefeldt, D.J. Production and properties of Staphylococcus aureus (strain Newman D2C) with uniform clumping factor activity. Thromb. Res. 1977, 10, 203–211. [Google Scholar] [CrossRef]
- Klumpp, J.; Dorscht, J.; Lurz, R.; Bielmann, R.; Wieland, M.; Zimmer, M.; Calendar, R.; Loessner, M.J. The terminally redundant, nonpermuted genome of Listeria bacteriophage A511: A model for the SPO1-like myoviruses of gram-positive bacteria. J. Bacteriol. 2008, 190, 5753–5765. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Benitez Quintana, A.D.; Bosch, M.A.; Coll De Pena, A.; Aguilera, E.; Coulibaly, A.; Wu, W.; Osier, M.V.; Hudson, A.O.; Weintraub, S.T.; et al. Identification of essential genes in the Salmonella phage SPN3US reveals novel insights into giant phage head structure and assembly. J. Virol. 2016, 90, 10284–10298. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Weintraub, S.T.; Wu, W.; Winkler, D.C.; Cheng, N.; Steven, A.C.; Black, L.W. Extensive proteolysis of head and inner body proteins by a morphogenetic protease in the giant Pseudomonas aeruginosa phage phiKZ. Mol. Microbiol. 2012, 84, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, J.; Shao, F.; Wang, P.; Duan, G.; Yang, H. Analysis of the features of 45 identified CRISPR loci in 32 Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2015, 464, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Heikinheimo, A.; Johler, S.; Karvonen, L.; Julmi, J.; Fredriksson-Ahomaa, M.; Stephan, R. New dominant spa type t2741 in livestock-associated MRSA (CC398-MRSA-V) in finnish fattening pigs at slaughter. Antimicrob. Resist. Infect. Control 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Cisek, A.A.; Dabrowska, I.; Gregorczyk, K.P.; Wyzewski, Z. Phage therapy in bacterial infections treatment: One hundred years after the discovery of bacteriophages. Curr. Microbiol. 2017, 74, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Endersen, L.; O’Mahony, J.; Hill, C.; Ross, R.P.; McAuliffe, O.; Coffey, A. Phage therapy in the food industry. Annu. Rev. Food Sci. Technol. 2014, 5, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, D.; Rodriguez-Rubio, L.; Martinez, B.; Rodriguez, A.; Garcia, P. Bacteriophages as weapons against bacterial biofilms in the food industry. Front. Microbiol. 2016, 7, 825. [Google Scholar] [CrossRef] [PubMed]
- Kazi, M.; Annapure, U.S. Bacteriophage biocontrol of foodborne pathogens. J. Food Sci. Technol. 2016, 53, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.M.; Kuhl, S.J.; Abedon, S.T. Re-establishing a place for phage therapy in western medicine. Future Microbiol. 2015, 10, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Pirnay, J.P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, T.; Verbeken, G.; Vos, D.D.; Merabishvili, M.; Vaneechoutte, M.; Lavigne, R.; Jennes, S.; Zizi, M.; Pirnay, J.P. Experimental phage therapy of burn wound infection: Difficult first steps. Int. J. Burns Trauma 2014, 4, 66–73. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Name | No of AA | MW * [kDa] | pI * (calc.) |
|---|---|---|---|---|
| RS_018 | TreR, terminal repeat encoded protein R | 156 | 17.8 | 3.78 |
| RS_033 | phage structural protein | 105 | 11.8 | 6.76 |
| RS_036 | phage structural protein | 64 | 7.6 | 4.65 |
| RS_037 | phage structural protein | 245 | 28.6 | 6.58 |
| RS_041 | phage structural protein | 57 | 6.8 | 5.26 |
| RS_042 | phage structural protein | 160 | 18.8 | 4.64 |
| RS_046 | putative membrane protein MbpR | 91 | 10.9 | 5.01 |
| RS_048 | phage structural protein | 372 | 42.2 | 4.84 |
| RS_050 | phage structural protein | 138 | 16.0 | 5.22 |
| RS_051 | HmzG, DNA-binding protein | 100 | 11.3 | 4.91 |
| RS_055 | phage structural protein | 87 | 10.1 | 5.91 |
| RS_059 | Lig, putative DNA or RNA ligase | 298 | 35.0 | 5.57 |
| RS_061 | Phr, putative PhoH-related protein | 246 | 28.6 | 5.29 |
| RS_063 | Rbn, phage ribonuclease H | 141 | 15.8 | 7.27 |
| RS_067 | phage structural protein | 75 | 9.2 | 9.95 |
| RS_070 | putative membrane protein MbpS | 263 | 29.3 | 8.82 |
| RS_072 | LysK.1, phage lysin | 209 | 23.1 | 9.66 |
| RS_074 | LysK.2, phage lysin | 267 | 29.8 | 9.45 |
| RS_075 | HolA, phage holin | 167 | 18.1 | 4.25 |
| RS_078 | DmcB | 69 | 8.0 | 5.97 |
| RS_080 | putative membrane protein MbpC | 108 | 13.0 | 5.54 |
| RS_082 | putative membrane protein MbpD | 88 | 10.3 | 8.31 |
| RS_085 | Ter.1, phage terminase | 65 | 7.7 | 9.60 |
| RS_087 | Ter.2, phage terminase | 515 | 59.7 | 6.10 |
| RS_088 | phage structural protein | 266 | 29.8 | 5.30 |
| RS_094 | Prt, portal protein | 563 | 64.0 | 6.42 |
| RS_095 | Pro, prohead protease | 257 | 28.6 | 5.01 |
| RS_096 | phage structural protein | 318 | 35.9 | 4.46 |
| RS_097 | Mcp, major capsid protein | 463 | 51.2 | 5.24 |
| RS_098 | phage structural protein | 98 | 11.3 | 9.42 |
| RS_099 | phage structural protein | 302 | 34.1 | 5.24 |
| RS_100 | phage structural protein | 292 | 33.7 | 5.82 |
| RS_101 | phage structural protein | 206 | 23.7 | 10.32 |
| RS_102 | phage structural protein | 278 | 31.7 | 4.79 |
| RS_104 | Tsp, major tail sheath protein | 587 | 64.5 | 4.98 |
| RS_105 | TmpA, tail tube protein | 142 | 15.9 | 5.54 |
| RS_109 | phage structural protein | 103 | 12.2 | 6.13 |
| RS_110 | phage structural protein | 152 | 18.1 | 4.79 |
| RS_111 | TmpB, tail morphogenic protein | 178 | 20.9 | 4.40 |
| RS_112 | TmpC, phage DNA transfer protein | 1351 | 143.7 | 9.11 |
| RS_113 | TmpD, tail murein hydrolase | 808 | 91.2 | 6.74 |
| RS_114 | TmpE, putative peptidoglycan hydrolase | 295 | 34.6 | 4.60 |
| RS_115 | Glycerophosphoryl diester phosphodiesterase | 848 | 96.0 | 4.96 |
| RS_116 | phage structural protein | 263 | 29.3 | 8.19 |
| RS_117 | phage structural protein | 174 | 19.9 | 4.61 |
| RS_118 | BmpA, baseplate morphogenetic protein | 234 | 26.6 | 4.77 |
| RS_119 | BmpB, baseplate morphogenetic protein | 348 | 39.2 | 4.86 |
| RS_120 | TmpF, tail morphogenetic protein | 1019 | 116.2 | 5.08 |
| RS_121 | BmpC, baseplate morphogenetic protein | 173 | 19.2 | 5.39 |
| RS_122 | TmpG, tail morphogenetic protein | 1152 | 129.0 | 5.19 |
| RS_124 | receptor binding protein | 640 | 72.6 | 7.39 |
| RS_126 | receptor binding protein | 458 | 50.3 | 6.27 |
| RS_127 | DhlA, DNA helicase | 582 | 67.2 | 5.85 |
| RS_129 | DhlB, DNA helicase | 480 | 54.5 | 5.72 |
| RS_132 | RncB, recombination nuclease B | 639 | 73.4 | 5.19 |
| RS_133 | Asf, anti-sigma factor | 198 | 23.2 | 6.81 |
| RS_137 | phage structural protein | 202 | 23.6 | 5.72 |
| RS_139 | NrdE, ribonucleotide reductase | 704 | 80.1 | 5.64 |
| RS_140 | NrdF, ribonucleotide reductase | 349 | 40.4 | 4.78 |
| RS_141 | phage structural protein | 109 | 12.4 | 4.68 |
| RS_143 | phage structural protein | 179 | 21.1 | 6.95 |
| RS_152 | phage structural protein | 423 | 46.8 | 4.75 |
| RS_153 | Rec.1, phage recombinase | 74 | 7.9 | 6.61 |
| RS_155 | Rec.2, phage recombinase | 315 | 35.7 | 5.16 |
| RS_157 | Sig, sigma factor | 220 | 26.6 | 5.36 |
| RS_158 | phage structural protein | 210 | 23.2 | 4.84 |
| RS_159 | TmpH, phage major tail protein | 73 | 7.9 | 4.54 |
| RS_160 | phage structural protein | 86 | 10.3 | 5.91 |
| RS_163 | putative membrane protein MbpG | 122 | 14.0 | 5.95 |
| RS_165 | phage structural protein | 178 | 20.8 | 7.47 |
| RS_168 | phage structural protein | 287 | 32.3 | 5.76 |
| RS_169 | phage structural protein | 243 | 28.3 | 5.34 |
| RS_170 | phage structural protein | 152 | 17.8 | 4.98 |
| RS_173 | putative membrane protein MbpH | 132 | 15.4 | 8.94 |
| RS_175 | phage structural protein | 80 | 9.4 | 9.31 |
| RS_181 | phage structural protein | 98 | 11.3 | 7.24 |
| RS_196 | phage structural protein | 87 | 9.9 | 10.05 |
| RS_206 | NadV, nicotinamide phosphoribosyltransferase | 489 | 56.1 | 5.44 |
| fRuSau02 Infectivity | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bacterial Hosts | Infected * | Intermediate * | Resistant * | ||||||
| Coagulase-Positive Human Isolates (n = 51) | |||||||||
| S. aureus | 49 | (96%) | 2 | (4%) | 0 | (0%) | |||
| Coagulase-Negative Human Isolates (n = 30) | |||||||||
| S. intermedius | 0 | 3 | 2 | ||||||
| S. lugdunensis | 1 | 4 | 0 | ||||||
| S. epidermidis | 0 | 1 | 4 | ||||||
| S. haemolyticus | 0 | 2 | 3 | ||||||
| S. saprophyticus | 1 | 2 | 2 | ||||||
| S. pseudointer | 0 | 4 | 1 | ||||||
| ALL | 2 | (7%) | 16 | (53%) | 12 | (40%) | |||
| Coagulase-Positive Porcine Isolates (n = 54) | |||||||||
| S. aureus | 18 | (33%) | 3 | (6%) | 33 | (61%) | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a Twort-Like Bacteriophage Isolated from a Therapeutic Phage Cocktail. Viruses 2017, 9, 258. https://doi.org/10.3390/v9090258
Leskinen K, Tuomala H, Wicklund A, Horsma-Heikkinen J, Kuusela P, Skurnik M, Kiljunen S. Characterization of vB_SauM-fRuSau02, a Twort-Like Bacteriophage Isolated from a Therapeutic Phage Cocktail. Viruses. 2017; 9(9):258. https://doi.org/10.3390/v9090258
Chicago/Turabian StyleLeskinen, Katarzyna, Henni Tuomala, Anu Wicklund, Jenni Horsma-Heikkinen, Pentti Kuusela, Mikael Skurnik, and Saija Kiljunen. 2017. "Characterization of vB_SauM-fRuSau02, a Twort-Like Bacteriophage Isolated from a Therapeutic Phage Cocktail" Viruses 9, no. 9: 258. https://doi.org/10.3390/v9090258
APA StyleLeskinen, K., Tuomala, H., Wicklund, A., Horsma-Heikkinen, J., Kuusela, P., Skurnik, M., & Kiljunen, S. (2017). Characterization of vB_SauM-fRuSau02, a Twort-Like Bacteriophage Isolated from a Therapeutic Phage Cocktail. Viruses, 9(9), 258. https://doi.org/10.3390/v9090258