
Regulation of Telomere Homeostasis during Epstein-Barr virus Infection and Immortalization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
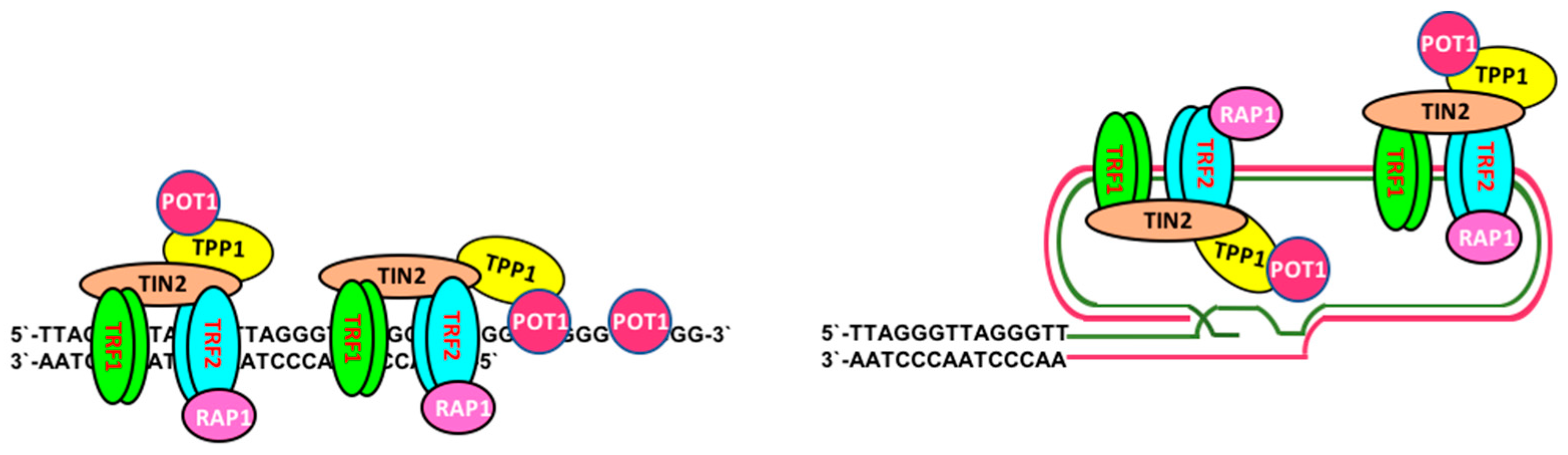
2. Telomere Maintenance and Cell Immortalization
2.1. Telomerase-Dependent Telomere Maintenance
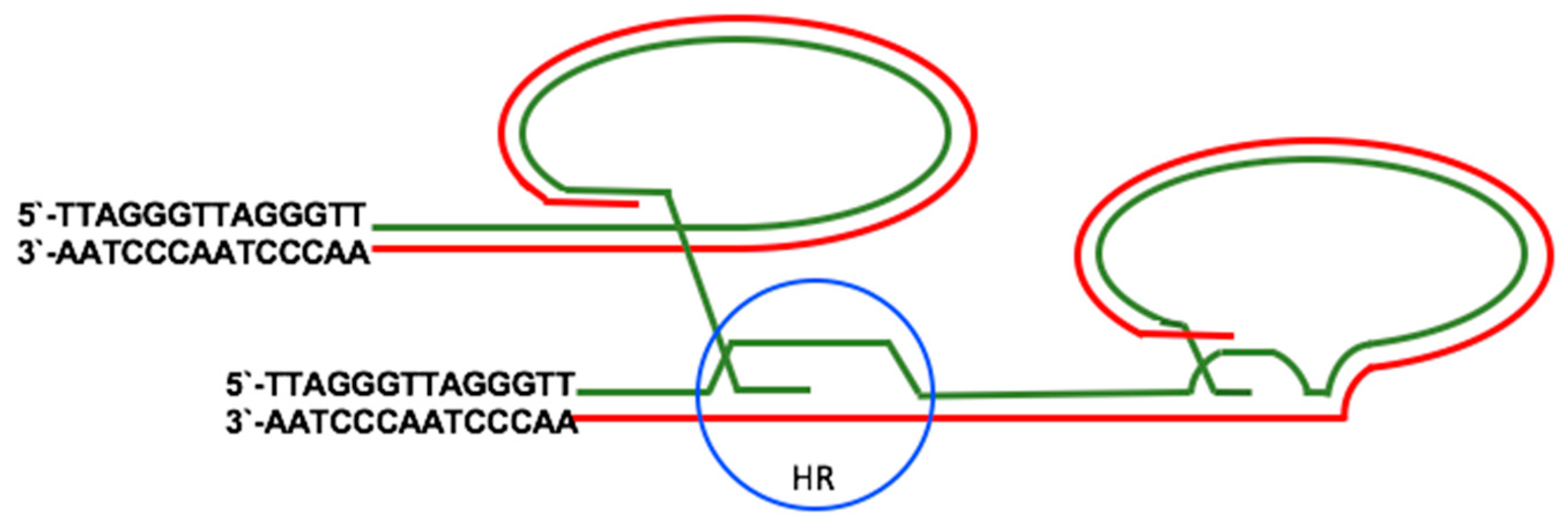
2.2. Telomerase-Independent Telomere Maintenance (ALT)
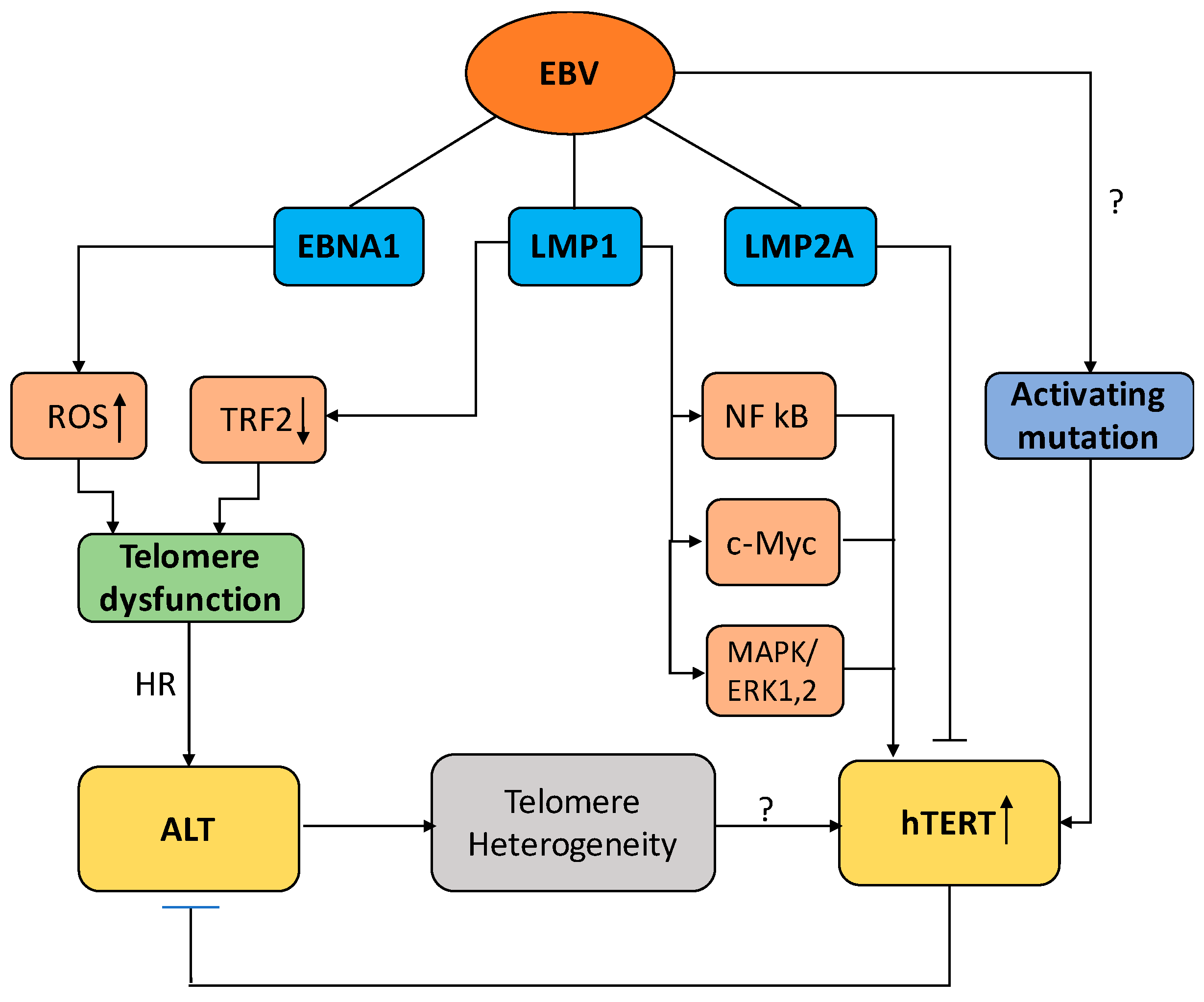
3. Telomere Maintenance in EBV-Infected Cells
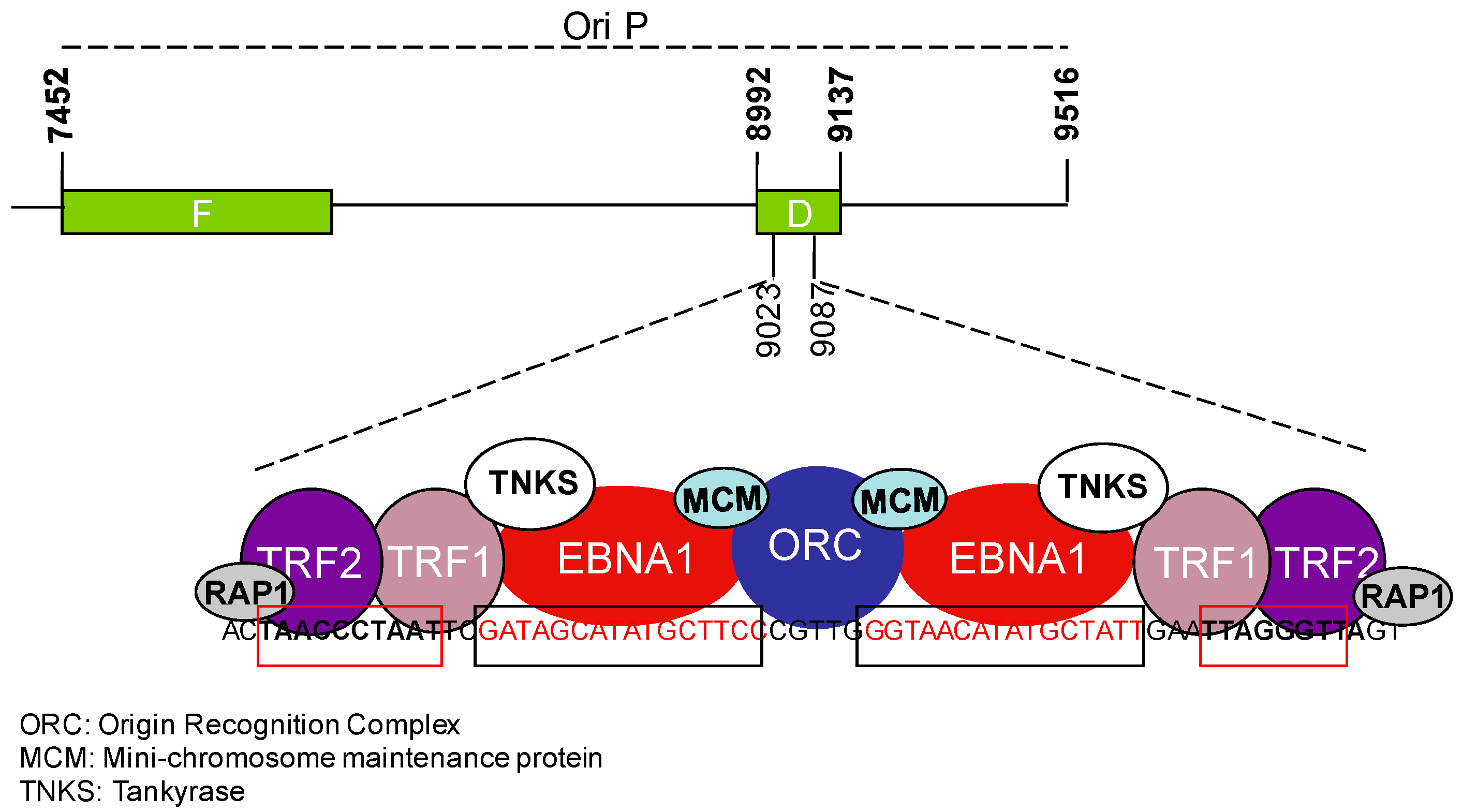
3.1. EBV and the Regulation of Telomerase Activity
3.2. EBV and the Activation of ALT
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Sitki-Green, D.L.; Edwards, R.H.; Covington, M.M.; Raab-Traub, N. Biology of Epstein-Barr virus during infectious mononucleosis. J. Infect. Dis. 2004, 189, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, R.; Dal Col, J.; Martorelli, D.; Carbone, A.; Klein, E. Interplay among viral antigens, cellular pathways and tumor microenvironment in the pathogenesis of EBV-driven lymphomas. Semin. Cancer Biol. 2013, 23, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Murray, P.G. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Yajima, M.; Kanda, T.; Takada, K. Critical role of Epstein-Barr Virus (EBV)-encoded RNA in efficient EBV-induced B-lymphocyte growth transformation. J. Virol. 2005, 79, 4298–4307. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Tahara, H.; Ide, T.; Furuichi, Y. Steps involved in immortalization and tumorigenesis in human B-lymphoblastoid cell lines transformed by Epstein-Barr virus. Cancer Res. 2004, 64, 3361–3364. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.P.; Nam, H.Y.; Shim, S.M.; Han, B.G. Sustained viral activity of epstein-Barr virus contributes to cellular immortalization of lymphoblastoid cell lines. Mol. Cells 2009, 27, 143–148. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, D.; Songyang, Z. The telosome/shelterin complex and its functions. Genome Biol. 2008, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Sfeir, A.J.; Shay, J.W.; Wright, W.E.; de Lange, T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005, 24, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Chen, Y.; Lei, M.; Chang, S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun. 2016, 7, 10881. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; van Steensel, B.; Bianchi, A.; Oelmann, S.; Schaefer, M.R.; Schnapp, G.; de Lange, T. Control of human telomere length by TRF1 and TRF2. Mol. Cell. Biol. 2000, 20, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L. Hunting G-quadruplexes. Biochimie 2008, 90, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, R.C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.V.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10114–10118. [Google Scholar] [CrossRef] [PubMed]
- Verdun, R.E.; Karlseder, J. Replication and protection of telomeres. Nature 2007, 447, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Mitchell, M.; Gillis, A.; Futahashi, M.; Fujiwara, H.; Skordalakes, E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat. Struct. Mol. Biol. 2010, 17, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Liu, D.; Songyang, Z. Telomere maintenance through spatial control of telomeric proteins. Mol. Cell. Biol. 2007, 27, 5898–5909. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.W.; Sokoloski, J.A.; Perez, J.R.; Maltese, J.Y.; Sartorelli, A.C.; Stein, C.A.; Nichols, G.; Khaled, Z.; Telanq, N.T.; Narayanan, R. Differentiation of immortal cells inhibits telomerase activity. Proc. Natl. Acad. Sci. USA 1995, 92, 12343–12346. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes (Basel) 2016, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Cable, P.L.; Afshari, C.; Barrett, J.C. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999, 59, 826–830. [Google Scholar] [PubMed]
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariqa, H.; Inoue, M. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000, 28, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Chois, S.; Honq, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, M.A.; Chandriani, S.; Park, J.; Kotenko, I.; Matheos, D.; Johnsson, A.; McMahon, S.B.; Cole, M.D. TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol. Cell. Biol. 2002, 22, 5054–5063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kyo, S.; Takakura, M.; Tanaka, M.; Yatabe, N.; Maida, Y.; Fujiwara, M.; Hayakawa, J.; Ohmichi, M.; Koike, K.; et al. Progesterone regulates human telomerase reverse transcriptase gene expression via activation of mitogen-activated protein kinase signaling pathway. Cancer Res. 2000, 60, 5376–5381. [Google Scholar] [PubMed]
- Ge, Z.; Liu, C.; Bjorkholm, M.; Gruber, A.; Xu, D. Mitogen-activated protein kinase cascade-mediated histone H3 phosphorylation is critical for telomerase reverse transcriptase expression/telomerase activation induced by proliferation. Mol. Cell. Biol. 2006, 26, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, L.; Yang, Z.; Funder, J.W.; Liu, J.P. Telomerase is controlled by protein kinase Cα in human breast cancer cells. J. Biol. Chem. 1998, 273, 33436–33442. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 1999, 274, 13085–13090. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Hideshima, T.; Hayashi, T.; Tai, Y.T.; Mitsiades, C.S.; Mitsiades, N.; Chauhan, D.; Richardson, P.; Munshi, N.C.; Anderson, K.C. Nuclear factor-κB p65 mediates tumor necrosis factor α-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res. 2003, 63, 18–21. [Google Scholar] [PubMed]
- Li, H.; Zhao, L.L.; Funder, J.W.; Liu, J.P. Protein phosphatase 2A inhibits nuclear telomerase activity in human breast cancer cells. J. Biol. Chem. 1997, 272, 16729–16732. [Google Scholar] [CrossRef] [PubMed]
- Takakura, M.; Kyo, S.; Inoue, M.; Wright, W.E.; Shay, J.W. Function of AP-1 in transcription of the telomerase reverse transcriptase gene (TERT) in human and mouse cells. Mol. Cell. Biol. 2005, 25, 8037–8043. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-De Matte, M.Y.; Yang, H.; Evans, M.S.; Cheng, J.Q.; Kruk, P.A. Telomerase is regulated by c-Jun NH2-terminal kinase in ovarian surface epithelial cells. Cancer Res. 2002, 62, 4575–4578. [Google Scholar] [PubMed]
- Chang, J.T.; Yang, H.T.; Wang, T.C.; Cheng, A.J. Upstream stimulatory factor (USF) as a transcriptional suppressor of human telomerase reverse transcriptase (hTERT) in oral cancer cells. Mol. Carcinog. 2005, 44, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Fan, S.; Meng, Q.; Schramm, L.; Wang, C.; Bouzahza, B.; Zhou, J.; Zafonte, B.; Goldberq, I.D.; Haddad, B.R.; et al. BRCA1 inhibition of telomerase activity in cultured cells. Mol. Cell. Biol. 2003, 23, 8668–8690. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Z.; Lu, K.P. The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell 2001, 107, 347–359. [Google Scholar] [CrossRef]
- Lin, S.Y.; Elledge, S.J. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003, 113, 881–889. [Google Scholar] [CrossRef]
- Li, H.; Xu, D.; Li, J.; Berndt, M.C.; Liu, J.P. Transforming growth factor β suppresses human telomerase reverse transcriptase (hTERT) by Smad3 interactions with c-Myc and the hTERT gene. J. Biol. Chem. 2006, 281, 25588–25600. [Google Scholar] [CrossRef] [PubMed]
- Shats, I.; Milyavsky, M.; Tang, X.; Stambolsky, P.; Erez, N.; Brosh, R.; Koqan, I.; Braunstein, I.; Tzukerman, M.; Ginsberg, D.; et al. p53-dependent down-regulation of telomerase is mediated by p21waf1. J. Biol. Chem. 2004, 279, 50976–50985. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, V.; Lingner, J. TERRA promotes telomere shortening through exonuclease 1-mediated resection of chromosome ends. PLoS Genet. 2012, 8, e1002747. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Smogorzewska, A.; de Lange, T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 2004, 119, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Slatter, T.L.; Tan, X.; Yuen, Y.C.; Gunningham, S.; Ma, S.S.; Daly, E.; Packer, S.; Devenish, C.; Royds, J.A.; Hunq, N.A. The alternative lengthening of telomeres pathway may operate in non-neoplastic human cells. J. Pathol. 2012, 226, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Jiang, X.; Lee, W.H.; Chen, P.L. Assembly of functional ALT-associated promyelocytic leukemia bodies requires Nijmegen Breakage Syndrome 1. Cancer Res. 2003, 63, 2589–2595. [Google Scholar] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.K.; Lin, T.; Tsai, R.Y. Nucleostemin prevents telomere damage by promoting PML-IV recruitment to SUMOylated TRF1. J. Cell Biol. 2012, 197, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Worz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J. Cell Sci. 2015, 128, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Chen, Y.C.; Spector, D.L.; de Lange, T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008, 456, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londono-Vallejo, A.; Decottiqnies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef] [PubMed]
- Galati, A.; Micheli, E.; Alicata, C.; Ingegnere, T.; Cicconi, A.; Pusch, M.C.; Giraud-Panis, M.J.; Gilson, E.; Cacchione, S. TRF1 and TRF2 binding to telomeres is modulated by nucleosomal organization. Nucleic Acids Res. 2015, 43, 5824–5837. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Betteqowda, C.; Rodriquez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef] [PubMed]
- Conomos, D.; Reddel, R.R.; Pickett, H.A. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat. Struct. Mol. Biol. 2014, 21, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Marzec, P.; Armenise, C.; Perot, G.; Roumelioti, F.M.; Basyuk, E.; Gagos, S.; Chibon, F.; Dejardin, J. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 2015, 160, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Nicot, C. Regulation of telomerase and telomeres: Human tumor viruses take control. J. Natl. Cancer Inst. 2008, 100, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Nosek, J.; Kosa, P.; Tomaska, L. On the origin of telomeres: A glimpse at the pre-telomerase world. Bioessays 2006, 28, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Lieberman, P.M. Telomeres and viruses: Common themes of genome maintenance. Front. Oncol. 2012, 2, 201. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, I.; Elias, P. Knockdown of DNA ligase IV/XRCC4 by RNA interference inhibits herpes simplex virus type I DNA replication. J. Biol. Chem. 2007, 282, 10865–10872. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, W.; Sugden, B. Replication of Epstein-Barr viral DNA. Cold Spring Harb. Perspect. Biol. 2013, 5, a013029. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, C.; Deng, Z.; Wiedmer, A.; Norseen, J.; Lieberman, P.M. ORC binding to TRF2 stimulates OriP replication. EMBO Rep. 2006, 7, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.M.; Zhang, X.Y.; McMahon, S.B.; Lieberman, P.M. Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J. Virol. 2006, 80, 5723–5732. [Google Scholar] [CrossRef] [PubMed]
- Holdorf, M.M.; Cooper, S.B.; Yamamoto, K.R.; Miranda, J.J. Occupancy of chromatin organizers in the Epstein-Barr virus genome. Virology 2011, 415, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Stong, N.; Plasschaert, R.; Moczan, A.; Chen, H.S.; Wikramasinqhe, P.; Davuluri, R.V.; Bartolomei, M.S.; Riethman, H.; et al. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J. 2012, 31, 4165–4178. [Google Scholar] [CrossRef] [PubMed]
- Pentland, I.; Parish, J.L. Targeting CTCF to Control Virus Gene Expression: A Common Theme amongst Diverse DNA Viruses. Viruses 2015, 7, 3574–3585. [Google Scholar] [CrossRef] [PubMed]
- Snudden, D.K.; Hearing, J.; Smith, P.R.; Grasser, F.A.; Griffin, B.E. EBNA-1, the major nuclear antigen of Epstein-Barr virus, resembles ‘RGG’ RNA binding proteins. EMBO J. 1994, 13, 4840–4847. [Google Scholar] [PubMed]
- Nayyar, V.K.; Shire, K.; Frappier, L. Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2). J. Cell Sci. 2009, 122, 4341–4350. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.; Ujihara, M.; Wong, S.; Ott, C.; Middeldorp, J.; Aiyar, A. The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol. 2004, 78, 11487–11505. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Wu, C.W.; Chang, S.C.; Chen, T.Y.; Hu, C.R.; Yeh, M.Y.; Chen, J.Y.; Chen, M.R. Epstein-Barr virus nuclear antigen 1 is a DNA-binding protein with strong RNA-binding activity. J. Gen. Virol. 2004, 85, 2755–2765. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Atanasiu, C.; Zhao, K.; Marmorstein, R.; Sbodio, J.I.; Chi, N.W.; Lieberman, P.M. Inhibition of Epstein-Barr virus OriP function by tankyrase, a telomere-associated poly-ADP ribose polymerase that binds and modifies EBNA1. J. Virol. 2005, 79, 4640–4650. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Lieberman, P.M. The replisome pausing factor Timeless is required for episomal maintenance of latent Epstein-Barr virus. J. Virol. 2011, 85, 5853–5863. [Google Scholar] [CrossRef] [PubMed]
- Leman, A.R.; Dheekollu, J.; Deng, Z.; Lee, S.W.; Das, M.M.; Lieberman, P.M.; Noquchi, E. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 2012, 11, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Leman, A.R.; Noguchi, C.; Lee, C.Y.; Noguchi, E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 2010, 123, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Orr, A.; Everett, R.D. Stimulation of the Replication of ICP0-Null Mutant Herpes Simplex Virus 1 and pp71-Deficient Human Cytomegalovirus by Epstein-Barr Virus Tegument Protein BNRF1. J. Virol. 2016, 90, 9664–9673. [Google Scholar] [CrossRef] [PubMed]
- Terrin, L.; Dolcetti, R.; Corradini, I.; Indraccolo, S.; Dal Col, J.; Bertorelle, R.; Bonaldi, L.; Esposito, G.; De Rossi, A. hTERT inhibits the Epstein-Barr virus lytic cycle and promotes the proliferation of primary B lymphocytes: Implications for EBV-driven lymphomagenesis. Int. J. Cancer 2007, 121, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Giunco, S.; Celeghin, A.; Gianesin, K.; Dolcetti, R.; Indraccolo, S.; De Rossi, A. Cross talk between EBV and telomerase: The role of TERT and NOTCH2 in the switch of latent/lytic cycle of the virus. Cell Death Dis. 2015, 6, e1774. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; Deppmann, C.D.; Erickson, K.D.; Coffin, W.F., 3rd; Thornton, T.M.; Humphrey, S.E.; Martin, J.M.; Taparowsky, E.J. EBNA2 and activated Notch induce expression of BATF. J. Virol. 2003, 77, 6029–6040. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Tumor viruses and replicative immortality--avoiding the telomere hurdle. Semin. Cancer Biol. 2014, 26, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, S.; Wiechec, E.; Dos Santos Silva, A.G.; Guffei, A.; Williams, G.; Lowbeer, M.; Benedek, K.; Henriksson, M.; Klein, G.; Mai, S. Chromosomal rearrangements after ex vivo Epstein-Barr virus (EBV) infection of human B cells. Oncogene 2010, 29, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Ide, T.; Goto, M.; Furuichi, Y. Reconsideration of senescence, immortalization and telomere maintenance of Epstein-Barr virus-transformed human B-lymphoblastoid cell lines. Mech. Ageing Dev. 1999, 107, 51–60. [Google Scholar] [CrossRef]
- Kamranvar, S.A.; Chen, X.; Masucci, M.G. Telomere dysfunction and activation of alternative lengthening of telomeres in B-lymphocytes infected by Epstein-Barr virus. Oncogene 2013, 32, 5522–5530. [Google Scholar] [CrossRef] [PubMed]
- Mochida, A.; Gotoh, E.; Senpuku, H.; Harada, S.; Kitamura, R.; Takahashi, T.; Yanaqi, K. Telomere size and telomerase activity in Epstein-Barr virus (EBV)-positive and EBV-negative Burkitt’s lymphoma cell lines. Arch. Virol. 2005, 150, 2139–2150. [Google Scholar] [CrossRef] [PubMed]
- Terrin, L.; Dal Col, J.; Rampazzo, E.; Zancai, P.; Pedrotti, M.; Ammirabile, G.; Bergamin, S.; Rizzo, S.; Dolcetti, R.; De Rossi, A. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J. Virol. 2008, 82, 10175–10187. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Deng, X.; Deng, L.; Gu, H.; Fan, W.; Cao, Y. Telomerase activation by Epstein-Barr virus latent membrane protein 1 is associated with c-Myc expression in human nasopharyngeal epithelial cells. J. Exp. Clin. Cancer Res. 2004, 23, 495–506. [Google Scholar] [PubMed]
- Mei, Y.P.; Zhu, X.F.; Zhou, J.M.; Huang, H.; Deng, R.; Zeng, Y.X. siRNA targeting LMP1-induced apoptosis in EBV-positive lymphoma cells is associated with inhibition of telomerase activity and expression. Cancer Lett. 2006, 232, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Young, L.S. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 1998, 16, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, L.L.; Yang, J.; Tao, Y.G.; Ye, M.; Shi, Y.; Tang, M.; Yi, W.; Li, X.L.; Gong, J.P.; et al. Epstein-Barr virus encoded latent membrane protein 1 modulates nuclear translocation of telomerase reverse transcriptase protein by activating nuclear factor-κB p65 in human nasopharyngeal carcinoma cells. Int. J. Biochem. Cell Biol. 2005, 37, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, C.; Lindvall, C.; Xu, D.; Ernberg, I. Epstein-Barr virus latent membrane 2A (LMP2A) down-regulates telomerase reverse transcriptase (hTERT) in epithelial cell lines. Int. J. Cancer 2005, 113, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Gordadze, A.V.; Onunwor, C.W.; Peng, R.; Poston, D.; Kremmer, E.; Ling, P.D. EBNA2 amino acids 3 to 30 are required for induction of LMP-1 and immortalization maintenance. J. Virol. 2004, 78, 3919–3929. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 2011, 25, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Gruhne, B.; Szeles, A.; Masucci, M.G. Epstein-Barr virus promotes genomic instability in Burkitt’s lymphoma. Oncogene 2007, 26, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mactner, J. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, V.; Lemieux, B.; Sawan, B.; Lichtensztejn, D.; Lichtensztejn, Z.; Wellinger, R.; Mai, S.; Knecht, H. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 2015, 125, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M., 3rd; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell. Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, E.; Buonsante, R.; Leone, S.; Asmar, A.J.; Miller, K.L.; Cimini, D.; Sgura, A. Transient ALT activation protects human primary cells from chromosome instability induced by low chronic oxidative stress. Sci. Rep. 2017, 7, 43309. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Oxidative stress enables Epstein-Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 2016, 35, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 2000, 908, 99–110. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamranvar, S.A.; Masucci, M.G. Regulation of Telomere Homeostasis during Epstein-Barr virus Infection and Immortalization. Viruses 2017, 9, 217. https://doi.org/10.3390/v9080217
Kamranvar SA, Masucci MG. Regulation of Telomere Homeostasis during Epstein-Barr virus Infection and Immortalization. Viruses. 2017; 9(8):217. https://doi.org/10.3390/v9080217
Chicago/Turabian StyleKamranvar, Siamak A., and Maria G. Masucci. 2017. "Regulation of Telomere Homeostasis during Epstein-Barr virus Infection and Immortalization" Viruses 9, no. 8: 217. https://doi.org/10.3390/v9080217
APA StyleKamranvar, S. A., & Masucci, M. G. (2017). Regulation of Telomere Homeostasis during Epstein-Barr virus Infection and Immortalization. Viruses, 9(8), 217. https://doi.org/10.3390/v9080217