
Telomerase Induction in HPV Infection and Oncogenesis


Abstract
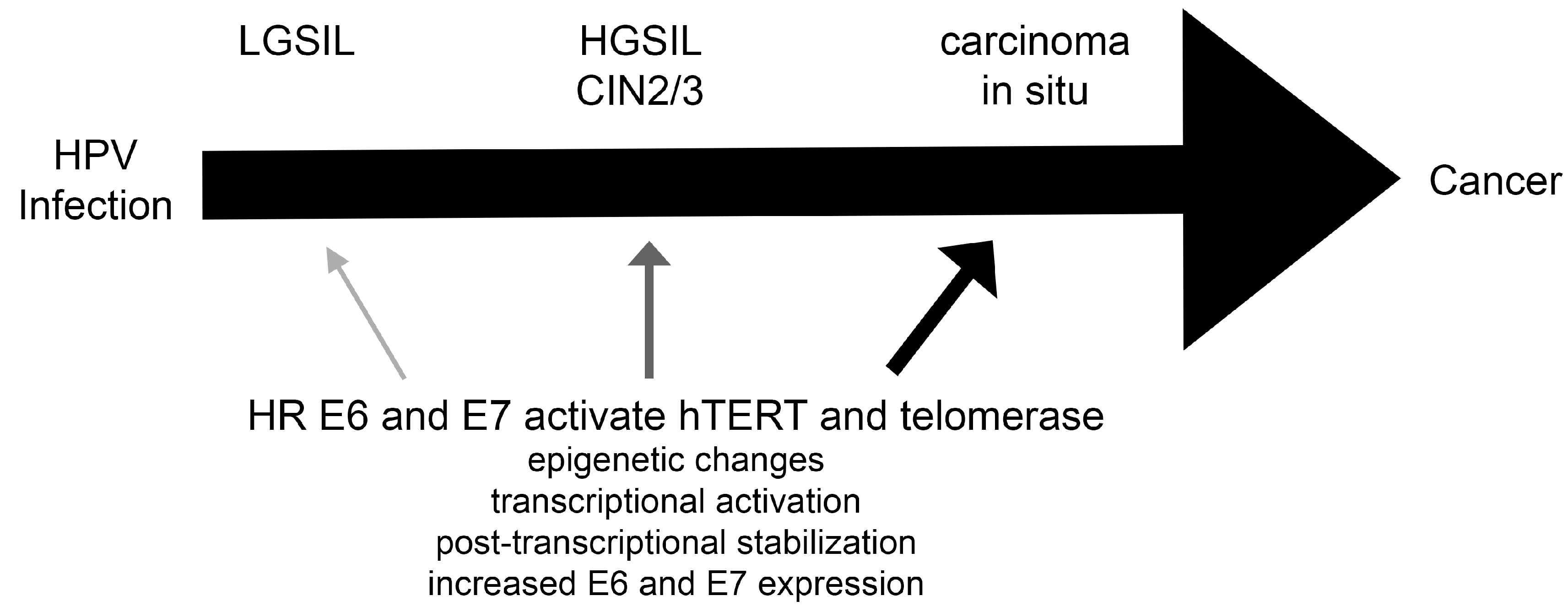
:1. Introduction: Human Papillomavirus Infections
Human Papillomavirus Infection and Life Cycle in Stratified Squamous Epithelium
2. Telomeric DNA and Telomerase
3. Telomerase and hTERT Activity Driven by HR E6 and E7
3.1. hTERT: Promoter Regulation
3.2. hTERT: Epigenetic Regulation
3.3. hTERT: Post-Transcriptional Regulation
3.4. hTERT: Beta HPV E6
4. Telomerase in HPV-Associated Cancers
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Yamamoto, Y.; Laury, A.; Wang, X.; Nucci, M.R.; McLaughlin-Drubin, M.E.; Munger, K.; Feldman, S.; McKeon, F.D.; Xian, W.; et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Longuespee, R.; Quick, C.M.; Roncarati, P.; Suarez-Carmona, M.; Hubert, P.; Lebeau, A.; Bruyere, D.; Mazzucchelli, G.; Smargiasso, N.; et al. Proteomic signatures reveal a dualistic and clinically relevant classification of anal canal carcinoma. J. Pathol. 2017, 241, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Manipulation of the innate immune response by human papillomaviruses. Virus Res. 2017, 231, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, A.K.; Riemer, A.B. The invisible enemy—How human papillomaviruses avoid recognition and clearance by the host immune system. Open Virol. J. 2012, 6, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Hawley-Nelson, P.; Vousden, K.H.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989, 8, 3905–3910. [Google Scholar] [PubMed]
- Munger, K.; Phelps, W.C.; Bubb, V.; Howley, P.M.; Schlegel, R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989, 63, 4417–4421. [Google Scholar] [PubMed]
- Wells, S.I.; Francis, D.A.; Karpova, A.Y.; Dowhanick, J.J.; Benson, J.D.; Howley, P.M. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21CIP-dependent pathways. EMBO J. 2000, 19, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Francis, D.A.; Schmid, S.I.; Howley, P.M. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J. Virol. 2000, 74, 2679–2686. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Glaunsinger, B.; Mantovani, F.; Banks, L.; Javier, R.T. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 2000, 74, 9680–9693. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Thomas, M.; Banks, L.; Roberts, S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 2003, 116, 4925–4934. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Nguyen, M.M.; Lee, D.; Griep, A.E.; Lambert, P.F. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J. Virol. 2003, 77, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R. Beginning to understand the end of the chromosome. Cell 2004, 116, 273–279. [Google Scholar] [CrossRef]
- Schmidt, J.C.; Cech, T.R. Human telomerase: Biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015, 29, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Hahn, W.C.; Wei, W.; Caddle, S.D.; Beijersbergen, R.L.; Lansdorp, P.M.; Sedivy, J.M.; Weinberg, R.A. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. USA 1998, 95, 14723–14728. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Meyerson, M.; Eaton, E.N.; Ellisen, L.W.; Caddle, S.D.; Haber, D.A.; Weinberg, R.A. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene 1998, 16, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Counter, C.M.; Eaton, E.N.; Ellisen, L.W.; Steiner, P.; Caddle, S.D.; Ziaugra, L.; Beijersbergen, R.L.; Davidoff, M.J.; Liu, Q.; et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997, 90, 785–795. [Google Scholar] [CrossRef]
- Cohen, S.B.; Graham, M.E.; Lovrecz, G.O.; Bache, N.; Robinson, P.J.; Reddel, R.R. Protein composition of catalytically active human telomerase from immortal cells. Science 2007, 315, 1850–1853. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998, 58, 4168–4172. [Google Scholar] [PubMed]
- Nandakumar, J.; Cech, T.R. Finding the end: Recruitment of telomerase to telomeres. Nat. Rev. Mol. Cell Biol. 2013, 14, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Plug-DeMaggio, A.W.; Sundsvold, T.; Wurscher, M.A.; Koop, J.I.; Klingelhutz, A.J.; McDougall, J.K. Telomere erosion and chromosomal instability in cells expressing the HPV oncogene 16E6. Oncogene 2004, 23, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Gabet, A.S.; Accardi, R.; Bellopede, A.; Popp, S.; Boukamp, P.; Sylla, B.S.; Londono-Vallejo, J.A.; Tommasino, M. Impairment of the telomere/telomerase system and genomic instability are associated with keratinocyte immortalization induced by the skin human papillomavirus type 38. FASEB J. 2008, 22, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Verdun, R.E.; Karlseder, J. Replication and protection of telomeres. Nature 2007, 447, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R. On the road to immortality: hTERT upregulation in cancer cells. FEBS Lett. 2004, 564, 9–13. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, T.; Foster, S.A.; Koop, J.I.; McDougall, J.K.; Galloway, D.A.; Klingelhutz, A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396, 84–88. [Google Scholar] [PubMed]
- Liu, X.; Roberts, J.; Dakic, A.; Zhang, Y.; Schlegel, R. HPV E7 contributes to the telomerase activity of immortalized and tumorigenic cells and augments E6-induced hTERT promoter function. Virology 2008, 375, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.E.; Jung, K.H.; Jae, L.C.; Tae, K.H.; Seong, H.E. The role of HPV oncoproteins and cellular factors in maintenance of hTERT expression in cervical carcinoma cells. Gynecol. Oncol. 2004, 94, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Burk, R.D. Association between hTERT activation by HPV E6 proteins and oncogenic risk. Virology 2012, 433, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-Rich Sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dakic, A.; Zhang, Y.; Dai, Y.; Chen, R.; Schlegel, R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc. Natl. Acad. Sci. USA 2009, 106, 18780–18785. [Google Scholar] [CrossRef] [PubMed]
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. HPV-16-E6 associated hTERT promoter acetylation is E6AP dependent, increased in later passage cells and enhanced by loss of p300. Int. J. Cancer 2006, 119, 1878–1885. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yuan, H.; Fu, B.; Disbrow, G.L.; Apolinario, T.; Tomaic, V.; Kelley, M.L.; Baker, C.C.; Huibregtse, J.; Schlegel, R. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J. Biol. Chem. 2005, 280, 10807–10816. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J. Virol. 2005, 79, 3737–3747. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Yugawa, T.; Narisawa-Saito, M.; Ohno, S.; Fujita, M.; Kiyono, T. E6AP-dependent degradation of DLG4/PSD95 by high-risk human papillomavirus type 18 E6 protein. J. Virol. 2007, 81, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Laura, R.; Hepner, K.; Guccione, E.; Sawyers, C.; Lasky, L.; Banks, L. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 2002, 21, 5088–5096. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.M.; Phillips, B.L.; Chan, E.K. miR-375 activates p21 and suppresses telomerase activity by coordinately regulating HPV E6/E7, E6AP, CIP2a, and 14–3-3ζ. Mol. Cancer 2014, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.S.; Wen, J.; Bacchetti, S. The human telomerase catalytic subunit hTERT: Organization of the gene and characterization of the promoter. Hum. Mol. Genet. 1999, 8, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Wick, M.; Zubov, D.; Hagen, G. Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT). Gene 1999, 232, 97–106. [Google Scholar] [CrossRef]
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999, 59, 551–557. [Google Scholar] [PubMed]
- Fujimoto, K.; Kyo, S.; Takakura, M.; Kanaya, T.; Kitagawa, Y.; Itoh, H.; Takahashi, M.; Inoue, M. Identification and characterization of negative regulatory elements of the human telomerase catalytic subunit (hTERT) gene promoter: Possible role of MZF-2 in transcriptional repression of hTERT. Nucleic Acids Res. 2000, 28, 2557–2562. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Song, Y.H.; Yim, J.; Kim, T.K. Identification of Mad as a repressor of the human telomerase (hTERT) gene. Oncogene 2000, 19, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Gunes, C.; Lichtsteiner, S.; Vasserot, A.P.; Englert, C. Expression of the hTERT gene is regulated at the level of transcriptional initiation and repressed by Mad1. Cancer Res. 2000, 60, 2116–2121. [Google Scholar] [PubMed]
- Horikawa, I.; Cable, P.L.; Mazur, S.J.; Appella, E.; Afshari, C.A.; Barrett, J.C. Downstream E-box-mediated regulation of the human telomerase reverse transcriptase (hTERT) gene transcription: Evidence for an endogenous mechanism of transcriptional repression. Mol. Biol. Cell 2002, 13, 2585–2597. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Yim, J.; Kim, T.K. Sp1 and Sp3 recruit histone deacetylase to repress transcription of human telomerase reverse transcriptase (hTERT) promoter in normal human somatic cells. J. Biol. Chem. 2002, 277, 38230–38238. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res. 2005, 33, 6850–6860. [Google Scholar] [CrossRef] [PubMed]
- Racek, T.; Mise, N.; Li, Z.; Stoll, A.; Putzer, B.M. C-terminal p73 isoforms repress transcriptional activity of the human telomerase reverse transcriptase (hTERT) promoter. J. Biol. Chem. 2005, 280, 40402–40405. [Google Scholar] [CrossRef] [PubMed]
- Lebel, R.; McDuff, F.O.; Lavigne, P.; Grandbois, M. Direct visualization of the binding of c-Myc/Max heterodimeric b-HLH-LZ to E-box sequences on the hTERT promoter. Biochemistry 2007, 46, 10279–10286. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Katzenellenbogen, R.A.; Grandori, C.; Galloway, D.A. An unbiased in vivo screen reveals multiple transcription factors that control HPV E6-regulated hTERT in keratinocytes. Virology 2013, 446, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dakic, A.; Chen, R.; Disbrow, G.L.; Zhang, Y.; Dai, Y.; Schlegel, R. Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc. J. Virol. 2008, 82, 11568–11576. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Yang, H.T.; Wang, T.C.; Cheng, A.J. Upstream stimulatory factor (USF) as a transcriptional suppressor of human telomerase reverse transcriptase (hTERT) in oral cancer cells. Mol. Carcinog. 2005, 44, 183–192. [Google Scholar] [CrossRef] [PubMed]
- McMurray, H.R.; McCance, D.J. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol. 2003, 77, 9852–9861. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.; Galloway, D.A. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-Myc. J. Virol. 2001, 75, 7198–7201. [Google Scholar] [CrossRef] [PubMed]
- Sekaric, P.; Cherry, J.J.; Androphy, E.J. Binding of human papillomavirus type 16 E6 to E6AP is not required for activation of hTERT. J. Virol. 2008, 82, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Shai, A.; Pitot, H.C.; Lambert, P.F. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010, 70, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Luo, W.; Elzi, D.J.; Grandori, C.; Galloway, D.A. NFX1 interacts with mSin3a/histone deacetylase to repress hTERT transcription in keratinocytes. Mol. Cell. Biol. 2008, 28, 4819–4828. [Google Scholar] [CrossRef] [PubMed]
- Schutze, D.M.; Kooter, J.M.; Wilting, S.M.; Meijer, C.J.; Quint, W.; Snijders, P.J.; Steenbergen, R.D. Longitudinal assessment of DNA methylation changes during HPVE6E7-induced immortalization of primary keratinocytes. Epigenetics 2015, 10, 73–81. [Google Scholar] [CrossRef] [PubMed]
- De Wild, J.; Kooter, J.M.; Overmeer, R.M.; Claassen-Kramer, D.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.D. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer 2010, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhao, L.J.; Zhao, C.; Zhang, G.; Zhao, Y.; Li, J.R.; Li, X.P.; Wei, L.H. Hypomethylated CpG around the transcription start site enables TERT expression and HPV16 E6 regulates TERT methylation in cervical cancer cells. Gynecol. Oncol. 2012, 124, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Zinn, R.L.; Pruitt, K.; Eguchi, S.; Baylin, S.B.; Herman, J.G. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007, 67, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, P.; Messinis, I.; Tsezou, A. DNA methylation is not likely to be responsible for hTERT expression in premalignant cervical lesions. Exp. Biol. Med. 2007, 232, 881–886. [Google Scholar]
- Schutze, D.M.; Snijders, P.J.; Bosch, L.; Kramer, D.; Meijer, C.J.; Steenbergen, R.D. Differential in vitro immortalization capacity of eleven, probable high-risk human papillomavirus types. J. Virol. 2014, 88, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Kilian, A.; Bowtell, D.D.; Abud, H.E.; Hime, G.R.; Venter, D.J.; Keese, P.K.; Duncan, E.L.; Reddel, R.R.; Jefferson, R.A. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum. Mol. Genet. 1997, 6, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, R.A.; Vliet-Gregg, P.; Xu, M.; Galloway, D.A. Nfx1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J. Virol. 2009, 83, 6446–6456. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, A.; Kalthoff, H.; Schuermann, M.; Schafer, B.; Boukamp, P. Dual regulation of telomerase activity through c-Myc-dependent inhibition and alternative splicing of hTERT. J. Cell Sci. 2002, 115, 1305–1312. [Google Scholar] [PubMed]
- Mole, S.; McFarlane, M.; Chuen-Im, T.; Milligan, S.G.; Millan, D.; Graham, S.V. RNA splicing factors regulated by HPV16 during cervical tumour progression. J. Pathol. 2009, 219, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, R.A.; Egelkrout, E.M.; Vliet-Gregg, P.; Gewin, L.C.; Gafken, P.R.; Galloway, D.A. Nfx1-123 and poly(A) binding proteins synergistically augment activation of telomerase in human papillomavirus type 16E6 expressing cells. J. Virol. 2007, 81, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V. The R3H motif: A domain that binds single-stranded nucleic acids. Trends Biochem. Sci. 1998, 23, 329–330. [Google Scholar] [CrossRef]
- Mangus, D.A.; Evans, M.C.; Jacobson, A. Poly(A)-binding proteins: Multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 2003, 4, 223. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, R.A.; Vliet-Gregg, P.; Xu, M.; Galloway, D.A. Cytoplasmic poly(A) binding proteins regulate telomerase activity and cell growth in human papillomavirus type 16 E6-expressing keratinocytes. J. Virol. 2010, 84, 12934–12944. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.M.; Underbrink, M.P.; Howie, H.L.; Galloway, D.A. The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J. Virol. 2008, 82, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Mutirangura, A.; Sriuranpong, V.; Termrunggraunglert, W.; Tresukosol, D.; Lertsaguansinchai, P.; Voravud, N.; Niruthisard, S. Telomerase activity and human papillomavirus in malignant, premalignant and benign cervical lesions. Br. J. Cancer 1998, 78, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Ley, C.; Bauer, H.M.; Reingold, A.; Schiffman, M.H.; Chambers, J.C.; Tashiro, C.J.; Manos, M.M. Determinants of genital human papillomavirus infection in young women. J. Natl. Cancer Inst. 1991, 83, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Branca, M.; Giorgi, C.; Ciotti, M.; Santini, D.; Di, B.L.; Costa, S.; Benedetto, A.; Bonifacio, D.; Di, B.P.; Paba, P.; et al. Upregulation of telomerase (hTERT) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus, virus persistence, or disease outcome in cervical cancer. Diagn. Cytopathol. 2006, 34, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Nachajova, M.; Brany, D.; Dvorska, D. Telomerase and the process of cervical carcinogenesis. Tumour Biol. 2015, 36, 7335–7338. [Google Scholar] [CrossRef] [PubMed]
- Snijders, P.J.; van Duin, M.; Walboomers, J.M.; Steenbergen, R.D.; Risse, E.K.; Helmerhorst, T.J.; Verheijen, R.H.; Meijer, C.J. Telomerase activity exclusively in cervical carcinomas and a subset of cervical intraepithelial neoplasia grade III lesions: Strong association with elevated messenger RNA levels of its catalytic subunit and high-risk human papillomavirus DNA. Cancer Res. 1998, 58, 3812–3818. [Google Scholar] [PubMed]
- Miller, J.; Dakic, A.; Chen, R.; Palechor-Ceron, N.; Dai, Y.; Kallakury, B.; Schlegel, R.; Liu, X. HPV16 E7 protein and hTERT proteins defective for telomere maintenance cooperate to immortalize human keratinocytes. PLoS. Pathog. 2013, 9, e1003284. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Reuschenbach, M.; Huebbers, C.U.; Prigge, E.S.; Bermejo, J.L.; Kalteis, M.S.; Preuss, S.F.; Seuthe, I.M.; Kolligs, J.; Speel, E.J.; Olthof, N.; et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer 2015, 121, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Baege, A.C.; Berger, A.; Schlegel, R.; Veldman, T.; Schlegel, R. Cervical epithelial cells transduced with the papillomavirus E6/E7 oncogenes maintain stable levels of oncoprotein expression but exhibit progressive, major increases in hTERT gene expression and telomerase activity. Am. J. Pathol. 2002, 160, 1251–1257. [Google Scholar] [CrossRef]

{kind=link}
| HPV Gene | Effect on hTERT | Cellular Protein Target |
|---|---|---|
| Chromatin Effects | ||
| E6 and E7 | Promoter methylation changes | |
| E6 | Increase promoter acetylation | HATs and HDACs, mSin3A |
| Transcription Effects | ||
| E6 | Increase transcriptional activators | c-Myc/Max, Sp1 |
| E6 | Decrease transcriptional repressors | c-Myc/Mad, Maz, USF1, NFX1-91 |
| E7 | Increase expression with E6 | |
| RNA Effects | ||
| E6 | Increase transcript stability | NFX1-123, PABPCs |
| E6 | Increase active spliced isoform of hTERT | c-Myc |
| Protein Effects | ||
| E6 | Binds hTERT | hTERT |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katzenellenbogen, R. Telomerase Induction in HPV Infection and Oncogenesis. Viruses 2017, 9, 180. https://doi.org/10.3390/v9070180
Katzenellenbogen R. Telomerase Induction in HPV Infection and Oncogenesis. Viruses. 2017; 9(7):180. https://doi.org/10.3390/v9070180
Chicago/Turabian StyleKatzenellenbogen, Rachel. 2017. "Telomerase Induction in HPV Infection and Oncogenesis" Viruses 9, no. 7: 180. https://doi.org/10.3390/v9070180
APA StyleKatzenellenbogen, R. (2017). Telomerase Induction in HPV Infection and Oncogenesis. Viruses, 9(7), 180. https://doi.org/10.3390/v9070180