
Somatic Host Cell Alterations in HPV Carcinogenesis



Abstract
1. Introduction
2. Mechanisms of HPV-Mediated Mutagenesis
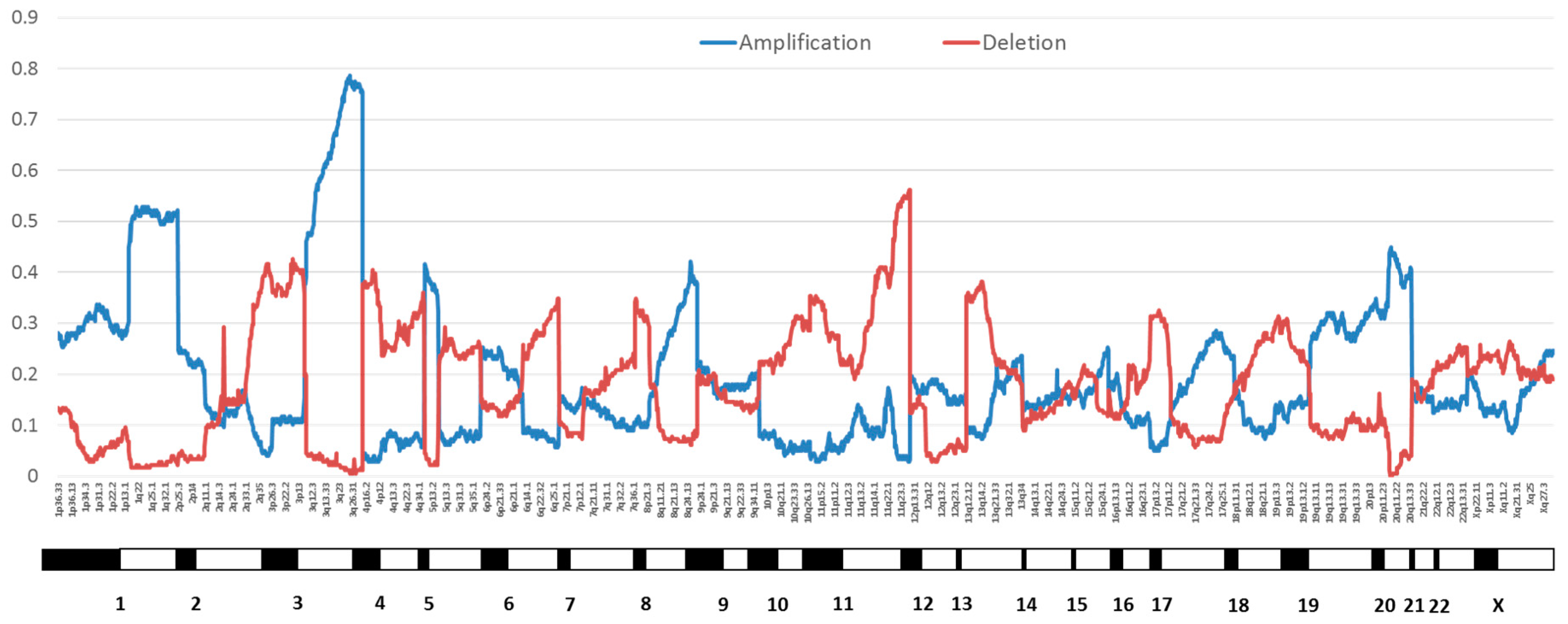
2.1. Genomic Instability
2.2. Mutational Signatures
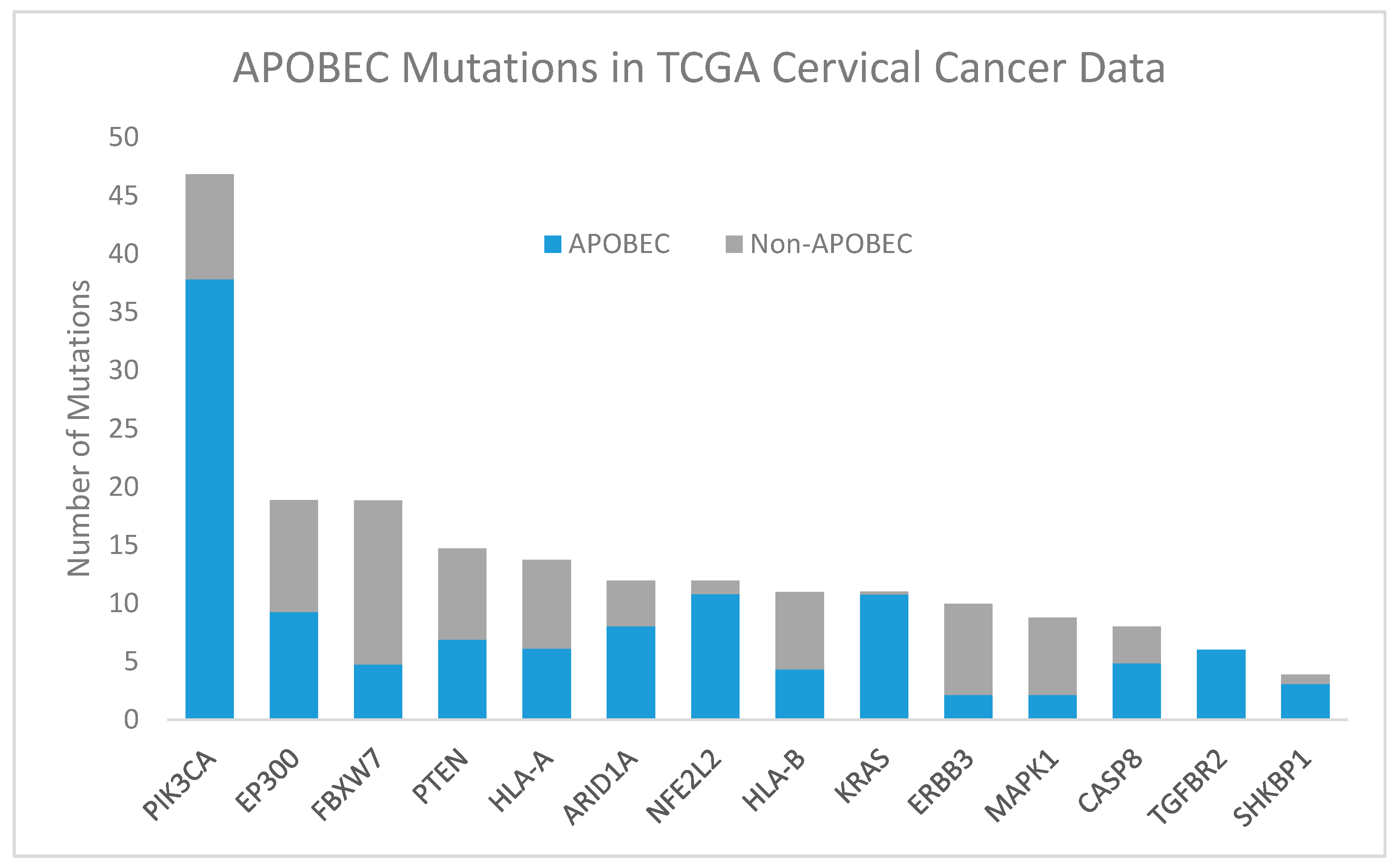
2.2.1. APOBEC
2.2.2. Other Mutational Signatures
3. Genes and Pathways
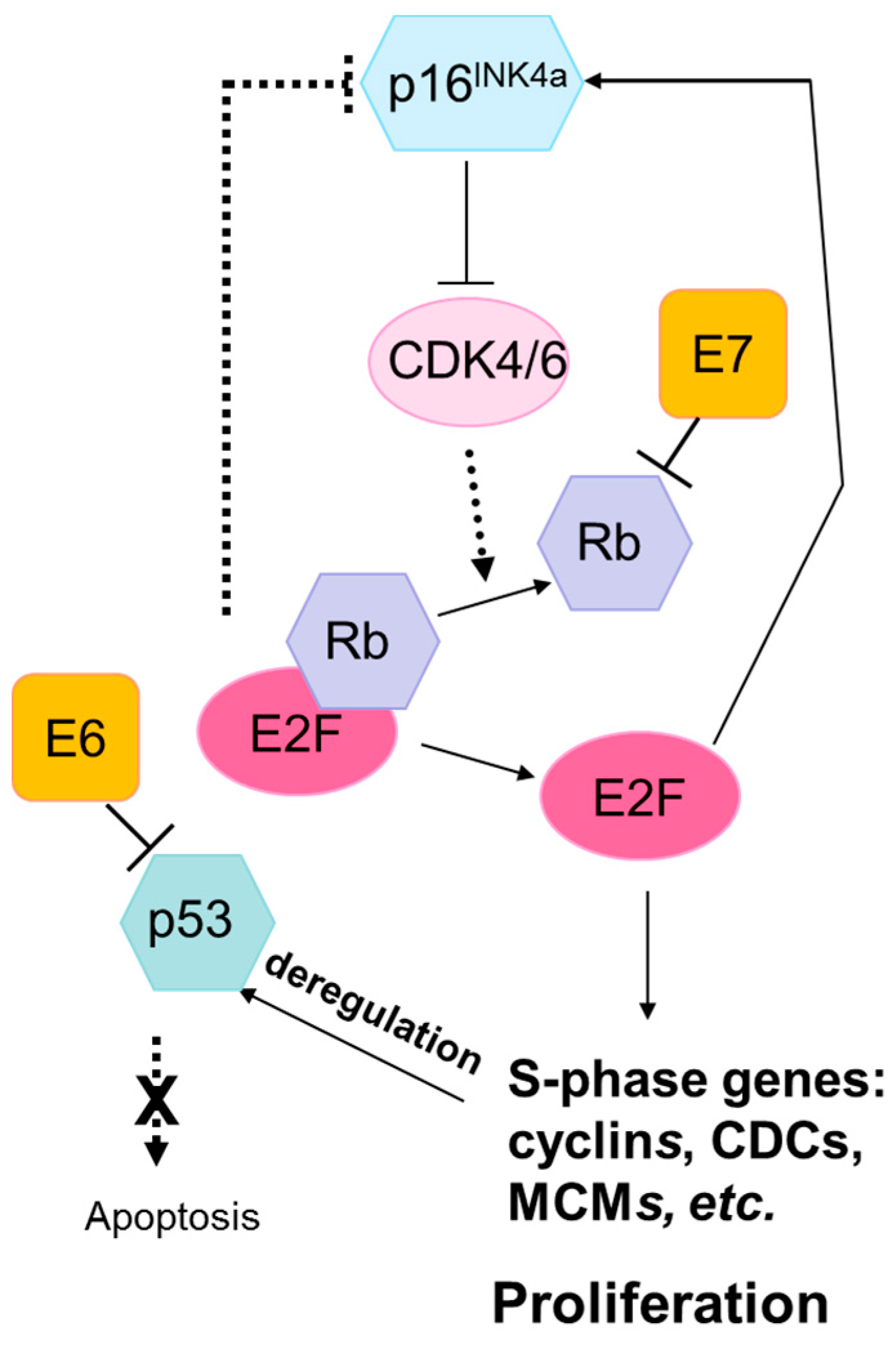
3.1. Lack of Mutations in TP53 and RB1
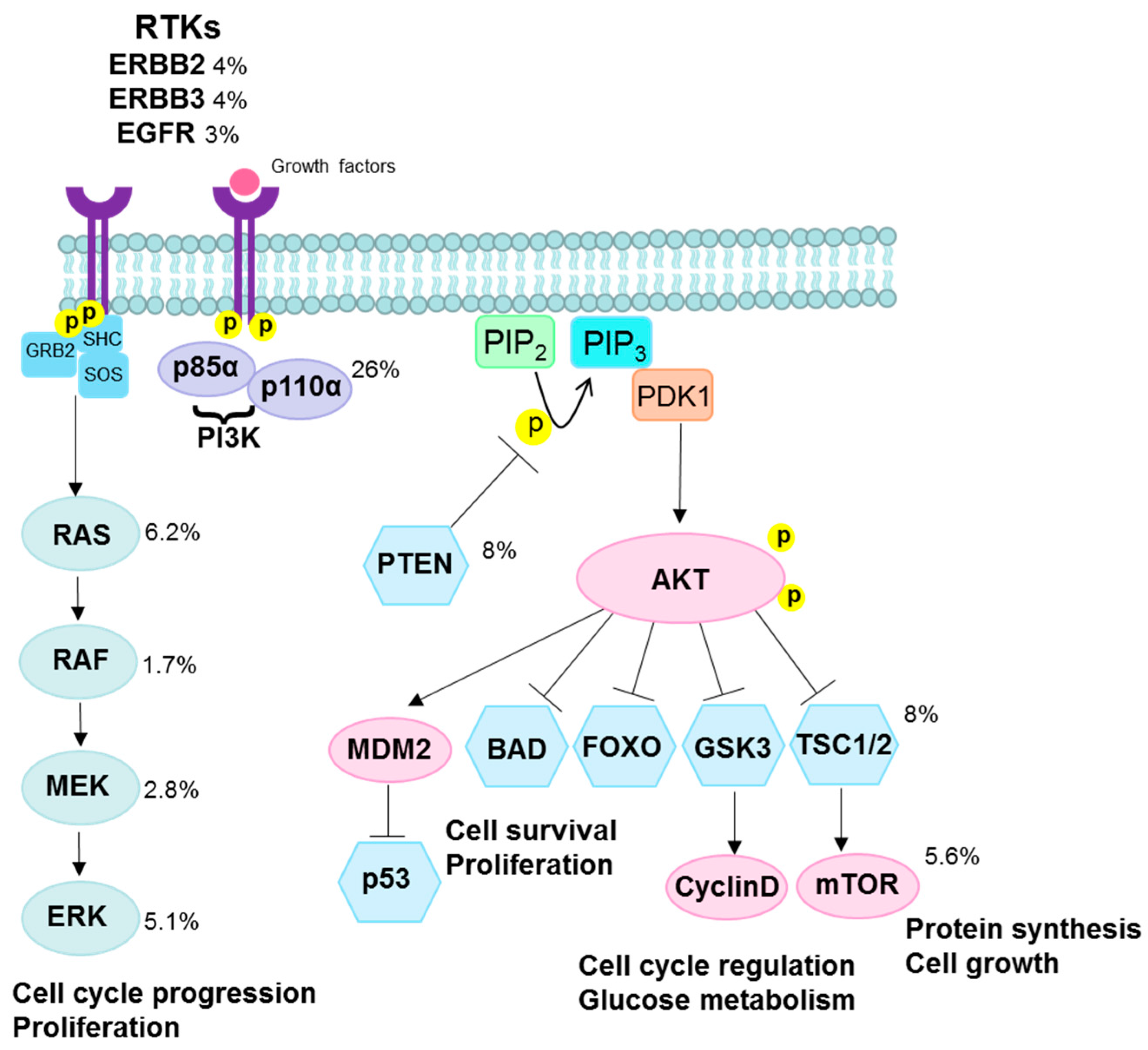
3.2. PI3K/AKT Pathway
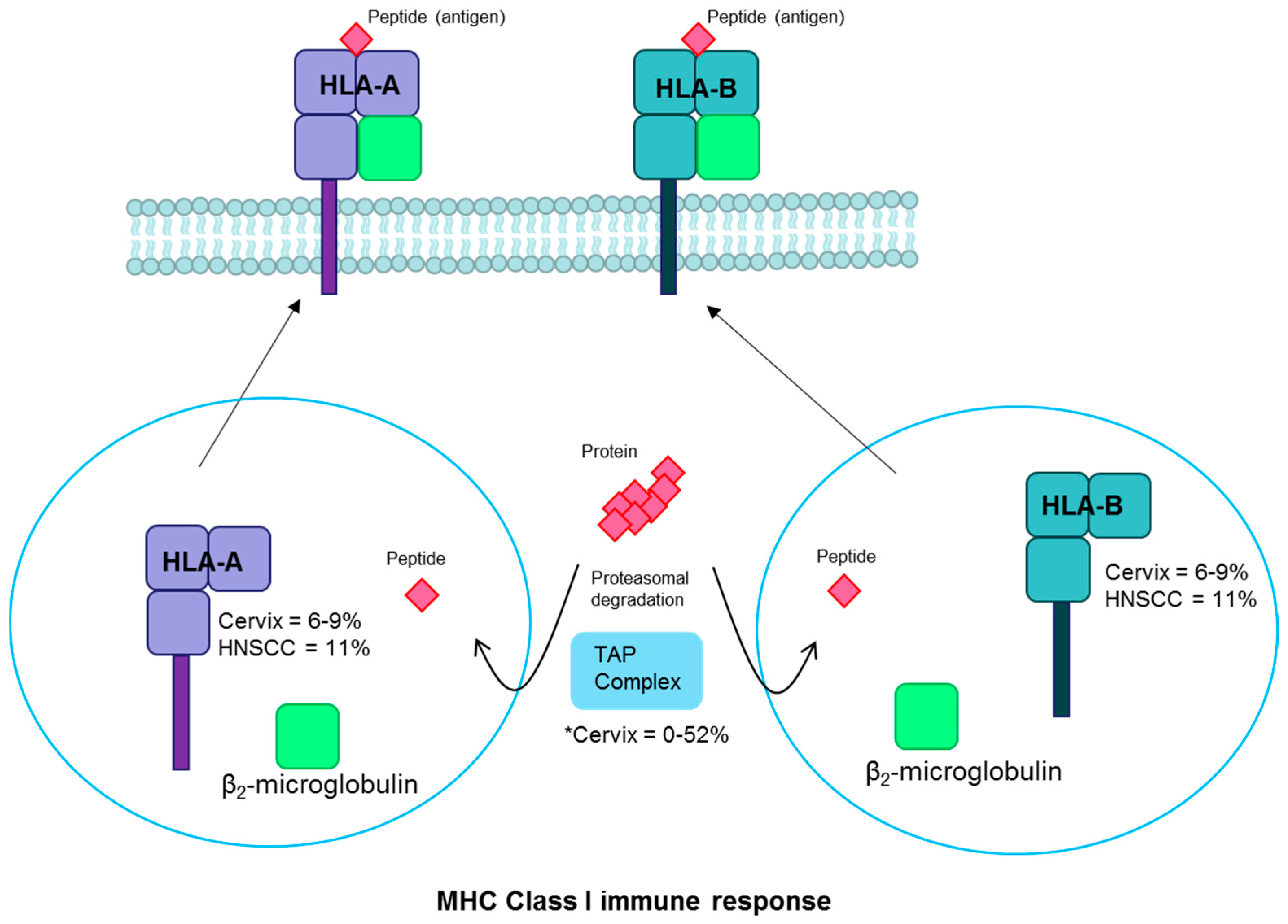
3.3. Human Leukocyte Antigen
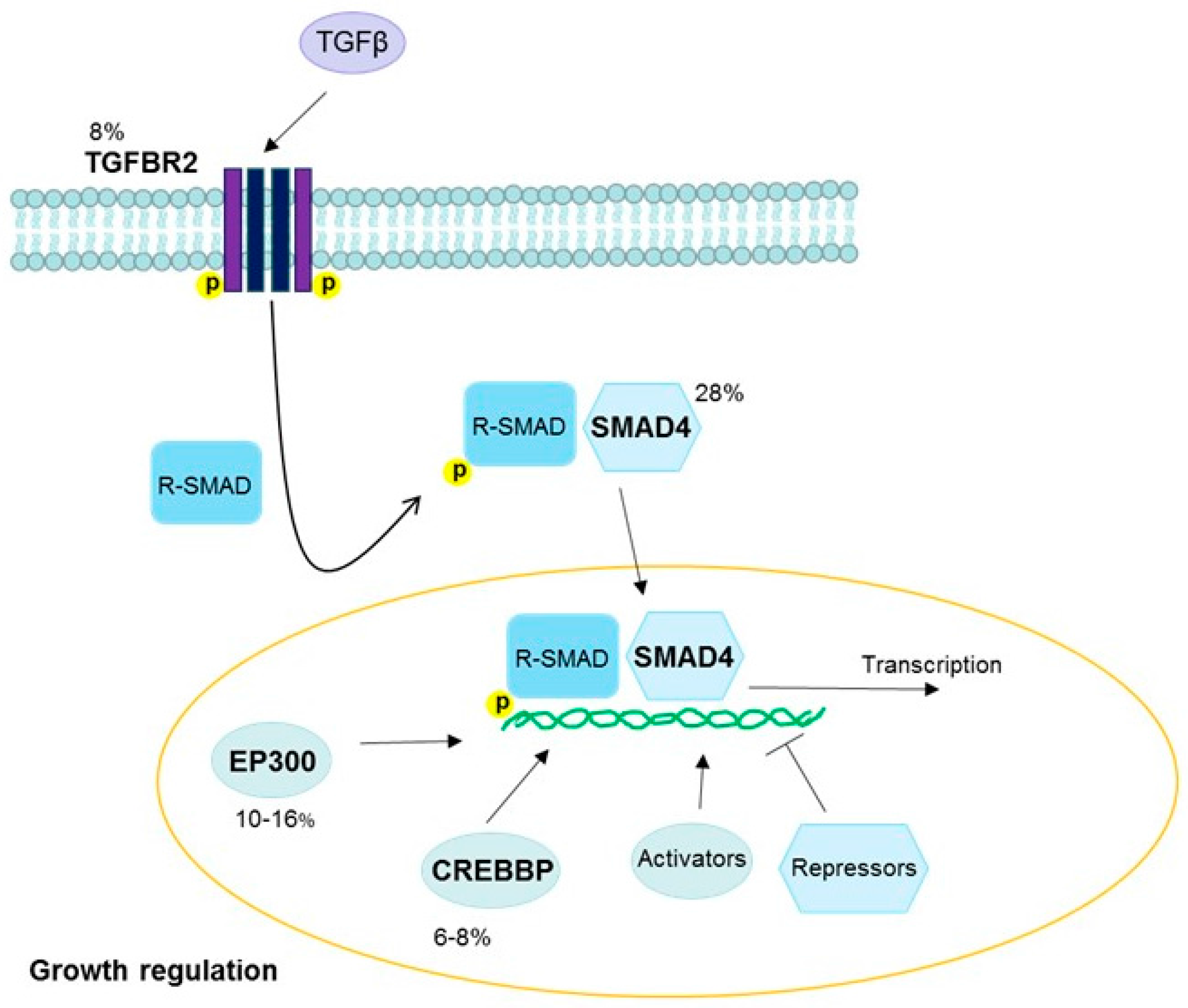
3.4. Transforming Growth Factor Beta Pathway
3.5. Notch Pathway
3.6. RAS/EGFR/ERK Pathway
3.7. Other Genes
4. Discussion
Acknowledgments
Conflicts of Interest
References
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsague, X.; Laporte, L.; Bosch, F.X.; de Sanjose, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4A detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Hartwig, S.; Baldauf, J.-J.; Dominiak-Felden, G.; Simondon, F.; Alemany, L.; de Sanjosé, S.; Castellsagué, X. Estimation of the epidemiological burden of HPV-related anogenital cancers, precancerous lesions, and genital warts in women and men in europe: Potential additional benefit of a nine-valent second generation HPV vaccine compared to first generation HPV vaccines. Papillomavirus Res. 2015, 1, 90–100. [Google Scholar]
- Backes, D.M.; Kurman, R.J.; Pimenta, J.M.; Smith, J.S. Systematic review of human papillomavirus prevalence in invasive penile cancer. Cancer Causes Control 2009, 20, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Miralles-Guri, C.; Bruni, L.; Cubilla, A.L.; Castellsague, X.; Bosch, F.X.; de Sanjose, S. Human papillomavirus prevalence and type distribution in penile carcinoma. J. Clin. Pathol. 2009, 62, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Ho, G.Y.; Bierman, R.; Beardsley, L.; Chang, C.J.; Burk, R.D. Natural history of cervicovaginal papillomavirus infection in young women. N. Engl. J. Med. 1998, 338, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Kjaer, S.K. Chapter 2: Natural history of anogenital human papillomavirus infection and neoplasia. J. Natl. Cancer Inst. Monogr. 2003, 2003, 14–19. [Google Scholar] [CrossRef]
- Rositch, A.F.; Koshiol, J.; Hudgens, M.G.; Razzaghi, H.; Backes, D.M.; Pimenta, J.M.; Franco, E.L.; Poole, C.; Smith, J.S. Patterns of persistent genital human papillomavirus infection among women worldwide: A literature review and meta-analysis. Int. J. Cancer 2013, 133, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Moscicki, A.B.; Schiffman, M.; Kjaer, S.; Villa, L.L. Chapter 5: Updating the natural history of HPV and anogenital cancer. Vaccine 2006, 2, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- McCredie, M.R.; Sharples, K.J.; Paul, C.; Baranyai, J.; Medley, G.; Jones, R.W.; Skegg, D.C. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: A retrospective cohort study. Lancet Oncol. 2008, 9, 425–434. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjose, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Arbyn, M.; Berkhof, J.; Bower, M.; Canfell, K.; Einstein, M.; Farley, C.; Monsonego, J.; Franceschi, S. Eurogin 2016 roadmap: How HPV knowledge is changing screening practice. Int. J. Cancer 2017, 140, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Rusan, M.; Li, Y.Y.; Hammerman, P.S. Genomic landscape of human papillomavirus-associated cancers. Clin. Cancer Res. 2015, 21, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar]
- Chen, D.; Gyllensten, U. Lessons and implications from association studies and post-gwas analyses of cervical cancer. Trends Genet. 2015, 31, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Nava, G.A.; Fernández-Niño, J.A.; Madrid-Marina, V.; Torres-Poveda, K. Cervical cancer genetic susceptibility: A systematic review and meta-analyses of recent evidence. PLoS ONE 2016, 11, e0157344. [Google Scholar] [CrossRef] [PubMed]
- Lesseur, C.; Diergaarde, B.; Olshan, A.F.; Wunsch-Filho, V.; Ness, A.R.; Liu, G.; Lacko, M.; Eluf-Neto, J.; Franceschi, S.; Lagiou, P.; et al. Genome-wide association analyses identify new susceptibility loci for oral cavity and pharyngeal cancer. Nat. Genet. 2016, 48, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.; Clifford, G.M. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int. J. Cancer 2011, 128, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Prigge, E.S.; von Knebel Doeberitz, M.; Reuschenbach, M. Clinical relevance and implications of HPV-induced neoplasia in different anatomical locations. Mutat. Res. 2017, 772, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. Centrosome abnormalities, genomic instability and carcinogenic progression. Biochim. Biophys. Acta 2001, 1471, M81–M88. [Google Scholar] [CrossRef]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- McDaniel, A.S.; Hovelson, D.H.; Cani, A.K.; Liu, C.J.; Zhai, Y.; Zhang, Y.; Weizer, A.Z.; Mehra, R.; Feng, F.Y.; Alva, A.S.; et al. Genomic profiling of penile squamous cell carcinoma reveals new opportunities for targeted therapy. Cancer Res. 2015, 75, 5219–5227. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.K.; Bermejo, J.L.; Vinokurova, S.; Jensen, K.; Bierkens, M.; Steenbergen, R.; Bergmann, M.; von Knebel Doeberitz, M.; Reuschenbach, M. Chromosomal gains and losses in human papillomavirus-associated neoplasia of the lower genital tract—A systematic review and meta-analysis. Eur. J. Cancer 2014, 50, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Vinokurova, S.; Sampson, J.N.; den Boon, J.A.; Walker, J.L.; Horswill, M.A.; Korthauer, K.; Schiffman, M.; Sherman, M.E.; Zuna, R.E.; et al. Chromosomal copy number alterations and HPV integration in cervical precancer and invasive cancer. Carcinogenesis 2016, 37, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Busso-Lopes, A.F.; Marchi, F.A.; Kuasne, H.; Scapulatempo-Neto, C.; Trindade-Filho, J.C.; de Jesus, C.M.; Lopes, A.; Guimaraes, G.C.; Rogatto, S.R. Genomic profiling of human penile carcinoma predicts worse prognosis and survival. Cancer Prev. Res. 2015, 8, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Heselmeyer, K.; du Manoir, S.; Blegen, H.; Friberg, B.; Svensson, C.; Schrock, E.; Veldman, T.; Shah, K.; Auer, G.; Ried, T. A recurrent pattern of chromosomal aberrations and immunophenotypic appearance defines anal squamous cell carcinomas. Br. J. Cancer 1997, 76, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Ridder, R.; Klaes, R.; Vinokurova, S.; Schaefer, U.; Doeberitz, M. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002, 21, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Durst, M.; Schneider, A.; von Knebel Doeberitz, M. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Driesch, C.; Jansen, L.; Runnebaum, I.B.; Durst, M. Non-random integration of the HPV genome in cervical cancer. PLoS ONE 2012, 7, e39632. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Vinokurova, S.; von Knebel Doeberitz, M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004, 64, 3878–3884. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Simonelli, V.; Narciso, L.; Dogliotti, E.; Fortini, P. Base excision repair intermediates are mutagenic in mammalian cells. Nucleic Acids Res. 2005, 33, 4404–4411. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3b mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Feber, A.; Worth, D.C.; Chakravarthy, A.; de Winter, P.; Shah, K.; Arya, M.; Saqib, M.; Nigam, R.; Malone, P.R.; Tan, W.S.; et al. CSN1 somatic mutations in penile squamous cell carcinoma. Cancer Res. 2016, 76, 4720–4727. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2013, 2, e00534. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; Hart, S.N.; Burns, M.B.; Carpenter, M.A.; Temiz, N.A.; Rathore, A.; Vogel, R.I.; Nikas, J.B.; Law, E.K.; Brown, W.L.; et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013, 73, 7222–7231. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.B.; Wu, J.S.; Craighead, P.S.; Phan, T.; Kobel, M.; Lees-Miller, S.P.; Ghatage, P.; Magliocco, A.M.; Doll, C.M. PIK3CA mutational status and overall survival in patients with cervical cancer treated with radical chemoradiotherapy. Gynecol. Oncol. 2013, 128, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Villagran, G.; Boland, J.F.; Im, K.M.; Polo, S.; Zhou, W.; Odey, U.; Juarez-Torres, E.; Medina-Martinez, I.; Roman-Basaure, E.; et al. Genome analysis of latin american cervical cancer: Frequent activation of the PIK3CA pathway. Clin. Cancer Res. 2015, 21, 5360–5370. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.A.; Howitt, B.E.; Myers, A.P.; Dahlberg, S.E.; Palescandolo, E.; Van Hummelen, P.; MacConaill, L.E.; Shoni, M.; Wagle, N.; Jones, R.T.; et al. Oncogenic mutations in cervical cancer: Genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer 2013, 119, 3776–3783. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Zheng, B.; Zhang, X.; Stendahl, U.; Andersson, S.; Wallin, K.L. Mutation of PIK3CA: Possible risk factor for cervical carcinogenesis in older women. Int. J. Oncol. 2009, 34, 409–416. [Google Scholar] [PubMed]
- Rashmi, R.; DeSelm, C.; Helms, C.; Bowcock, A.; Rogers, B.E.; Rader, J.L.; Grigsby, P.W.; Schwarz, J.K. Akt inhibitors promote cell death in cervical cancer through disruption of mtor signaling and glucose uptake. PLoS ONE 2014, 9, e92948. [Google Scholar] [CrossRef] [PubMed]
- Spaans, V.M.; Trietsch, M.D.; Crobach, S.; Stelloo, E.; Kremer, D.; Osse, E.M.; Haar, N.T.; van Eijk, R.; Muller, S.; van Wezel, T.; et al. Designing a high-throughput somatic mutation profiling panel specifically for gynaecological cancers. PLoS ONE 2014, 9, e93451. [Google Scholar] [CrossRef] [PubMed]
- Spaans, V.M.; Trietsch, M.D.; Peters, A.A.; Osse, M.; Ter Haar, N.; Fleuren, G.J.; Jordanova, E.S. Precise classification of cervical carcinomas combined with somatic mutation profiling contributes to predicting disease outcome. PLoS ONE 2015, 10, e0133670. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Annunziata, C.; Buonaguro, L.; Losito, S.; Greggi, S.; Buonaguro, F.M. TP53 and PIK3CA gene mutations in adenocarcinoma, squamous cell carcinoma and high-grade intraepithelial neoplasia of the cervix. J. Transl. Med. 2014, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.M.; Liu, X.; Wheler, J.; Naing, A.; Hong, D.; Coleman, R.L.; Tsimberidou, A.; Janku, F.; Zinner, R.; Lu, K.; et al. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: A phase I clinical experience. Oncotarget 2014, 5, 11168–11179. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.K.H.; Cheung, T.H.; Yim, S.F.; Yu, M.Y.; Chiu, R.W.K.; Lo, K.W.K.; Lee, I.P.C.; Wong, R.R.Y.; Lau, K.K.M.; Wang, V.W.; et al. Liquid biopsy of PIK3CA mutations in cervical cancer in Hong Kong Chinese women. Gynecol. Oncol. 2017, 146, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Arora, H.; Biswas, S.; Perwez, A.; Naseem, A.; Wajid, S.; Gandhi, G.; Rizvi, M.A. Mutation analysis of EGFR and its correlation with the HPV in indian cervical cancer patients. Tumour Biol. 2016, 37, 9089–9098. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Ishikawa, M.; Katagiri, A.; Yeasmin, S.; Otsuki, Y.; Kobayashi, H.; Nakayama, S.; et al. EGFR gene amplification is related to adverse clinical outcomes in cervical squamous cell carcinoma, making the EGFR pathway a novel therapeutic target. Br. J. Cancer 2011, 105, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Westra, W.H.; Taube, J.M.; Poeta, M.L.; Begum, S.; Sidransky, D.; Koch, W.M. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2008, 14, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Kashofer, K.; Regauer, S. Analysis of full coding sequence of the TP53 gene in invasive vulvar cancers: Implications for therapy. Gynecol. Oncol. 2017, 146, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Trietsch, M.D.; Nooij, L.S.; Gaarenstroom, K.N.; van Poelgeest, M.I. Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: A review of the current literature. Gynecol. Oncol. 2015, 136, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, H.S.; Seo, S.S.; Park, S.Y.; Sidransky, D.; Dong, S.M. Inverse correlation between RASSF1A hypermethylation, kras and braf mutations in cervical adenocarcinoma. Gynecol. Oncol. 2007, 105, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Stenzinger, A.; Eder, T.; Konschak, R.; Niehr, F.; Endris, V.; Distel, L.; Hautmann, M.G.; Mandic, R.; Stromberger, C.; et al. Targeted next-generation sequencing identifies molecular subgroups in squamous cell carcinoma of the head and neck with distinct outcome after concurrent chemoradiation. Ann. Oncol. 2016, 27, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Koopman, L.A.; Corver, W.E.; van der Slik, A.R.; Giphart, M.J.; Fleuren, G.J. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J. Exp. Med. 2000, 191, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Banister, C.E.; Liu, C.; Pirisi, L.; Creek, K.E.; Buckhaults, P.J. Identification and characterization of HPV-independent cervical cancers. Oncotarget 2017, 8, 13375–13386. [Google Scholar] [CrossRef] [PubMed]
- Salam, M.; Rockett, J.; Morris, A. The prevalence of different human papillomavirus types and p53 mutations in laryngeal carcinomas: Is there a reciprocal relationship? Eur. J. Surg. Oncol. 1995, 21, 290–296. [Google Scholar] [CrossRef]
- Rajendra, S.; Wang, B.; Merrett, N.; Sharma, P.; Humphris, J.; Lee, H.C.; Wu, J. Genomic analysis of HPV-positive versus HPV-negative oesophageal adenocarcinoma identifies a differential mutational landscape. J. Med. Genet. 2016, 53, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Edwards, A.; Fang, Z.; Flemington, E.K.; Zhang, K. Integrative genomics and transcriptomics analysis reveals potential mechanisms for favorable prognosis of patients with HPV-positive head and neck carcinomas. Sci. Rep. 2016, 6, 24927. [Google Scholar] [CrossRef] [PubMed]
- Serup-Hansen, E.; Linnemann, D.; Skovrider-Ruminski, W.; Hogdall, E.; Geertsen, P.F.; Havsteen, H. Human papillomavirus genotyping and p16 expression as prognostic factors for patients with american joint committee on cancer stages i to iii carcinoma of the anal canal. J. Clin. Oncol. 2014, 32, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Lont, A.P.; Kroon, B.K.; Horenblas, S.; Gallee, M.P.; Berkhof, J.; Meijer, C.J.; Snijders, P.J. Presence of high-risk human papillomavirus DNA in penile carcinoma predicts favorable outcome in survival. Int. J. Cancer 2006, 119, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Djajadiningrat, R.S.; Jordanova, E.S.; Kroon, B.K.; van Werkhoven, E.; de Jong, J.; Pronk, D.T.; Snijders, P.J.; Horenblas, S.; Heideman, D.A. Human papillomavirus prevalence in invasive penile cancer and association with clinical outcome. J. Urol. 2015, 193, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, A.L.; Lopes, A.; Santiago, G.H.; Ribeiro, K.C.; Latorre, M.R.; Villa, L.L. Human papillomavirus as a prognostic factor in carcinoma of the penis: Analysis of 82 patients treated with amputation and bilateral lymphadenectomy. Cancer 2001, 91, 2315–2321. [Google Scholar] [CrossRef]
- Bezerra, S.M.; Chaux, A.; Ball, M.W.; Faraj, S.F.; Munari, E.; Gonzalez-Roibon, N.; Sharma, R.; Bivalacqua, T.J.; Burnett, A.L.; Netto, G.J. Human papillomavirus infection and immunohistochemical p16(INK4A) expression as predictors of outcome in penile squamous cell carcinomas. Hum. Pathol. 2015, 46, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Pillai, S.; Chellappan, S.P. The RB-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv. Cancer Res. 2014, 121, 147–182. [Google Scholar] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Felsani, A.; Mileo, A.M.; Paggi, M.G. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 2006, 25, 5277–5285. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, A.K.; Westra, W.H. p16 expression as a surrogate marker for HPV-related oropharyngeal carcinoma: A guide for interpretative relevance and consistency. Head Neck 2012, 34, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Klaes, R.; Friedrich, T.; Spitkovsky, D.; Ridder, R.; Rudy, W.; Petry, U.; Dallenbach-Hellweg, G.; Schmidt, D.; von Knebel Doeberitz, M. Overexpression of p16INK4A as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int. J. Cancer 2001, 92, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Fetterman, B.; Castle, P.E.; Schiffman, M.; Wood, S.N.; Stiemerling, E.; Tokugawa, D.; Bodelon, C.; Poitras, N.; Lorey, T.; et al. p16/KI-67 dual stain cytology for detection of cervical precancer in HPV-positive women. J. Natl. Cancer Inst. 2015, 107, djv257. [Google Scholar] [CrossRef] [PubMed]
- Cuschieri, K.; Wentzensen, N. Human papillomavirus mRNA and p16 detection as biomarkers for the improved diagnosis of cervical neoplasia. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.G.; Wagner, A.J.; Conzen, S.D.; Jordan, J.; Bellacosa, A.; Tsichlis, P.N.; Hay, N. The PI 3-kinase/AKT signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997, 11, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Klippel, A.; Escobedo, M.A.; Wachowicz, M.S.; Apell, G.; Brown, T.W.; Giedlin, M.A.; Kavanaugh, W.M.; Williams, L.T. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol. Cell Biol. 1998, 18, 5699–5711. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Wei, S.J.; Lin, Y.C.; Lung, J.C.; Chang, T.C.; Whang-Peng, J.; Liu, J.M.; Yang, D.M.; Yang, W.K.; Shen, C.Y. PIK3CA as an oncogene in cervical cancer. Oncogene 2000, 19, 2739–2744. [Google Scholar] [CrossRef] [PubMed]
- Koncar, R.F.; Feldman, R.; Bahassi, E.M.; Hashemi Sadraei, N. Comparative molecular profiling of HPV-induced squamous cell carcinomas. Cancer Med. 2017, 6, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Ericson, K. Oncogenic PI3K and its role in cancer. Curr. Opin. Oncol. 2006, 18, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K.; Kang, S.; Elsliger, M.A.; Gymnopoulos, M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem. Sci. 2007, 32, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Frampton, G.M.; Fenton, T.; Feber, A.; Palmer, G.; Jay, A.; Pillay, N.; Forster, M.; Cronin, M.T.; Lipson, D.; et al. Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV− tumors. Genome Med. 2013, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Verlaat, W.; Snijders, P.J.; van Moorsel, M.I.; Bleeker, M.; Rozendaal, L.; Sie, D.; Ylstra, B.; Meijer, C.J.; Steenbergen, R.D.; Heideman, D.A. Somatic mutation in PIK3CA is a late event in cervical carcinogenesis. J. Pathol. Clin. Res. 2015, 1, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Husain, R.S.; Ramakrishnan, V. Global variation of human papillomavirus genotypes and selected genes involved in cervical malignancies. Ann. Glob. Health 2015, 81, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.; Gatalica, Z.; Knezetic, J.; Reddy, S.; Nathan, C.A.; Javadi, N.; Teknos, T. Molecular profiling of head and neck squamous cell carcinoma. Head Neck 2016, 38, E1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Millis, S.Z.; Ikeda, S.; Reddy, S.; Gatalica, Z.; Kurzrock, R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19,784 diverse solid tumors. JAMA Oncol. 2016, 2, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Shayesteh, L.; Lu, Y.; Kuo, W.L.; Baldocchi, R.; Godfrey, T.; Collins, C.; Pinkel, D.; Powell, B.; Mills, G.B.; Gray, J.W. PIK3CA is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Pawalowska, M.; Lubin, J.; Markowska, J. Signalling pathways in endometrial cancer. Contemp. Oncol. 2014, 18, 143–148. [Google Scholar]
- Hewitt, E.W. The MHC class I antigen presentation pathway: Strategies for viral immune evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, C.F.; Jordanova, E.S.; Zomerdijk-Nooijen, Y.A.; ter Haar, N.T.; Peters, A.A.; Fleuren, G.J. Frequent HLA class I loss is an early event in cervical carcinogenesis. Hum. Immunol. 2005, 66, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.L.; Frazer, I.H. Mutations in TAP genes are common in cervical carcinomas. Gynecol. Oncol. 2004, 92, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, C.F.; Jordanova, E.S.; ter Haar, N.T.; Kolkman-Uljee, S.M.; de Miranda, N.F.; Ferrone, S.; Peters, A.A.; Fleuren, G.J. Expression and genetic analysis of transporter associated with antigen processing in cervical carcinoma. Gynecol. Oncol. 2007, 105, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-beta in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Tsao, S.W.; Kwok, Y.K.; Wong, E.; Huang, X.R.; Liu, S.; Tsang, C.M.; Ngan, H.Y.; Cheung, A.N.; Lan, H.Y.; et al. Transforming growth factor beta1 promotes chromosomal instability in human papillomavirus 16 E6E7-infected cervical epithelial cells. Cancer Res. 2008, 68, 7200–7209. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Shen, Z.; Hu, X.; Sun, L.; Zhu, X. Transforming growth factor-beta1 in carcinogenesis, progression, and therapy in cervical cancer. Tumour Biol. 2016, 37, 7075–7083. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.S.; Lin, C.H.; Yang, C.H.; Liang, Y.J.; Yu, W.C. The human papillomavirus-16 (HPV-16) oncoprotein E7 conjugates with and mediates the role of the transforming growth factor-beta inducible early gene 1 (TIEG1) in apoptosis. Int. J. Biochem. Cell Biol. 2010, 42, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Habig, M.; Smola, H.; Dole, V.S.; Derynck, R.; Pfister, H.; Smola-Hess, S. E7 proteins from high- and low-risk human papillomaviruses bind to TGF-beta-regulated smad proteins and inhibit their transcriptional activity. Arch. Virol. 2006, 151, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Murvai, M.; Borbely, A.A.; Konya, J.; Gergely, L.; Veress, G. Effect of human papillomavirus type 16 E6 and E7 oncogenes on the activity of the transforming growth factor-beta2 (TGF-beta2) promoter. Arch. Virol. 2004, 149, 2379–2392. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Kim, B.C.; Kim, I.Y.; Cho, E.A.; Satterwhite, D.J.; Kim, S.J. The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the smad complex to its target sequence. J. Biol. Chem. 2002, 277, 38557–38564. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Fertig, E.J.; Ozawa, H.; Hatakeyama, H.; Howard, J.D.; Perez, J.; Considine, M.; Thakar, M.; Ranaweera, R.; Krigsfeld, G.; et al. Decreased SMAD4 expression is associated with induction of epithelial-to-mesenchymal transition and cetuximab resistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2015, 16, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Sun, K.; Jones, S.; Brait, M.; Agrawal, N.; Koch, W.; McCord, C.L.; Riley, D.R.; Angiuoli, S.V.; Velculescu, V.E.; et al. NOTCH1 mutations are drivers of oral tumorigenesis. Cancer Prev. Res. 2015, 8, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Izumi, N.; Helker, C.; Ehling, M.; Behrens, A.; Herzog, W.; Adams, R.H. FBXW7 controls angiogenesis by regulating endothelial notch activity. PLoS ONE 2012, 7, e41116. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 sublineage associations with histology-specific cancer risk using HPV whole-genome sequences in 3200 women. J. Natl. Cancer Inst. 2016, 108, djw100. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 genetic conservation is critical to carcinogenesis. Cell 2017, in press. [Google Scholar]
- Pinto, A.P.; Miron, A.; Yassin, Y.; Monte, N.; Woo, T.Y.; Mehra, K.K.; Medeiros, F.; Crum, C.P. Differentiated vulvar intraepithelial neoplasia contains TP53 mutations and is genetically linked to vulvar squamous cell carcinoma. Mod. Pathol. 2010, 23, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Bettegowda, C.; Wang, Y.; Wu, J.; Agrawal, N.; Shih Ie, M.; Kurman, R.; Dao, F.; Levine, D.A.; Giuntoli, R.; et al. Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci. Transl. Med. 2013, 5, 167ra164. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Hong, D.S.; Stepanek, V.M.; Fu, S.; Piha-Paul, S.A.; Lee, J.J.; Luthra, R.; et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013, 73, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Godinho, M.F.; Wulfkuhle, J.D.; Look, M.P.; Sieuwerts, A.M.; Sleijfer, S.; Foekens, J.A.; Petricoin, E.F., 3rd; Dorssers, L.C.; van Agthoven, T. BCAR4 induces antioestrogen resistance but sensitises breast cancer to lapatinib. Br. J. Cancer 2012, 107, 947–955. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Arm or Location 1 | Gene | Pathway | Alteration | Cancer Site 2 | Fraction Altered in HPV+ Cancers (%) | References |
|---|---|---|---|---|---|---|
| 1p | -- | -- | Gain | Cervix squamous | 33 | [35] |
| 1q | -- | -- | Gain | Cervix squamous | 29–36 | [33,35] |
| -- | -- | Gain | Cervix adeno | 22–35 | [33,35] | |
| 2q | -- | -- | Loss | Cervix squamous | 22 | [33] |
| 3p | -- | -- | Loss | Cervix squamous | 36–51 | [33,34,35] |
| -- | -- | Loss | Vulva | 45 | [33] | |
| 3p24.1 | TGFBR2 | TGFβ | Gain | Cervix | 36 | [18] |
| 3p14.1 | FOXP1 | Transcription | Loss | Cervix squamous | 42 | [34] |
| 3q | -- | -- | Gain | Cervix | 66 | [18,33,35] |
| -- | -- | Gain | Cervix squamous | 44–62 | [33,34,35] | |
| -- | -- | Gain | Cervix adeno | 29–39 | [33,35] | |
| -- | -- | Gain | Vulva | 58 | [33] | |
| 3q25.32 | MLF1 | Phenotypic determination | Gain | Cervix squamous | 60 | [34] |
| 3q26.32 | PIK3CA | PI3K/AKT | Gain | Head and neck | 30–56 | [2,36] |
| 3q26.33 | SOX2 | Transcription-Sox2 | Gain | Head and neck | 11–28 | [2,36] |
| 3q27.1 | KLHL6 | Immune signaling | Gain | Head and neck | 1–25 | [2,36] |
| 3q27.3 | BCL6 | RTK–JAK–STAT | Gain | Head and neck | 1–25 | [2,36] |
| 3q28 | TP63 | p53 | Gain | Cervix | 77 | [18] |
| 3q28 | LPP | Cell-cell adhesion | Gain | Cervix squamous | 60 | [34] |
| 4p | -- | -- | Loss | Cervix squamous | 24–47 | [33,35] |
| -- | -- | Loss | Cervix adeno | 10–46 | [33,35] | |
| -- | -- | Loss | Vulva | 27 | [33] | |
| 4q | -- | -- | Loss | Cervix squamous | 21–34 | [33,35] |
| -- | -- | Loss | Cervix adeno | 17–42 | [33,35] | |
| 4q31.3 | FBXW7 | Notch | Loss | Head and neck | 3–12 | [2,36] |
| 5p | -- | -- | Gain | Cervix squamous | 27–28 | [33,35] |
| -- | -- | Gain | Vulva | 15 | [33] | |
| 5p13.1 | RICTOR | PI3K/AKT | Gain | Head and neck | 4–6 | [2,36] |
| 6p | -- | -- | Loss | Cervix squamous | 24 | [35] |
| 6q | -- | -- | Loss | Cervix squamous | 20–29 | [33,35] |
| 7p | EGFR | RAS/EGFR/ERK | Gain | Cervix | 17 | [18] |
| 8p | -- | -- | Loss | Cervix squamous | 27 | [35] |
| 8q | -- | -- | Gain | Cervix squamous | 25 | [34,35] |
| 8q24.21 | MYC | TGFβ | Gain | Head and neck | 3–6 | [2,36] |
| 9p | CD274 | Immune response | Gain | Cervix | 8 | [18] |
| 10q23.31 | PTEN | PI3K/AKT | Loss | Cervix | 31 | [2,18,36] |
| Loss | Head and neck | 3–15 | [2,36] | |||
| 11p | -- | -- | Loss | Cervix squamous | 32 | [35] |
| -- | -- | Loss | Cervix adeno | 35 | [35] | |
| 11q | -- | -- | Loss | Cervix squamous | 32–33 | [33,35] |
| -- | -- | Loss | Cervix adeno | 9–35 | [33,35] | |
| -- | -- | Loss | Vulva | 30 | [33] | |
| 11q13.3 | FGF19 | RAS/EGFR/ERK | Gain | Head and neck | 4–6 | [2,36] |
| 11q13.3 | FGF3 | RAS/EGFR/ERK | Gain | Head and neck | 4–6 | [2,36] |
| 11q13.3 | FGF4 | RAS/EGFR/ERK | Gain | Head and neck | 4–6 | [2,36] |
| 11q22.1 | YAP1 | Hippo | Gain | Cervix | 16 | [18] |
| 13q | -- | -- | Loss | Cervix squamous | 24–41 | [33,35] |
| -- | -- | Loss | Cervix adeno | 21 | [33] | |
| -- | -- | Loss | Vulva | 12 | [33] | |
| 13q14.2 | RB1 | Rb | Loss | Head and neck | 6–24 | [2,36] |
| 14q | -- | -- | Gain | Cervix squamous | 26 | [35] |
| 14q32.32 | TRAF3 | NF-κB | Loss | Head and neck | 14 | [2] |
| 14q32.33 | AKT1 | PI3K/AKT | Gain | Head and neck | 5 | [2] |
| 16p | -- | -- | Loss | Cervix adeno | 33 | [35] |
| 16p13.13 | BCAR4 | Hedgehog | Gain | Cervix | 7 | [18] |
| 16q | -- | -- | Loss | Cervix adeno | 45 | [35] |
| 17p | -- | -- | Loss | Cervix squamous | 34 | [35] |
| 18q | -- | -- | Loss | Cervix adeno | 54 | [35] |
| 18q21.2 | SMAD4 | TGFβ | Gain | Cervix | 28 | [18] |
| 19p | -- | -- | Loss | Cervix adeno | 30 | [35] |
| 19q | -- | -- | Gain | Cervix squamous | 23 | [35] |
| -- | -- | Gain | Cervix adeno | 32 | [35] | |
| 20p | -- | -- | Gain | Cervix squamous | 33 | [35] |
| -- | -- | Gain | Cervix adeno | 26 | [35] | |
| 20q | -- | -- | Gain | Cervix squamous | 31 | [35] |
| Xp11.3 | KDM6A | Chromatin organization | Loss | Head and neck | 3–7 | [2,36] |
| Gene 1 | Pathway | Mutation | Cancer Site 2 | Fraction Mutated in HPV+ Cancers (%) | References |
|---|---|---|---|---|---|
| PIK3CA3 | PI3K/AKT | Activating | Cervix squamous | 6–42 | [18,35,60,61,62,63,64,65,66,67,68,69] |
| Cervix adeno | 10–42 | [18,35,60,61,62,63,64,65,66,67,68,69] | |||
| Head and neck | 22–56 | [2,36,37] | |||
| EGFR | RAS/EGFR/ERK | Activating | Cervix squamous | 3–33 | [18,62,70,71] |
| Cervix adeno | 6 | [18,62,70] | |||
| SMAD4 | TGFβ | Inactivating | Cervix | 28 | [18] |
| ERBB2 | PI3K/AKT | Activating | Cervix squamous | 4 | [18] |
| Cervix adeno | 26 | [18] | |||
| TP53 | DNA repair | Inactivating | Cervix squamous | 5 | [35] |
| Head and neck | 0–25 | [2,3,36,37,72] | |||
| Vulvar | 8–16 | [73,74] | |||
| RB1 | Rb | Inactivating | Head and neck | 6–24 | [2,36,37] |
| FGFR2 & FGFR3 | RAS/EGFR/ERK | Activating | Head and neck | 1–24 | [2,36,37] |
| KRAS | RAS/EGFR/ERK | Activating | Cervix squamous | 4 | [18,35,62] |
| Cervix adeno | 8–23 | [18,35,62,75] | |||
| Head and neck | 6 | [37] | |||
| MLL2 | Chromatin organization | Activating | Head and neck | 10–20 | [2,36] |
| ASXL1 | Inactivating | Head and neck | 5–19 | [36] | |
| NOTCH1 | Notch | Activating, inactivating | Head and neck | 6–18 | [2,36,37,76,77] |
| EP300 | TGFβ | Inactivating | Cervix squamous | 13–16 | [18] |
| Cervix adeno | 10 | [18] | |||
| Head and neck | 10–14 | [36] | |||
| ERBB3 | PI3K/AKT | Activating | Cervix squamous | 4 | [18] |
| Cervix adeno | 16 | [18] | |||
| ATM | DNA repair | Inactivating | Head and neck | 1–16 | [36] |
| FBXW7 | Notch | Inactivating | Cervix | 11–15 | [18,35] |
| Head and neck | 3–12 | [36] | |||
| PTEN | PI3K/AKT | Inactivating | Cervix squamous | 6–13 | [18,35] |
| Cervix adeno | 13 | [18,35] | |||
| Head and neck | 3–15 | [2,36] | |||
| BRCA1 | DNA repair | Inactivating | Head and neck | 2–14 | [36] |
| NF1 | RAS/EGFR/ERK | Inactivating | Head and neck | 0–14 | [2,36] |
| ELF3 | PI3K/AKT | Inactivating | Cervix adeno | 13 | [35] |
| FLG | Head and neck | 12 | [2] | ||
| BRCA2 | DNA repair | Inactivating | Head and neck | 3–12 | [36] |
| LRP1B | RTK | Head and neck | 2–12 | [36] | |
| HRAS | RAS/EGFR/ERK | Activating | Head and neck | 1–12 | [36] |
| HLA-A/B | MHC | Inactivating | Cervix | 6–9 | [18,35,78] |
| Head and neck | 11 | [2,37] | |||
| MLL3 | Chromatin organization | Activating | Head and neck | 10 | [2] |
| TGFBR2 | TGFβ | Inactivating | Cervix squamous | 8 | [18] |
| CREBBP | TGFβ | Inactivating | Cervix squamous | 8 | [18] |
| Cervix adeno | 6 | [18] | |||
| TRAF3 | NF-κB | Truncating mutations | Head and neck | 8 | [2] |
| MAPK1 | MEK/ERK | Activating | Cervix squamous | 8 | [18,35] |
| CBFB | RUNX1/RUNX2 | Inactivating | Cervix adeno | 8 | [35] |
| DDX3X | Inactivating | Head and neck | 8 | [2] | |
| ARID1A | Chromatin organization | Inactivating | Cervix | 7 | [18] |
| NFE2L2 | Inactivating | Cervix squamous | 4–7 | [18,35] | |
| TPRX1 | Head and neck | 6 | [2] | ||
| CYLD | NF-κB | Inactivating | Head and neck | 6 | [2] |
| RIPK4 | NF-κB, Notch | Head and neck | 6 | [2] | |
| UBR5 | Proteolysis | Head and neck | 6 | [2] | |
| CASP8 | Fas apoptosis | Inactivating | Cervix | 4 | [18] |
| STK11 | PI3K/AKT | Inactivating | Cervix squamous | 4 | [35] |
| SHKBP1 | RAS/EGFR/ERK | Cervix | 2 | [18] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litwin, T.R.; Clarke, M.A.; Dean, M.; Wentzensen, N. Somatic Host Cell Alterations in HPV Carcinogenesis. Viruses 2017, 9, 206. https://doi.org/10.3390/v9080206
Litwin TR, Clarke MA, Dean M, Wentzensen N. Somatic Host Cell Alterations in HPV Carcinogenesis. Viruses. 2017; 9(8):206. https://doi.org/10.3390/v9080206
Chicago/Turabian StyleLitwin, Tamara R., Megan A. Clarke, Michael Dean, and Nicolas Wentzensen. 2017. "Somatic Host Cell Alterations in HPV Carcinogenesis" Viruses 9, no. 8: 206. https://doi.org/10.3390/v9080206
APA StyleLitwin, T. R., Clarke, M. A., Dean, M., & Wentzensen, N. (2017). Somatic Host Cell Alterations in HPV Carcinogenesis. Viruses, 9(8), 206. https://doi.org/10.3390/v9080206