Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis



{kind=link}
{kind=link}
Abstract
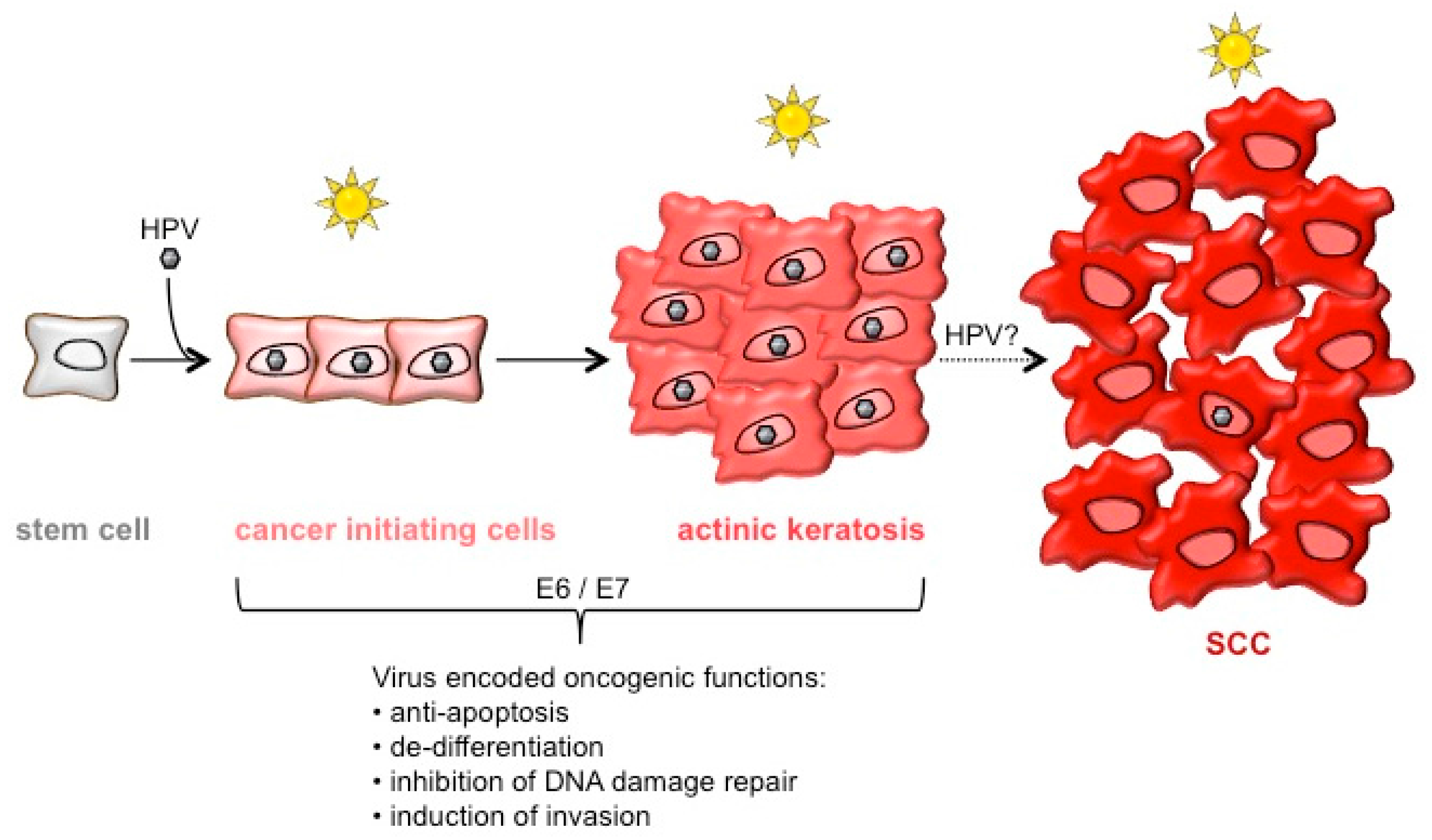
1. Introduction
2. Invasion is Regulated Both at the Level of the Tumor Cell and the Extracellular Matrix
3. Mouse Models
4. Do βHPV Affect Self-Renewal of Infected Keratinocytes?
5. Do Wound-Healing Processes Play a Role in βHPV-Mediated Cancer Initiation?
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rogers, H.W.; Weinstock, M.A.; Harris, A.R.; Hinckley, M.R.; Feldman, S.R.; Fleischer, A.B.; Coldiron, B.M. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch. Dermatol. 2010, 146, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.R.; Gheit, T.; Waterboer, T.; Giuliano, A.R.; Messina, J.L.; Fenske, N.A.; Cherpelis, B.S.; Sondak, V.K.; Roetzheim, R.G.; Michael, K.M.; Tommasino, M.; Pawlita, M.; Rollison, D.E. Case-control study of cutaneous human papillomaviruses in squamous cell carcinoma of the skin. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Neale, R.E.; Weissenborn, S.; Abeni, D.; Bavinck, J.N.; Euvrard, S.; Feltkamp, M.C.; Green, A.C.; Harwood, C.; de Koning, M.; Naldi, L.; Nindl, I.; Pawlita, M.; Proby, C.; Quint, W.G.; Waterboer, T.; Wieland, U.; Pfister, H. Human papillomavirus load in eyebrow hair follicles and risk of cutaneous squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.R.; Gheit, T.; Pfister, H.; Giuliano, A.R.; Messina, J.L.; Fenske, N.A.; Cherpelis, B.S.; Sondak, V.K.; Roetzheim, R.G.; Silling, S.; Pawlita, M.; Tommasino, M.; Rollison, D.E. Case-control study of genus-β human papillomaviruses in plucked eyebrow hairs and cutaneous squamous cell carcinoma. Int. J. Cancer 2014, 134, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Genders, R.E.; Mazlom, H.; Michel, A.; Plasmeijer, E.I.; Quint, K.D.; Pawlita, M.; van der Meijden, E.; Waterboer, T.; de Fijter, H.; Claas, F.H.; et al. The presence of βpapillomavirus antibodies around transplantation predicts the development of keratinocyte carcinoma in organ transplant recipients: A cohort study. J. Investig. Dermatol. 2015, 135, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Smola, S. Human papillomaviruses and skin cancer. Adv. Exp. Med. Biol. 2014, 810, 192–207. [Google Scholar] [PubMed]
- Tommasino, M. The biology of β human papillomaviruses. Virus Res. 2017, 231, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, S.J.; Nindl, I.; Purdie, K.; Harwood, C.; Proby, C.; Breuer, J.; Majewski, S.; Pfister, H.; Wieland, U. Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J. Investig. Dermatol. 2005, 125, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Hübbers, C.U.; Akgül, B. HPV and cancer of the oral cavity. Virulence 2015, 6, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Chow, L.T.; Broker, T.R. Retrovirus-mediated gene transfer to analyze HPV gene regulation and protein functions in organotypic "raft" cultures. Methods Mol. Med. 2005, 119, 187–202. [Google Scholar] [PubMed]
- Lee, D.; Norby, K.; Hayes, M.; Chiu, Y.F.; Sugden, B.; Lambert, P.F. Using Organotypic Epithelial Tissue Culture to Study the Human Papillomavirus Life Cycle. Curr. Protoc. Microbiol. 2016, 41, 14B-8-1–14B-8-19. [Google Scholar] [PubMed]
- Boxman, I.L.; Mulder, L.H.; Noya, F.; de Waard, V.; Gibbs, S.; Broker, T.R.; ten Kate, F.; Chow, L.T.; ter Schegget, J. Transduction of the E6 and E7 genes of epidermodysplasia-verruciformis-associated human papillomaviruses alters human keratinocyte growth and differentiation in organotypic cultures. J. Investig. Dermatol. 2001, 117, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Z.; South, A.P. Tumour-stroma crosstalk in the development of squamous cell carcinoma. Int. J. Biochem. Cell Biol. 2014, 53, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Fujiwara, H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Leverrier, S.; Bergamaschi, D.; Ghali, L.; Ola, A.; Warnes, G.; Akgül, B.; Blight, K.; Garcia-Escudero, R.; Penna, A.; Eddaoudi, A.; et al. Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis 2007, 12, 549–560. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, R.F.; Akgül, B.; Storey, A.; Pfister, H.; Harwood, C.A.; Byrne, C. Cutaneous human papillomaviruses down-regulate AKT1, whereas AKT2 up-regulation and activation associates with tumors. Cancer Res. 2007, 67, 8207–8215. [Google Scholar] [CrossRef] [PubMed]
- Akgül, B.; Garcia-Escudero, R.; Ghali, L.; Pfister, H.J.; Fuchs, P.G.; Navsaria, H.; Storey, A. The E7 protein of cutaneous human papillomavirus type 8 causes invasion of human keratinocytes into the dermis in organotypic cultures of skin. Cancer Res. 2005, 65, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Smola-Hess, S.; Pahne, J.; Mauch, C.; Zigrino, P.; Smola, H.; Pfister, H.J. Expression of membrane type 1 matrix metalloproteinase in papillomavirus-positive cells: Role of the human papillomavirus (HPV) 16 and HPV8 E7 gene products. J. Gen. Virol. 2005, 86, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells - what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Heuser, S.; Hufbauer, M.; Marx, B.; Tok, A.; Majewski, S.; Pfister, H.; Akgul, B. The levels of epithelial anchor proteins β-catenin and zona occludens-1 are altered by E7 of human papillomaviruses 5 and 8. J. Gen. Virol. 2016, 97, 463–472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Franceschi, N.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic—the update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Heuser, S.; Hufbauer, M.; Steiger, J.; Marshall, J.; Sterner-Kock, A.; Mauch, C.; Zigrino, P.; Akgul, B. The fibronectin/α3β1 integrin axis serves as molecular basis for keratinocyte invasion induced by βHPV. Oncogene 2016, 35, 4529–4539. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Secades, P.; van Hulst, L.; Kreft, M.; Song, J.Y.; Sonnenberg, A. Loss of integrin α3 prevents skin tumor formation by promoting epidermal turnover and depletion of slow-cycling cells. Proc. Natl. Acad. Sci. USA 2012, 109, 21468–21473. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Dysart, M.M.; Clarke, K.C.; Stabenfeldt, S.E.; Barker, T.H. Integrin α3β1 Binding to Fibronectin Is Dependent on the Ninth Type III Repeat. J. Biol. Chem. 2015, 290, 25534–25547. [Google Scholar] [CrossRef] [PubMed]
- Hodivala-Dilke, K.M.; DiPersio, C.M.; Kreidberg, J.A.; Hynes, R.O. Novel roles for α3β1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J. Cell Biol. 1998, 142, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Longmate, W.M.; Lyons, S.P.; Chittur, S.V.; Pumiglia, K.M.; Van De Water, L.; DiPersio, C.M. Suppression of integrin α3β1 by α9β1 in the epidermis controls the paracrine resolution of wound angiogenesis. J. Cell Biol. 2017, 216, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Schaper, I.D.; Marcuzzi, G.P.; Weissenborn, S.J.; Kasper, H.U.; Dries, V.; Smyth, N.; Fuchs, P.; Pfister, H. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005, 65, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Akgül, B.; Pfefferle, R.; Marcuzzi, G.P.; Zigrino, P.; Krieg, T.; Pfister, H.; Mauch, C. Expression of matrix metalloproteinase (MMP)-2, MMP-9, MMP-13, and MT1-MMP in skin tumors of human papillomavirus type 8 transgenic mice. Exp. Dermatol. 2006, 15, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Lazic, D.; Akgül, B.; Brandsma, J.L.; Pfister, H.; Weissenborn, S.J. Enhanced human papillomavirus type 8 oncogene expression levels are crucial for skin tumorigenesis in transgenic mice. Virology 2010, 403, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Lazic, D.; Reinartz, M.; Akgül, B.; Pfister, H.; Weissenborn, S.J. Skin tumor formation in human papillomavirus 8 transgenic mice is associated with a deregulation of oncogenic miRNAs and their tumor suppressive targets. J. Dermatol. Sci. 2011, 64, 7–15. [Google Scholar] [CrossRef] [PubMed]
- De Andrea, M.; Ritta, M.; Landini, M.M.; Borgogna, C.; Mondini, M.; Kern, F.; Ehrenreiter, K.; Baccarini, M.; Marcuzzi, G.P.; Smola, S.; et al. Keratinocyte-specific Stat3 heterozygosity impairs development of skin tumors in human papillomavirus 8 transgenic mice. Cancer Res. 2010, 70, 7938–7948. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, J.; Pofahl, R.; Pfister, H.; Haase, I. Deletion of epidermal Rac1 inhibits HPV-8 induced skin papilloma formation and facilitates HPV-8- and UV-light induced skin carcinogenesis. Oncotarget 2016, 7, 57841–57850. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, R.; Marcuzzi, G.P.; Akgül, B.; Kasper, H.U.; Schulze, F.; Haase, I.; Wickenhauser, C.; Pfister, H. The human papillomavirus type 8 E2 protein induces skin tumors in transgenic mice. J. Investig. Dermatol. 2008, 128, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
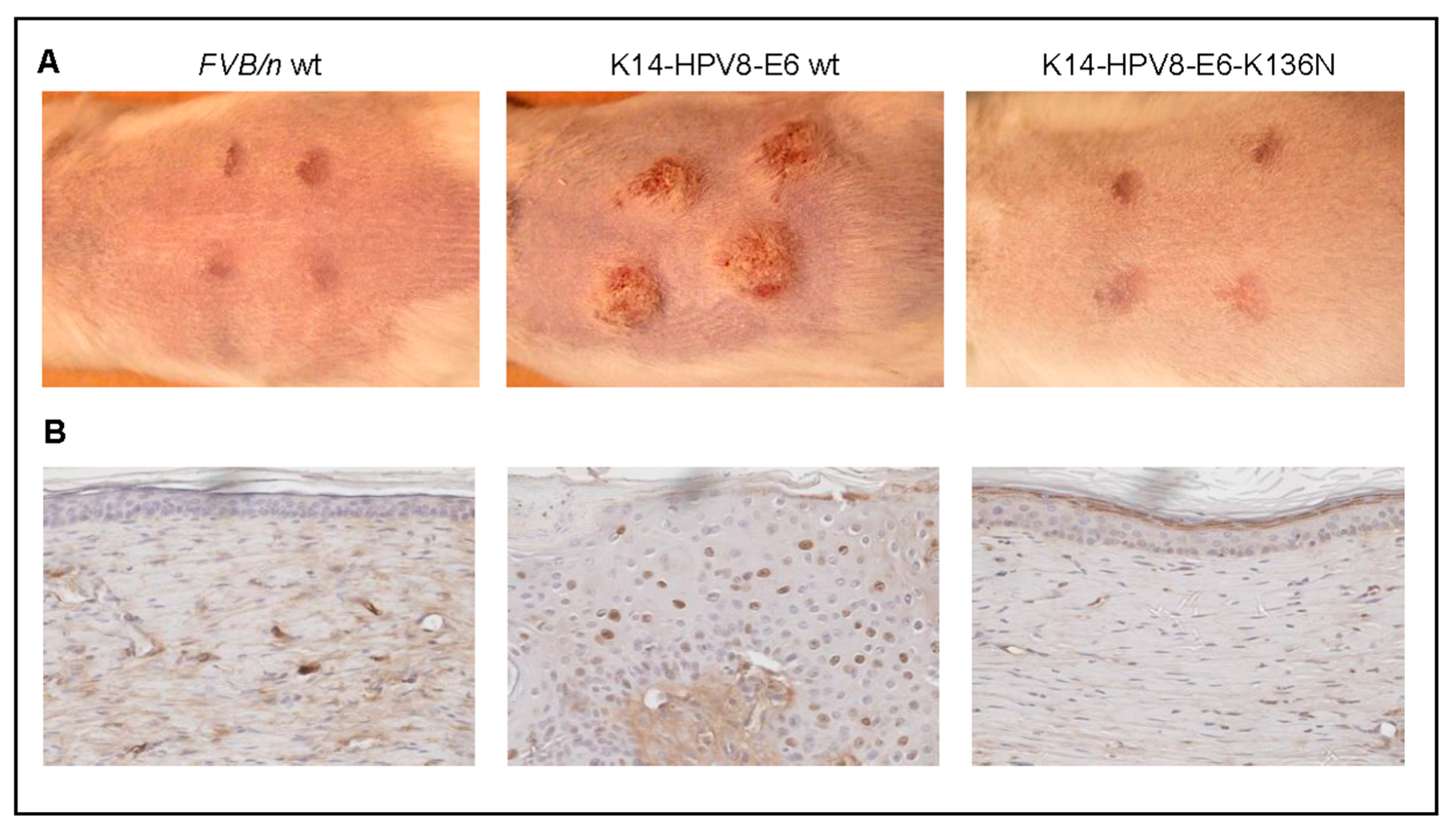
- Marcuzzi, G.P.; Hufbauer, M.; Kasper, H.U.; Weissenborn, S.J.; Smola, S.; Pfister, H. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 2009, 90, 2855–2864. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Cooke, J.; van der Horst, G.T.; Pfister, H.; Storey, A.; Akgül, B. Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol. Cancer 2015, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Mueller-Decker, K.; Kloz, U.; Aengeneyndt, B.; Kopp-Schneider, A.; Grone, H.J.; Gheit, T.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011, 7, e1002125. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Muller-Decker, K.; Hassel, J.C.; Alvarez, J.C.; Flechtenmacher, C.; Pawlita, M.; Gissmann, L.; Tommasino, M. The BRAF Inhibitor Vemurafenib Enhances UV-Induced Skin Carcinogenesis in β HPV38 E6 and E7 Transgenic Mice. J. Investig. Dermatol. 2017, 137, 261–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vinzon, S.E.; Braspenning-Wesch, I.; Muller, M.; Geissler, E.K.; Nindl, I.; Grone, H.J.; Schafer, K.; Rosl, F. Protective vaccination against papillomavirus-induced skin tumors under immunocompetent and immunosuppressive conditions: A preclinical study using a natural outbred animal model. PLoS Pathog. 2014, 10, e1003924. [Google Scholar] [CrossRef] [PubMed]
- Vinzon, S.E.; Rosl, F. HPV vaccination for prevention of skin cancer. Hum. Vaccines Immunother. 2015, 11, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Epithelial Skin Biology: Three Decades of Developmental Biology, a Hundred Questions Answered and a Thousand New Ones to Address. Curr. Top. Dev. Biol. 2016, 116, 357–374. [Google Scholar] [PubMed]
- Boxman, I.L.; Berkhout, R.J.; Mulder, L.H.; Wolkers, M.C.; Bouwes Bavinck, J.N.; Vermeer, B.J.; ter Schegget, J. Detection of human papillomavirus DNA in plucked hairs from renal transplant recipients and healthy volunteers. J. Investig. Dermatol. 1997, 108, 712–715. [Google Scholar] [CrossRef] [PubMed]
- De Koning, M.N.; Struijk, L.; Bavinck, J.N.; Kleter, B.; ter Schegget, J.; Quint, W.G.; Feltkamp, M.C. Betapapillomaviruses frequently persist in the skin of healthy individuals. J. Gen. Virol. 2007, 88, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Bouwes Bavinck, J.N.; Plasmeijer, E.I.; Feltkamp, M.C. β-papillomavirus infection and skin cancer. J. Investig. Dermatol. 2008, 128, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. beta-HPV 5 and 8 E6 disrupt homology dependent double strand break repair by attenuating BRCA1 and BRCA2 expression and foci formation. PLoS Pathog. 2015, 11, e1004687. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Harwood, C.; Thomas, M.; Banks, L.; Storey, A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000, 14, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Ghali, L.; Harwood, C.; Storey, A. Reduced apoptotic levels in squamous but not basal cell carcinomas correlates with detection of cutaneous human papillomavirus. Br. J. Cancer 2002, 87, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Lanfredini, S.; Olivero, C.; Borgogna, C.; Calati, F.; Powell, K.; Davies, K.J.; De Andrea, M.; Harries, S.; Tang, H.K.C.; Pfister, H.; et al. HPV8 Field Cancerization in a Transgenic Mouse Model is due to Lrig1+ Keratinocyte Stem Cell Expansion. J. Investig. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, K.; Watt, F.M. Markers of epidermal stem cell subpopulations in adult mammalian skin. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, A.M.; Podgorska, M.; Feld, P.; Fingerle, A.; Knerr-Rupp, K.; Grasser, F.; Smola, H.; Roemer, K.; Ebert, E.; Kim, Y.J.; et al. Identification of C/EBPα as a novel target of the HPV8 E6 protein regulating miR-203 in human keratinocytes. PLoS Pathog. 2017, 13, e1006406. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Spangle, J.M.; Munger, K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 2013, 87, 4762–4767. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Biddle, A.; Borgogna, C.; Gariglio, M.; Doorbar, J.; Storey, A.; Pfister, H.; Mackenzie, I.; Akgül, B. Expression of betapapillomavirus oncogenes increases the number of keratinocytes with stem cell-like properties. J. Virol. 2013, 87, 12158–12165. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.S.; Klingbeil, P.; Schnolzer, M.; Zoller, M. CD44 variant isoforms associate with tetraspanins and EpCAM. Exp. Cell. Res. 2004, 297, 329–347. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Holloway, A.; Simmonds, M.; Azad, A.; Fox, J.L.; Storey, A. Resistance to UV-induced apoptosis by β-HPV5 E6 involves targeting of activated BAK for proteolysis by recruitment of the HERC1 ubiquitin ligase. Int. J. Cancer 2015, 136, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semi. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lucas, T.; Marcuzzi, G.P.; Pfister, H.; Eming, S.A. Distinct functions of epidermal and myeloid-derived VEGF-A in skin tumorigenesis mediated by HPV8. Cancer Res. 2015, 75, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Fa, P.; Cui, Z.; Xia, Y.; Sun, L.; Li, Z.; Tang, A.; Gui, Y.; Cai, Z. The EDA-containing cellular fibronectin induces epithelial-mesenchymal transition in lung cancer cells through integrin α9β1-mediated activation of PI3-K/AKT and ERK1/2. Carcinogenesis 2014, 35, 184–191. [Google Scholar] [PubMed]
- Wen, T.; Zhang, Z.; Yu, Y.; Qu, H.; Koch, M.; Aumailley, M. Integrin α3 subunit regulates events linked to epithelial repair, including keratinocyte migration and protein expression. Wound Repair Regen. 2010, 18, 325–334. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Howley, P.M. Proteomic approaches to the study of papillomavirus-host interactions. Virology 2013, 435, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 Proteins Target the NOTCH and TGF-β Tumor Suppressors to Inhibit Differentiation and Sustain Keratinocyte Proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef] [PubMed]
- Akgül, B.; Ghali, L.; Davies, D.; Pfister, H.; Leigh, I.M.; Storey, A. HPV8 early genes modulate differentiation and cell cycle of primary human adult keratinocytes. Exp. Dermatol. 2007, 16, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Buitrago-Perez, A.; Hachimi, M.; Duenas, M.; Lloveras, B.; Santos, A.; Holguin, A.; Duarte, B.; Santiago, J.L.; Akgül, B.; Rodriguez-Peralto, J.L.; Storey, A.; Ribas, C.; Larcher, F.; del Rio, M.; Paramio, J.M.; Garcia-Escudero, R. A humanized mouse model of HPV-associated pathology driven by E7 expression. PLoS ONE 2012, 7, e41743. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hufbauer, M.; Akgül, B. Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis. Viruses 2017, 9, 187. https://doi.org/10.3390/v9070187
Hufbauer M, Akgül B. Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis. Viruses. 2017; 9(7):187. https://doi.org/10.3390/v9070187
Chicago/Turabian StyleHufbauer, Martin, and Baki Akgül. 2017. "Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis" Viruses 9, no. 7: 187. https://doi.org/10.3390/v9070187
APA StyleHufbauer, M., & Akgül, B. (2017). Molecular Mechanisms of Human Papillomavirus Induced Skin Carcinogenesis. Viruses, 9(7), 187. https://doi.org/10.3390/v9070187