
Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation


Abstract
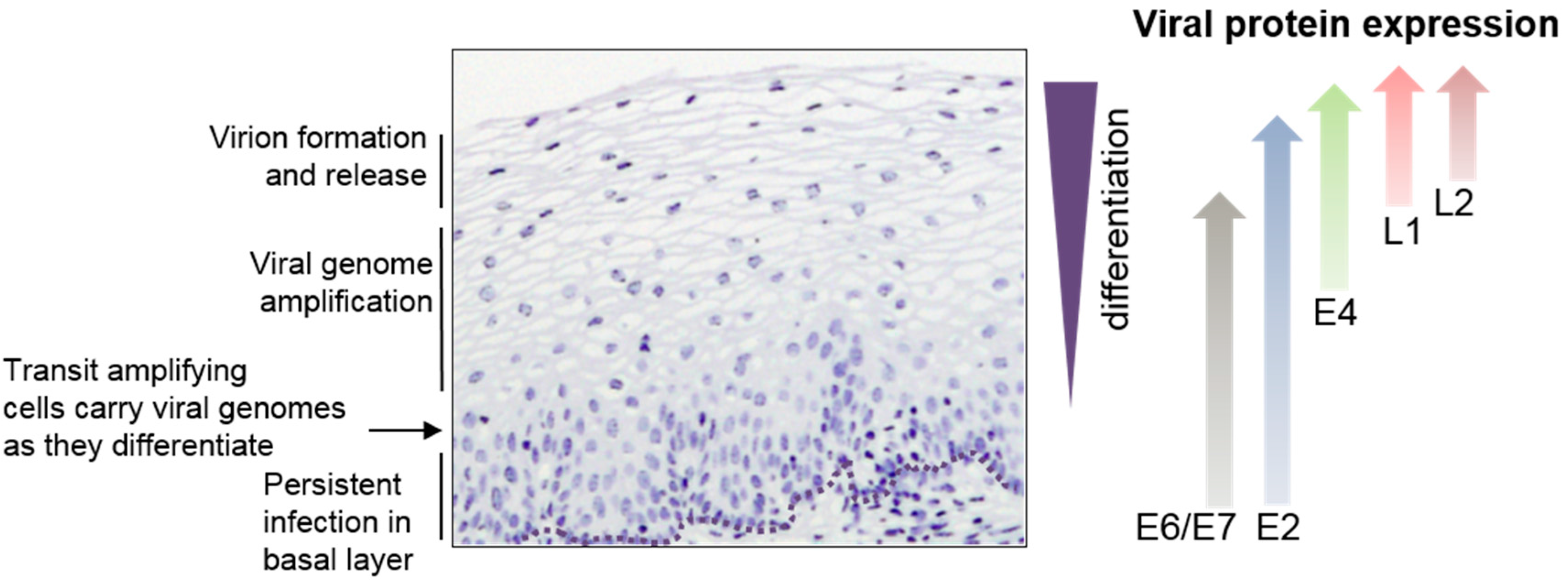
1. Introduction
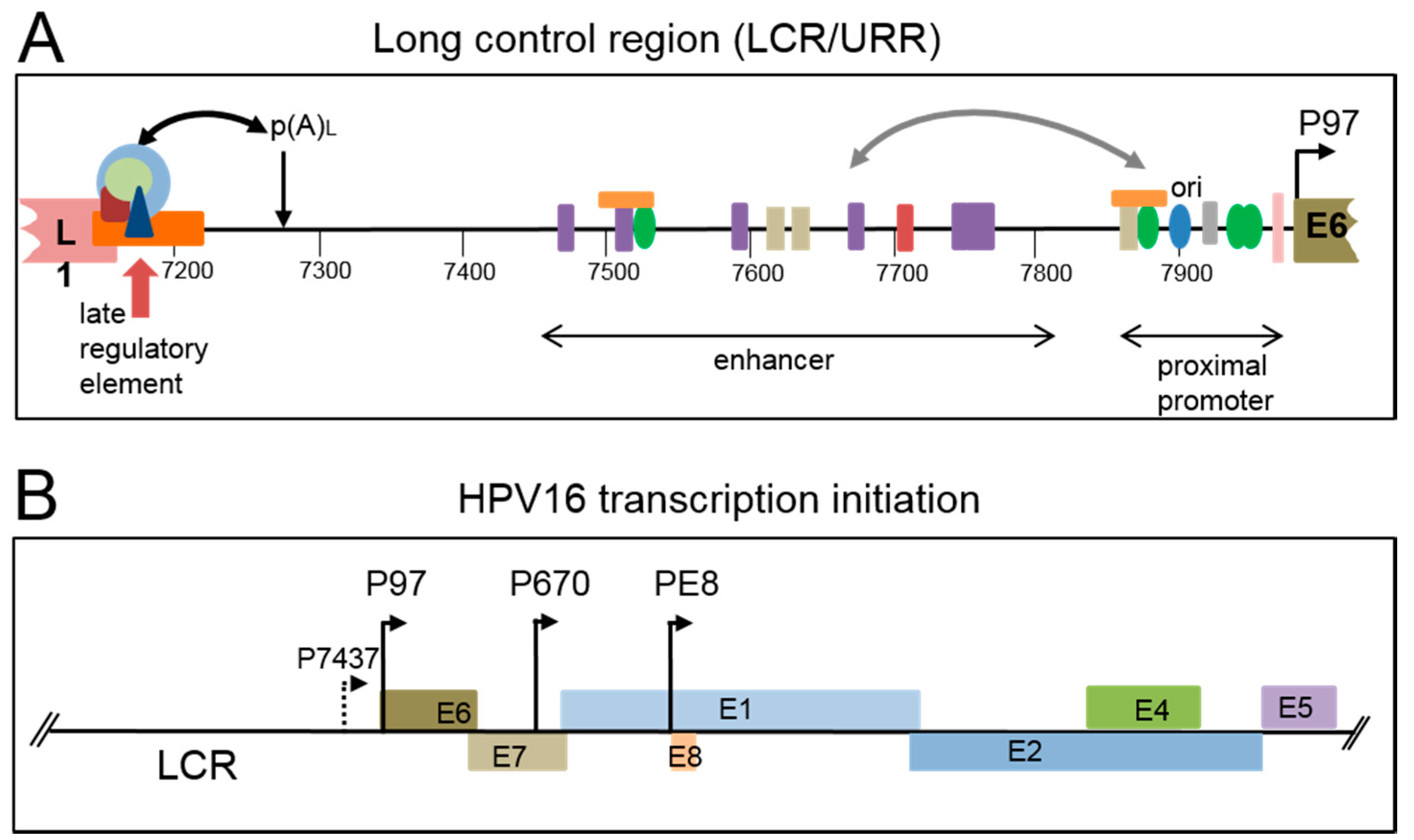
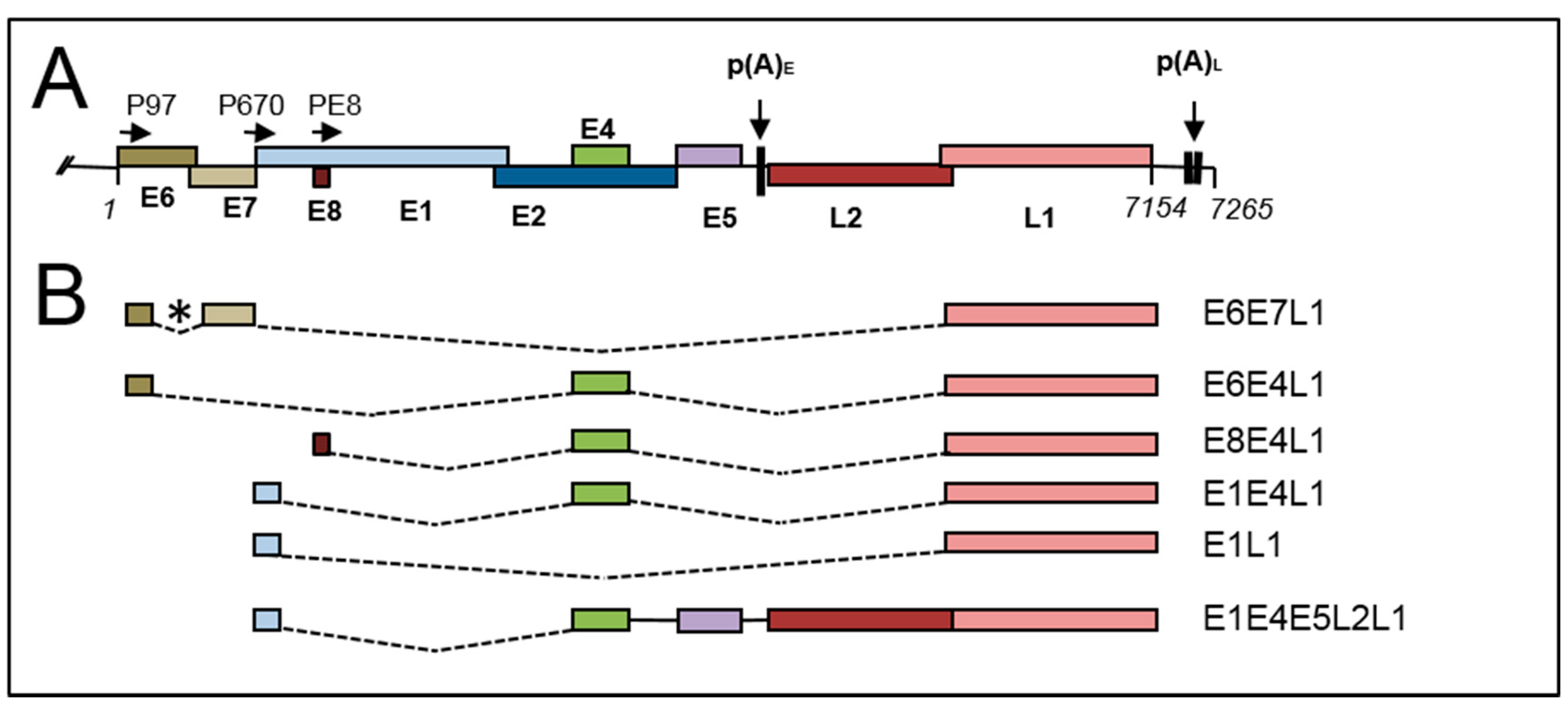
2. Viral Genome Organization
2.1. Early Gene Expression
2.2. Late Gene Expression and the Late Promoter
3. Viral Late Promoter Activity
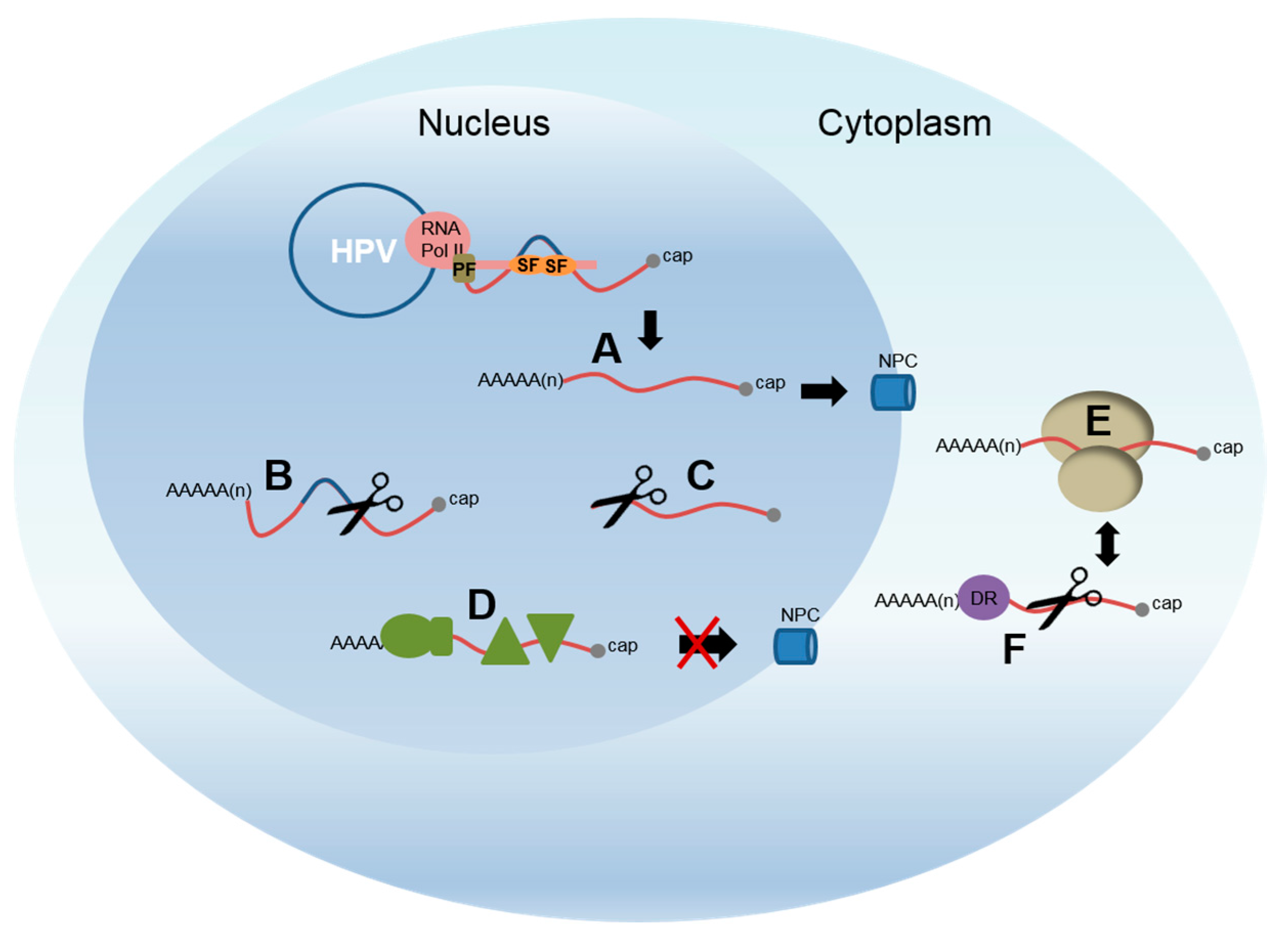
4. Differentiation-Dependent Regulation of Polyadenylation
4.1. Repression of the Early Polyadenylation Site and Late Gene Expression
4.2. Control of Late Gene Expression Occurs at Multiple Post-Transcriptional Levels
4.3. Control of Late Polyadenylation
4.4. Regulation of Late RNA Stability
4.5. Late RNA Splicing
4.6. Control of Late mRNA Splicing
4.7. Control of Capsid Protein Translation
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Tommasino, M.; Depuydt, C.; Dillner, J. Are 20 human papillomavirus types causing cervical cancer? J. Pathol. 2014, 234, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus—Positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human papillomaviruses; epithelial tropisms, and the development of neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Coleman, N. Pathogenesis of human papillomavirus-associated mucosal disease. J. Pathol. 2015, 235, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xue, Y.; Poidinger, M.; Lim, T.; Chew, S.H.; Pang, C.L.; Abastado, J.-P.; Thierry, F. Mapping of HPV transcripts in four human cervical lesions using RNAseq suggests quantitative rearrangements during carcinogenic progression. Virology 2014, 462–463, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meyers, C.; Wang, H.-K.; Chow, L.T.; Zheng, Z.-M. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef] [PubMed]
- Milligan, S.G.; Veerapraditsin, T.; Ahamat, B.; Mole, S.; Graham, S.V. Analysis of novel human papillomavirus type 16 late mRNAs in differentiated W12 cervical epithelial cells. Virology 2007, 360, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.N.; Nielsen, L.; Norrild, B. Activities of E7 promoters in the human papillomavirus type 16 genome during cell differentiation. Virus Res. 2010, 150, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, M.A.; Meyers, C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 1998, 72, 2715–2722. [Google Scholar] [PubMed]
- Ozbun, M.A.; Meyers, C. Two novel promoters in the upstream regulatory region of human papillomavirus type 31b are negatively regulated by epithelial differentiation. J. Virol. 1999, 73, 3505–3510. [Google Scholar] [PubMed]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Hummel, M.; Iftner, T.; Laimins, L.A. The E8^E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 2000, 74, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Griffin, H.M.; Southern, S.; Jackson, D.; Martin, A.; McIntosh, P.; Davy, C.; Masterson, P.J.; Walker, P.A.; Laskey, P.; et al. Functional analysis of the human papillomavirus type 16E1^E4 protein provides a mechanism for in vivo and in vitro keratin filament reorganization. J. Virol. 2004, 78, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Jackson, D.J.; Raj, K.; Peh, W.L.; Southern, S.A.; Das, P.; Sorathia, R.; Laskey, P.; Middleton, K.; Nakahara, T.; et al. Human Papillomavirus type 16 E1^E4-induced G2 arrest is associated with cytoplasmic retention of active Cdk1/Cyclin B1 complexes. J. Virol. 2005, 79, 3998–4011. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Middleton, K.; Peh, W.; Southern, S.A.; Griffin, H.M.; Sotlar, K.; Nakahara, T.; El-Sherif, A.; Morris, L.; Seth, R.; Hibma, M.; et al. Organisation of the human papillomavirus productive cycle during neoplastic progression provides a basis for the selection of diagnostic markers. J. Virol. 2003, 77, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Klumpp, D.J.; Laimins, L.A. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differnetiation. J. Virol. 2003, 77, 2819–2831. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Ryan, G.B.; Knight, G.L.; Laimins, L.A.; Roberts, S. The full-length E1^E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology 2007, 362, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Genther, S.M.; Sterling, S.; Duensing, S.; Munger, K.; Sattler, C.; Lambert, P.F. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 2003, 77, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human papillomavirus type 16 E1^E4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 2005, 79, 13150–13165. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Nishimura, A.; Tanaka, M.; Ueno, T.; Ishimoto, A.; Sakai, H. Modulation of the cell division cycle by human papillomavirus type 18 E4. J. Virol. 2002, 76, 10914–10920. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013, 9, e1003321. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Conway, S.; Wilkinson, N.S.; Ramsdale, J.; Morris, J.R.; Sanders, C.M.; Burns, J.E.; Stern, P.L.; Wells, M. Expression patterns of the human papillomavirus type 16 transcription factor E2 in low- and high-grade cervical intraepithelial neoplasia. J. Pathol. 1998, 186, 275–280. [Google Scholar] [CrossRef]
- Xue, Y.; Bellanger, S.; Zhang, W.; Lim, D.; Low, J.; Lunny, D.; Thierry, F. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Cancer Res. 2010, 70, 5316–5325. [Google Scholar] [CrossRef] [PubMed]
- Paris, C.; Pentland, I.; Groves, I.; Roberts, D.C.; Powis, S.J.; Coleman, N.; Roberts, S.; Parish, J.L. CCCTC-Binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J. Virol. 2015, 89, 4770–4785. [Google Scholar] [CrossRef] [PubMed]
- Peh, W.L.; Middleton, K.; Christensen, N.; Nicholls, P.; Egawa, K.; Sotlar, K.; Brandsma, J.; Percival, A.; Lewis, J.; Liu, W.J.; et al. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 2002, 76, 10411–10416. [Google Scholar] [CrossRef]
- Grassman, K.; Rapp, B.; Maschek, H.; Petry, K.U.; Iftner, T. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 1996, 70, 2339–2349. [Google Scholar]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Ruesch, M.N.; Stubenrauch, F.; Laimins, L.A. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin. J. Virol. 1998, 72, 5016–5024. [Google Scholar] [PubMed]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Ge, H.; Ajiro, M.; Sharma, N.R.; Meyers, C.; Morozov, P.; Tuschl, T.; Klar, A.; Court, D.; et al. Viral DNA replication orientation and hnRNPs regulate transcription of the human papillomavirus 18 late promoter. Microbiology 2017, 8, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Meyers, C. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J. Virol. 2005, 79, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Sankovski, E.; Männik, A.; Geimanen, J.; Ustav, E.; Ustav, M. Mapping of betapapillomavirus human papillomavirus 5 transcription and characterization of viral-genome replication function. J. Virol. 2014, 88, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Malejczyk, J.; Fuchs, P.G.; Pfister, H.J. Late promoter of human papillomavirus type 8 and its regualtion. J. Virol. 1992, 64, 3144–3149. [Google Scholar]
- Meyers, C.; Mayer, T.J.; Ozbun, M.A. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 1997, 71, 7381–7386. [Google Scholar] [PubMed]
- Del Mar Pena, L.; Laimins, L.A. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J. Virol. 2001, 75, 10005–10013. [Google Scholar] [CrossRef] [PubMed]
- Carson, A.; Khan, S.A. Characterisation of transcription factor binding to human papillomavirus type 16 DNA during cellular differentiation. J. Virol. 2006, 80, 4356–4362. [Google Scholar] [CrossRef] [PubMed]
- Wooldridge, T.R.; Laimins, L.A. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology 2008, 374, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Songock, W.K.; Scott, M.L.; Bodily, J.M. Regulation of the human papillomavirus type 16 late promoter by transcriptional elongation. Virology 2017, 507, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Alam, S.; Meyers, C. Regulation of human papillomavirus type 31 late promoter activation and genome amplification by protein kinase C. Virology 2006, 348, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Straub, E.; Fertey, J.; Dreer, M.; Iftner, T.; Stubenrauch, F. Characterization of the human papillomavirus 16 E8 promoter. J. Virol. 2015, 89, 7304–7313. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Hennigan, C.; Wrobel, G.A.; Rodriguez, C.M. Regulation of the human papillomavirus type 16 late promoter by E7 and the cell cycle. Virology 2013, 443, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Weschler, E.I.; Wang, Q.; Roberts, I.; Pagliarulo, E.; Jackson, D.; Untersperger, C.; Coleman, N.; Griffin, H.; Doorbar, J. Reconstruction of human papillomavirus type 16-mediated early-stage neoplasia implicated E6/E7 deregulation and the loss of contact inhibition in neoplastic progression. J. Virol. 2012, 86, 6358–6364. [Google Scholar]
- Stoler, M.H.; Wolinsky, S.M.; Whitbeck, A.; Broker, T.R.; Chow, L.T. Differentiation-linked human papillomavirus types 6 and 11 transcription in genital condylomata revealed by in situ hybridisation with message-specific RNA probes. Virology 1989, 172, 331–340. [Google Scholar] [CrossRef]
- Stoler, M.H.; Rhodes, C.R.; Whitbeck, A.; Wolinsky, S.M.; Chow, L.T.; Broker, T.R. Human papillomavirus type 16 and 18 gene expression in cervical neoplasia. Hum. Pathol. 1992, 23, 117–128. [Google Scholar] [CrossRef]
- Beyer-Finkler, E.; Stoler, M.H.; Girardi, F.; Pfister, H.J. Cell differentiation-related gene expression of human papillomavirus 33. Med. Microbiol. Immunol. 1990, 179, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P.; Nuovo, G.; Freidman, D.; Silverstein, S.J. Accumulation of RNA homologous to human papillomavirus type 16 open reading frames in genital precancers. J. Virol. 1988, 62, 84–90. [Google Scholar] [PubMed]
- Ozbun, M.A.; Meyers, C. Characterisation of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 1997, 71, 5161–5172. [Google Scholar] [PubMed]
- Neve, J.; Patel, R.; Wang, Z.; Louey, A.; Furger, A.M. Cleavage and polyadenylation: Ending the message expands gene regulation. RNA Biol. 2017, 14, 865–890. [Google Scholar] [CrossRef] [PubMed]
- Rohlfs, M.; Winkenbach, S.; Meyer, S.; Rupp, T.; Dürst, M. Viral transcription in human keratinocyte cell lines immortalized by human papillomvirus type-16. Virology 1991, 183, 331–342. [Google Scholar] [CrossRef]
- Higgins, G.D.; Uzelin, D.M.; Phillips, G.E.; McEvoy, P.; Burrel, C.J. Transcription patterns of human papillomavirus type 16 in genital intraepithelia neoplasia: Evidence for promoter usage within the E7 open reading frame during epithelial differentiation. J. Gen. Virol. 1992, 73, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Lim, H.B.; Laimins, L.A. Human papillomavirus type 31b late gene expression is regulated through protein kinase C-mediated changes in RNA processing. J. Virol. 1995, 69, 3381–3388. [Google Scholar] [PubMed]
- Terhune, S.S.; Milcarek, C.; Laimins, L.A. Regulation of human papillomavirus type 31 polyadenylation during the differentiation-dependent life cycle. J. Virol. 1999, 73, 7185–7192. [Google Scholar] [PubMed]
- Terhune, S.S.; Hubert, W.G.; Thomas, J.T.; Laimins, L.A. Early polyadenylation signals of human papillomavirus type 31 negatively regulates capsid gene expression. J. Virol. 2001, 75, 8147–8157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Oberg, D.; Rush, M.; Fay, J.; Lambkin, H.; Schwartz, S. A 57-nucleotide upstream early polyadenylation element in human papillomavirus type 16 interacts with hFip1, CstF-64, hnRNP C1/C2 and polypyrimidine tract binding protein. J. Virol. 2005, 79, 4270–4288. [Google Scholar] [CrossRef] [PubMed]
- Öberg, D.; Fay, J.; Lambkin, H.; Schwartz, S. A downstream polyadenylation element in human papillomavirus type 16 L2 encodes multiple GGG motifs and interacts with hnRNP H. J. Virol. 2005, 79, 9254–9269. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Repellin, C.E.; McPhillips, M.; Radford, J.C.; Clements, J.B.; Graham, S.V. The human papillomavirus type 31 late 3′ untranslated region contains a complex bipartite negative regulatory element. J. Virol. 2002, 76, 5993–6003. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, I.M.; Haddow, J.K.; Clements, J.B. Analysis of human papillomavirus type 16 late mRNA 3′ processing signals in vitro and in vivo. J. Virol. 1990, 64, 1825–1829. [Google Scholar] [PubMed]
- Öberg, D.; Collier, B.; Zhao, X.; Schwartz, S. Mutational inactivation of two distinct negative RNA elements in the human papillomavirus type 16 L2 coding region induces production of high levels of L2 in human cells. J. Virol. 2003, 77, 11674–11684. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Liu, X.; Tao, M.; Kruhlak, M.; Guo, M.; Meyers, C.; Baker, C.C.; Zheng, Z.M. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J. Virol. 2009, 83, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.; Zhao, X.; Schwartz, S. A splicing enhancer in the E4 coding region of human papillomavirus type 16 is required for early mRNA splicing and polyadenylation as well as inhibition of premature late gene expression. J. Virol. 2005, 79, 12002–12015. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Schwartz, S. Multiple ASF/SF2 sites in the human papillomavirus type 16 (HPV-16) E4-coding region promote splicing to the most commonly Used 3′-splice site on the HPV-16 genome. J. Virol. 2010, 84, 8219–8230. [Google Scholar] [CrossRef] [PubMed]
- Berget, S. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995, 270, 2411–2414. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.; Kelehan, P.; Lambkin, H.; Schwartz, S. Increased expression of cellular RNA-binding proteins in HPV-induced neoplasia and cervical cancer. J. Med. Virol. 2009, 81, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.; McFarlane, M.; Chuen-Im, T.; Milligan, S.G.; Millan, D.; Graham, S.V. RNA splicing factors regulated by HPV16 during cervical tumour progression. J. Pathol. 2009, 219, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Johannson, C.; Somberg, M.; Li, X.; Winquist, E.B.; Fay, J.; Ryan, F.; Pim, D.; Banks, L.; Schwartz, S. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J. 2012, 31, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Anderson, D.E.; van Doorslaer, K.; McBride, A.A. A proteomic approach to discover and compare interacting partners of papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics 2015, 15, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Green, R. Connections underlying translation and mRNA stability. J. Mol. Biol. 2016, 428, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Hall-Pogar, T.; Liang, S.; Hague, L.K.; Lutz, C.S. Specific trans-acting proteins interact with auxiliary RNA polyadenylation elements in the COX-2 3′-UTR. RNA 2007, 13, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Moriera, A.; Takagaki, Y.; Brackenridge, S.; Wollerton, M.; Manley, J.L.; Proudfoot, N.J. The upstream sequence element of the C2 complement poly(A) signal activates mRNA 3′ end formation by two distinct mechanisms. Genes Dev. 1998, 12, 2522–2534. [Google Scholar] [CrossRef]
- Natalizio, B.J.; Muñiz, L.C.; Arhin, G.K.; Wilusz, J.; Lutz, C.S. Upstream elements present in the 3′-untranslated region of collagen genes influence the processing efficiency of overlapping polyadenylation signals. J. Biol. Chem. 2002, 277, 42733–42740. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, G.M.; Fleming, E.S.; Oetjen, J.; Graveley, B.R. CPSF recognition of an HIV-1 mRNA 3′ processing enhancer: Multiple sequence contacts involved in poly(A) site definition. Genes Dev. 1995, 9, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.S.; Alwine, J.C. Direct interaction of the U1 snRNP-A protein with the upstream efficiency element of the SV40 late polyadenylation signal. Genes Dev. 1994, 8, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.S.; Murthy, K.G.K.; Schek, N.; O’Connor, J.P.; Manley, J.L.; Alwine, J.C. Interaction between the U1 snRNP-A protein and the 160 kD subunit of cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro. Genes Dev. 1996, 10, 325–337. [Google Scholar] [CrossRef] [PubMed]
- DeZazzo, J.D.; Imperiale, M.J. Sequences upstream of AAUAAA influence poly(A) site selection in a complex transcription unit. Mol. Cell. Biol. 1989, 9, 4951–4961. [Google Scholar] [CrossRef] [PubMed]
- Boelens, W.C.; Jansen, E.J.R.; van Venrooij, W.J.; Stripecke, R.; Mattaj, I.M.; Gunderson, S.I. The human U1 snRNP-specific U1A protein inhibits polyadenylation of its own pre-mRNA. Cell 1993, 72, 881–892. [Google Scholar] [CrossRef]
- Zhao, X.; Rush, M.; Carlsson, A.; Schwartz, S. The presence of inhibitory RNA elements in the late 3′-untranslated region is a conserved property of human papillomaviruses. Virus Res. 2007, 125, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Papillomavirus 3′UTR regulatory elements. Front. Biosci. 2008, 13, 5646–5663. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; McPhillips, M.G.; Veerapraditsin, T.; Milligan, S.G.; Graham, S.V. Activity of the human papillomavirus type 16 late negative regulatory element is partly due to four weak consensus 5′ splice sites that bind a U1 snRNP-like complex. J. Virol. 2003, 77, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Goraczniak, R.; Gunderson, S.I. The regulatory element in the 3′-untranslated region of human papillomavirus 16 inhibits expression by binding CUG-binding protein 1. J. Biol. Chem. 2008, 283, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Chuen-Im, T.; Zhang, J.; Graham, S.V. The RNA stability regulator HuR regulates L1 protein expression in vivo in differentiating cervical epithelial cells. Virology 2009, 383, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Eberle, A.B.; Visa, N. Quality control of mRNP biogenesis: Networking at the transcription site. Semin. Cell Dev. Biol. 2014, 32, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Celik, A.; He, F.; Jacobson, A. NMD monitors translational fidelity 24/7. Curr. Genet. 2017, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Schwartz, S. RNA binding proteins that control human papillomavirus gene expression. Biomolecules 2015, 5, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Koffa, M.D.; Graham, S.V.; Takagaki, Y.; Manley, J.L.; Clements, J.B. The human papillomavirus type 16 negative regulatory element interacts with three proteins that act at different posttranscriptional levels. Proc. Natl. Acad. Sci. USA 2000, 97, 4677–4682. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Hernandez-Lopez, H.; MacDonald, A.I.; Bodily, J.M.; Graham, S.V. Human papillomavirus E2 regulates SRSF3 (SRp20) to promote capsid protein expression in infected differentiated keratinocytes. J. Virol. 2016, 90, 5047–5058. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Sokolowski, M.; Tan, W.; Schwartz, S. Characterisation and partial purification of cellular factors interacting with a negative element on human papillomavirus type 1 late mRNAs. Virus Res. 1998, 55, 1–13. [Google Scholar] [CrossRef]
- Sokolowski, M.; Zhao, C.; Tan, W.; Schwartz, S. AU-rich mRNA instability elements on human papillomavirus type 1 late mRNAs and c-fos mRNAs interact with the same cellular factors. Oncogene 1997, 15, 2303–2319. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, M.; Furneaux, H.; Schwartz, S. The inhibitory activity of the AU-rich RNA element in the human papillomavirus type 1 late 3′ untranslated region correlates with its affinity for the elav-like HuR protein. J. Virol. 1999, 73, 1080–1091. [Google Scholar] [PubMed]
- Sokolowski, M.; Schwartz, S. Heterogeneous nuclear ribonucleoprotein C binds exclusively to the functionally important UUUUU-motifs in the human papillomavirus type-1 AU-rich inhibitory element. Virus Res. 2001, 73, 163–175. [Google Scholar] [CrossRef]
- Sokolowski, M.; Tan, W.; Jellne, M.; Schwartz, S. mRNA instability elements in the human papillomavirus type 16 L2 coding region. J. Virol. 1998, 72, 1504–1515. [Google Scholar] [PubMed]
- Hawkins, J.D. A survey on intron and exon lengths. Nucleic Acids Res. 1988, 16, 9893–9908. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, G.; Lafaille, C.; Petrillo, E.; Buggiano, V.; Gómez Acuña, L.I.; Fiszbein, A.; Godoy Herz, M.A.; Nieto Moreno, N.; Muñoz, M.J.; Alló, M.; et al. Transcriptional elongation and alternative splicing. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Atambayeva, S.A.; Khailenko, V.A.; Ivashchenko, A.T. Intron and exon length variation in Arabidopsis, rice, nematode, and human. Mol. Biol. 2008, 42, 352–361. [Google Scholar]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Hertel, K.J. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip. Rev. RNA 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eperon, I.C.; Makarova, O.V.; Mayeda, A.; Munroe, S.H.; Caceres, J.F.; Hayward, D.G.; Krainer, A.R. Selection of alternative 5′ splice sites: Role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol. Cell. Biol. 2000, 20, 8303–8318. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Li, X.; Johansson, C.; Orru, B.; Chang, R.; Rush, M.; Fay, J.; Ryan, F.; Schwartz, S. Serine/arginine-rich protein 30c activates human papillomavirus type 16 L1 mRNA expression via a bimodal mechanism. J. Gen. Virol. 2011, 92, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fay, J.; Lambkin, H.; Schwartz, S. Identification of a 17-nucleotide splicing enhancer in HPV-16 L1 that counteracts the effect of multiple hnRNP A1-binding splicing silencers. Virology 2007, 369, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rush, M.; Schwartz, S. Identification of an hnRNP A1-dependent splicing silencer in the human papillomavirus type 16 L1 coding region that prevents premature expression of the late L1 gene. J. Virol. 2004, 78, 10888–10905. [Google Scholar] [CrossRef] [PubMed]
- Chuen-Im, T.; Zhang, J.; Milligan, S.G.; McPhillips, M.G.; Graham, S.V. The alternative splicing factor hnRNP A1 is up-regulated during virus-infected epithelial cell differentiation and binds the human papillomavirus type 16 late regulatory element. Virus Res. 2008, 131, 189–198. [Google Scholar]
- Li, X.; Johansson, C.; Glahder, J.; Mossberg, A.-K.; Schwartz, S. Suppression of HPV-16 late L1 5′-splice site SD3632 by binding of hnRNP D proteins and hnRNP A2/B1 to upstream AUAGUA RNA motifs. Nucleic Acids Res. 2013, 41, 10488–10508. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Zhao, X.; Frõhlich, M.; Evander, M.; Schwartz, S. Polypyrimidine tract binding protein induces human papillomavirus type 16 late gene expression by interfering with splicing inhibitory elements at the major late 5′ splice site, SD3632. J. Virol. 2008, 82, 3665–3678. [Google Scholar] [CrossRef] [PubMed]
- Dhanjal, S.; Kajitani, N.; Glahder, J.; Mossberg, A.-K.; Johansson, C.; Schwartz, S. Heterogeneous nuclear ribonucleoprotein C proteins interact with the human papillomavirus type 16 (HPV16) early 3′-untranslated region and alleviate suppression of HPV16 late L1 mRNA splicing. J. Biol. Chem. 2015, 290, 13354–13371. [Google Scholar] [CrossRef] [PubMed]
- Collier, B.; Oberg, D.; Zhao, X.; Schwartz, S. Specific inactivation of inhibitory sequences in the 5′ end of the human papillomavirus type 16 L1 open reading frame results in production of high levels of L1 protein in human epithelial cells. J. Virol. 2002, 76, 2739–2752. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Some thoughts about translational regulation: Forward and backward glances. J. Cell. Biochem. 2007, 102, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.; Trus, B. The papillomavirus virion: A machine built to hide molecular achilles’ heels. In Viral Molecular Machines; Rossmann, M.G., Rao, V.B., Eds.; Springer: New York, NY, USA, 2012; Volume 726, pp. 403–422. [Google Scholar]
- Zhou, J.; Lui, W.J.; Peng, S.W.; Sun, X.Y.; Frazer, I.H. Papillomavirus capsid protein expression levels depends on the match between codon usage and tRNA availability. J. Virol. 1999, 73, 4972–4982. [Google Scholar] [PubMed]
- Zhao, K.N.; Lui, W.J.; Frazer, I.H. Codon usage bias and A+T content variation in human papillomavirus genomes. Virus Res. 2003, 98, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.-N.; Gu, W.; Fang, N.X.; Saunders, N.A.; Frazer, I.H. Gene codon composition determines differentiation-dependent expression of a viral capsid gene in keratinocytes in vitro and in vivo. Mol. Cell. Biol. 2005, 25, 8643–8655. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.-X.; Gu, W.; Ding, J.; Saunders, N.A.; Frazer, I.H.; Zhao, K.-N. Calcium enhances mouse keratinocyte differentiation in vitro to differentially regulate expression of papillomavirus authentic and codon modified L1 genes. Virology 2007, 365, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Ding, J.; Wang, X.; de Kluyyer, R.L.; Saunders, N.A.; Frazer, I.H.; Zhao, K.-N. Generalized substitution of isoencoding codons shortens the duration of papillomavirus L1 protein expression in transiently gene-transfected keratinocytes due to cell differentiation. Nucleic Acids Res. 2007, 35, 4820–4832. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Doorbar, J.; Li, B.; Zhou, F.; Gu, W.; Zhao, L.; Saunders, N.A.; Frazer, I.H.; Zhao, K.-N. Expression of papillomavirus L1 proteins regulated by authentic gene codon usage is favoured in G2/M-like cells in differentiating keratinocytes. Virology 2010, 399, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Collier, B.; Goobar, L.; Sokolowski, M.; Schwartz, S. Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogeneous ribonucleoprotein K and poly (rC)-binding proteins 1 and 2. J. Biol. Chem. 1998, 273, 22648–22656. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, L.; Sokolowski, M.; Carlsson, A.; Rush, M.; Schwartz, S. Inhibition of translation by UAUUUAU and UAUUUUUAU motifs of the AU-rich RNA instability element in the HPV-1 late 3′ untranslated region. J. Biol. Chem. 2002, 277, 40462–40471. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV Genotype | Protein | Function |
|---|---|---|
| HPV1 | HuR | mRNA stability, polyadenylation, nuclear export |
| hnRNPC1/C2 | Splicing, polyadenylation, mRNA stability, nuclear export | |
| PABP | Polyadenylation, translation | |
| HPV16 | U1snRNP | splicing |
| HuR 1 | mRNA stability, polyadenylation, nuclear export | |
| SRSF1 3 | Splicing regulation | |
| hnRNP A1 | Splicing regulation | |
| U2AF | Auxiliary splicing factor | |
| CstF-64 | Polyadenylation | |
| CUG-BP | Alternative splicing, translation | |
| HPV31 | U1snRNP 2 | splicing |
| HuR | mRNA stability, polyadenylation, nuclear export | |
| U2AF | Auxiliary splicing factor | |
| CstF-64 | Polyadenylation | |
| HPV18 | U1snRNP2 | splicing |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, S.V. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses 2017, 9, 245. https://doi.org/10.3390/v9090245
Graham SV. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses. 2017; 9(9):245. https://doi.org/10.3390/v9090245
Chicago/Turabian StyleGraham, Sheila V. 2017. "Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation" Viruses 9, no. 9: 245. https://doi.org/10.3390/v9090245
APA StyleGraham, S. V. (2017). Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses, 9(9), 245. https://doi.org/10.3390/v9090245