
PCR-DGGE Analysis: Unravelling Complex Mixtures of Badnavirus Sequences Present in Yam Germplasm


Abstract
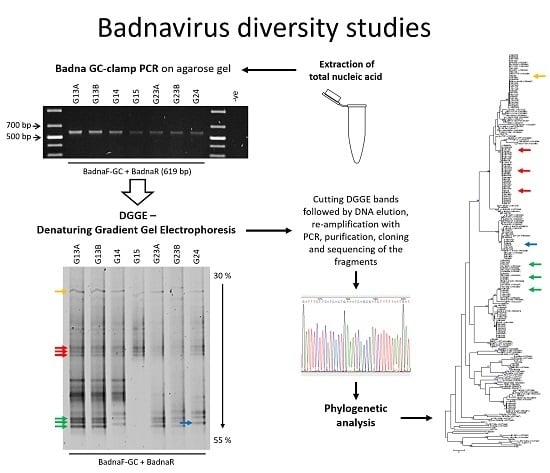
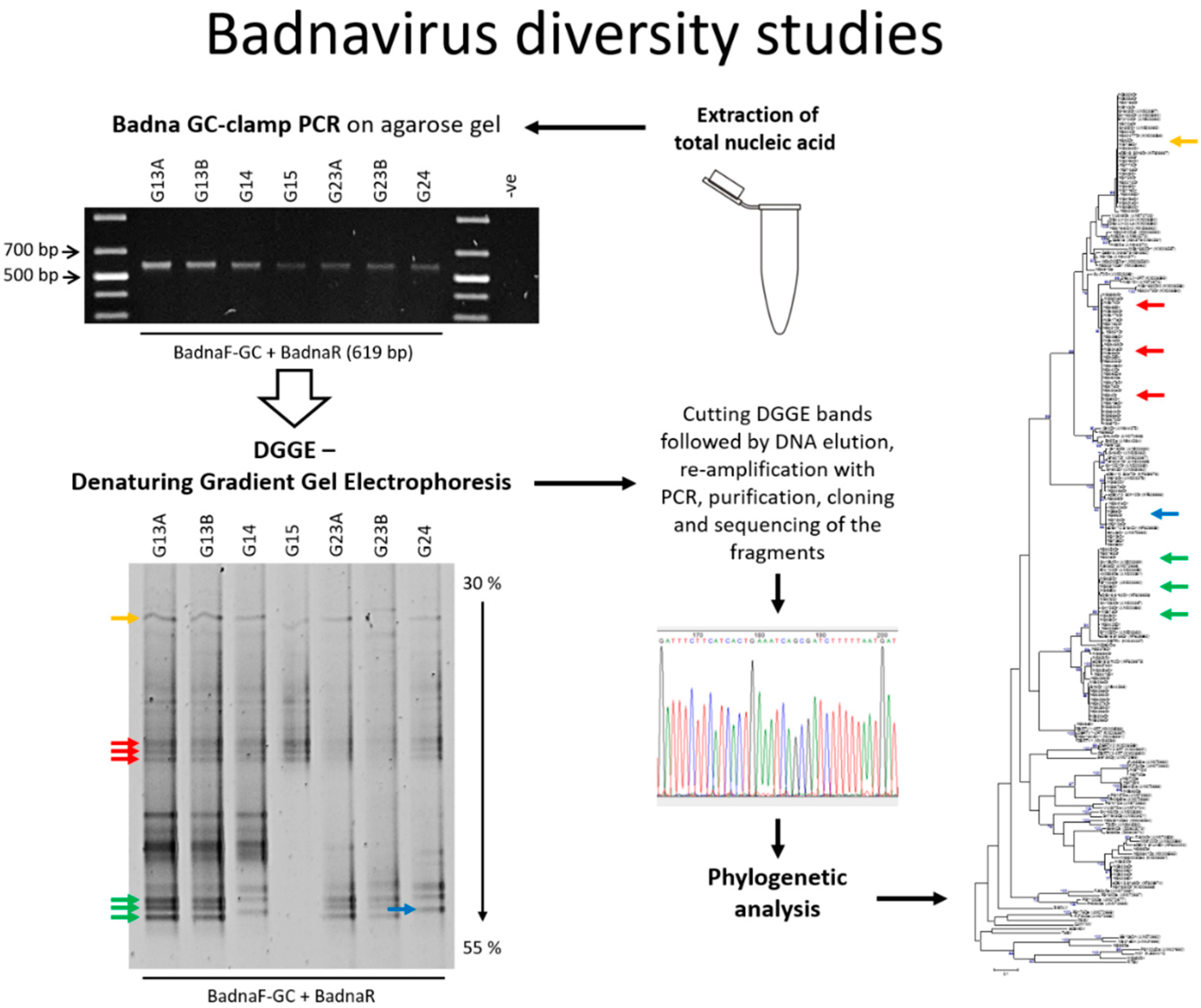
:
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Total Nucleic Acid Extraction from Yam Leaves and PCR Amplification of Badnavirus Sequences
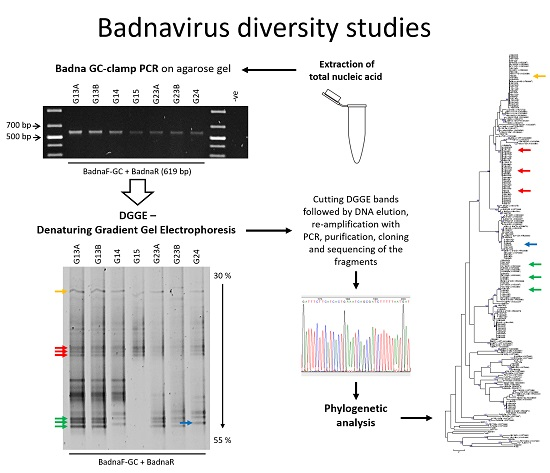
2.3. Denaturing Gradient Gel Electrophoresis (DGGE)
2.4. Sequence Analysis and Phylogeny
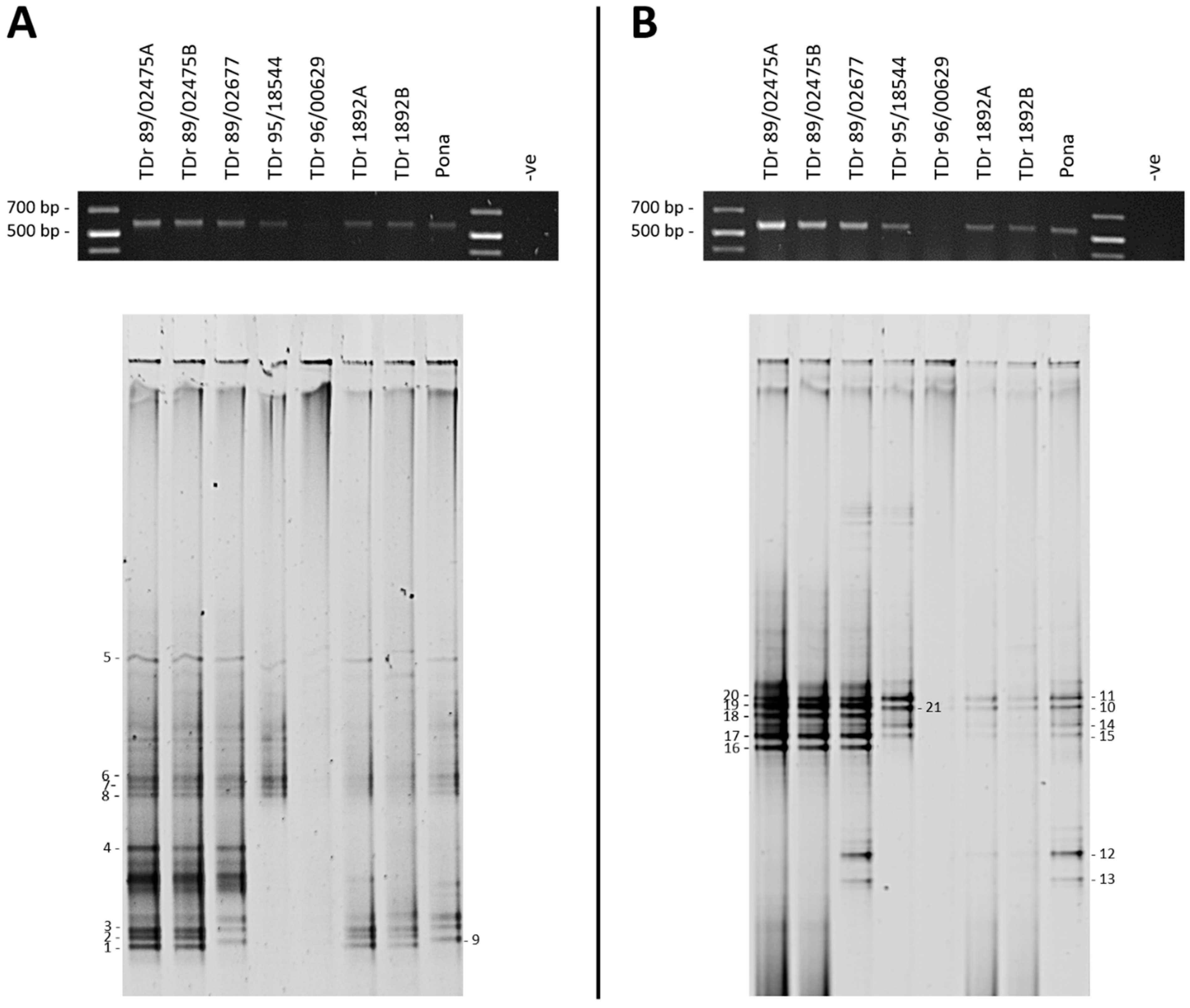
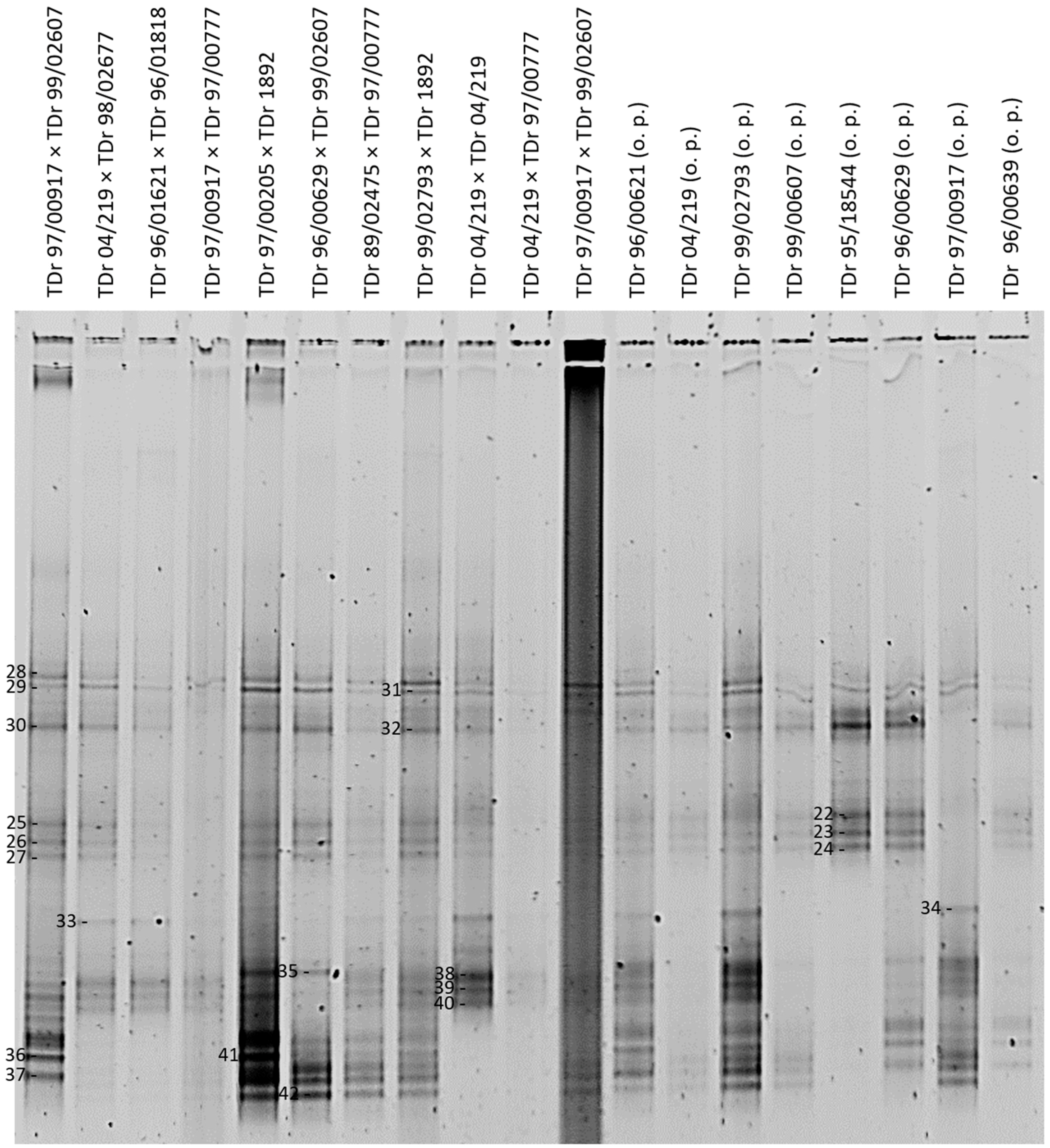
3. Results
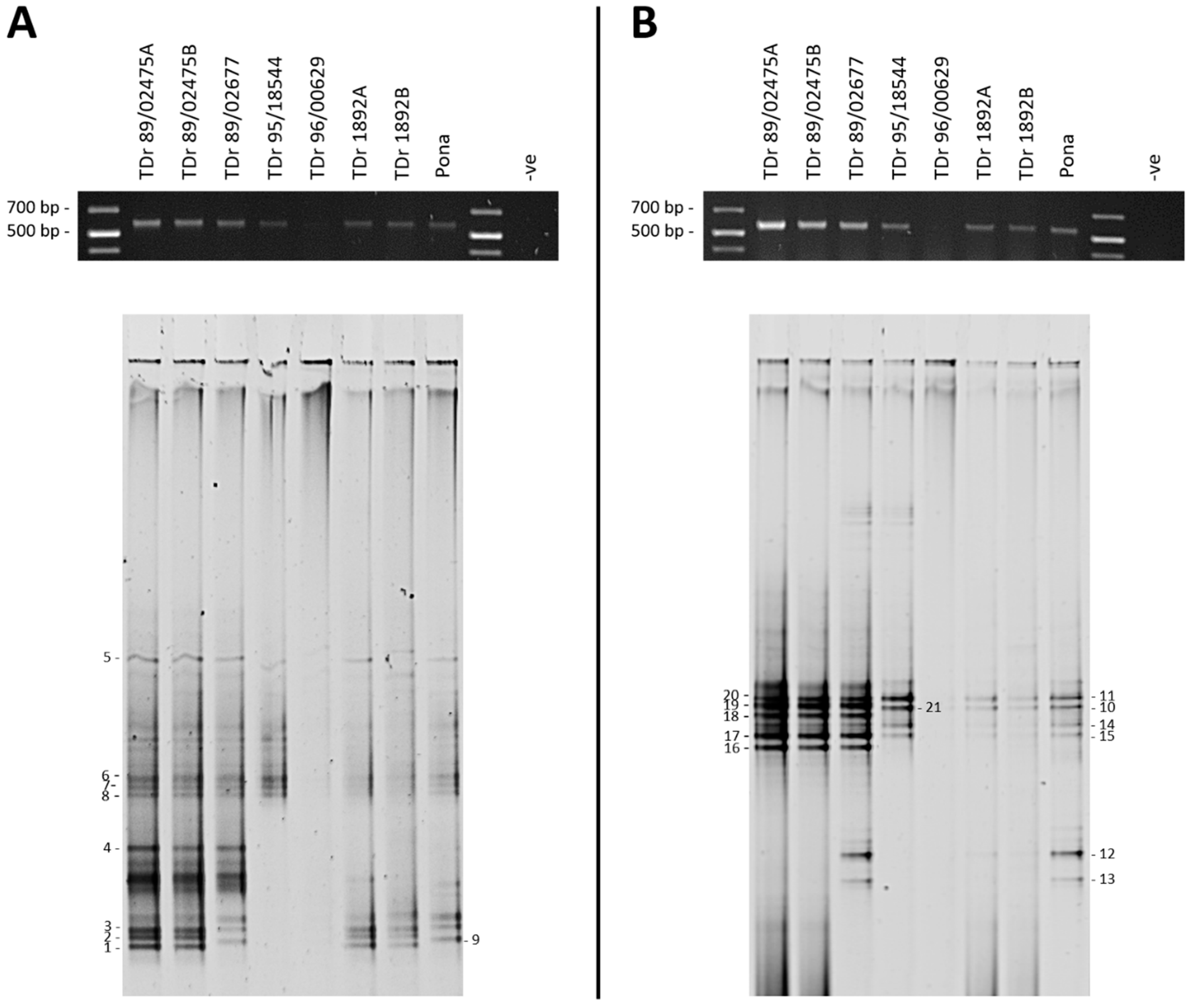
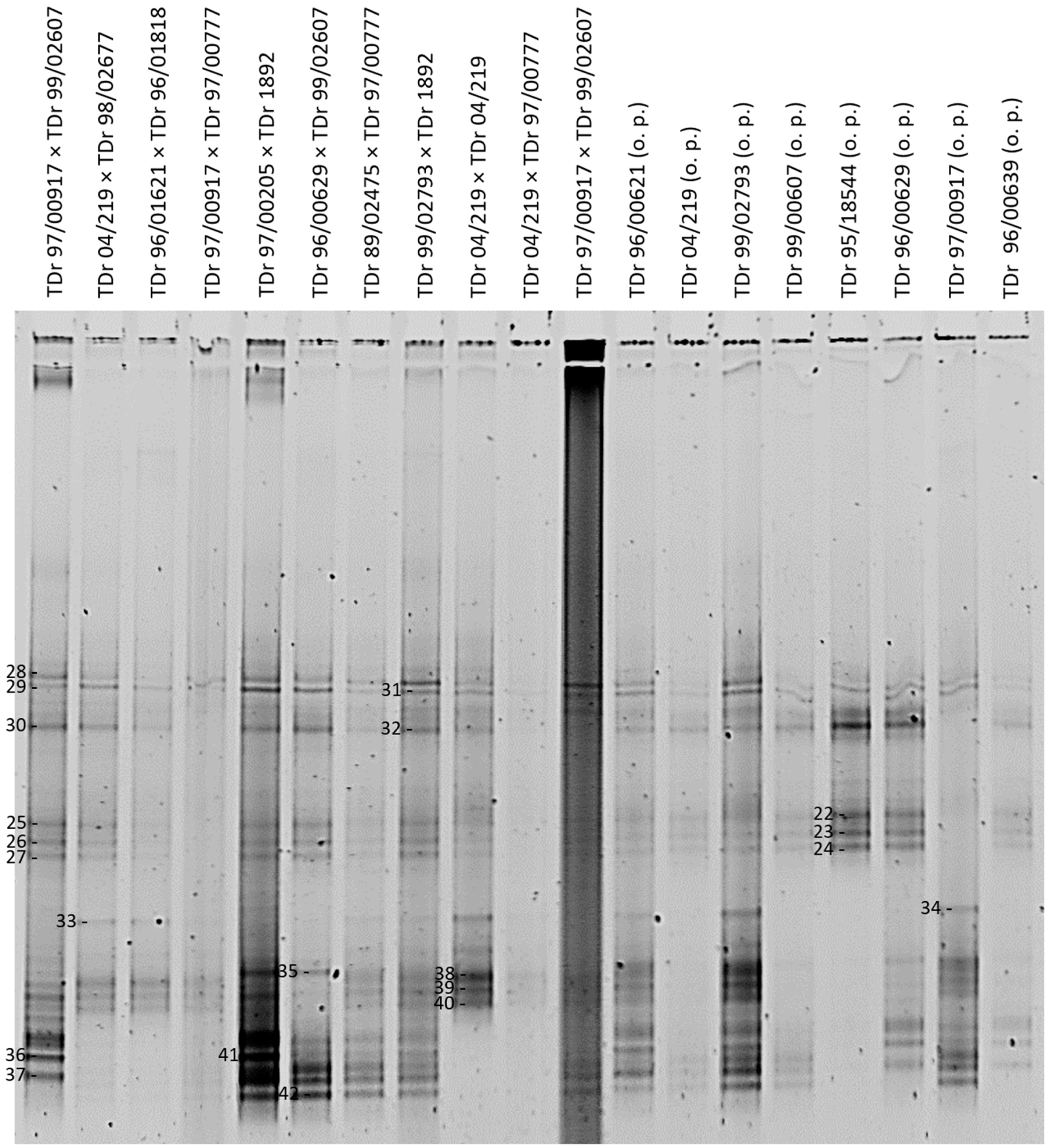
3.1. DGGE Resolves a Complex Mixture of Badnavirus Sequences Present in Dioscorea Species
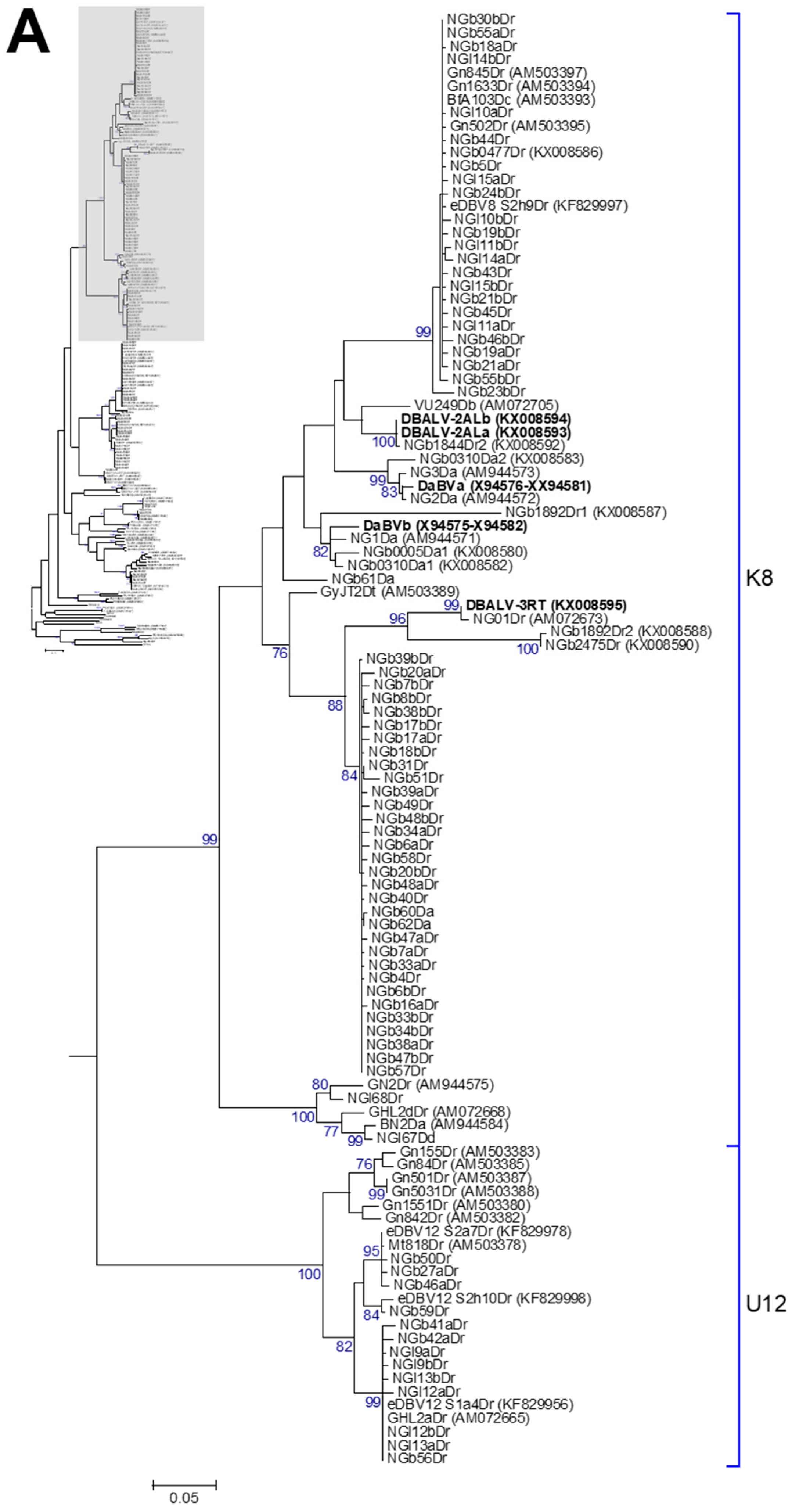
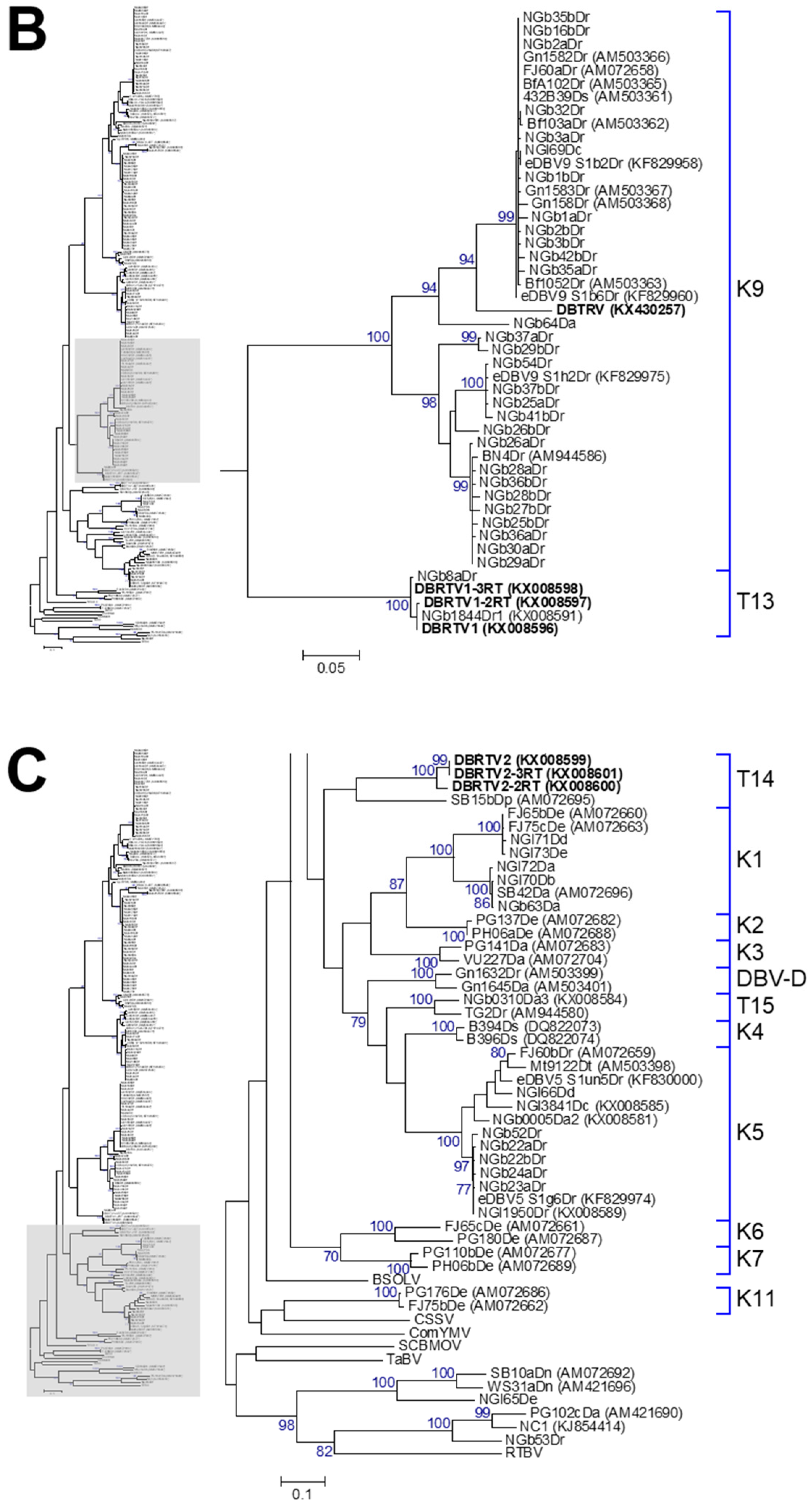
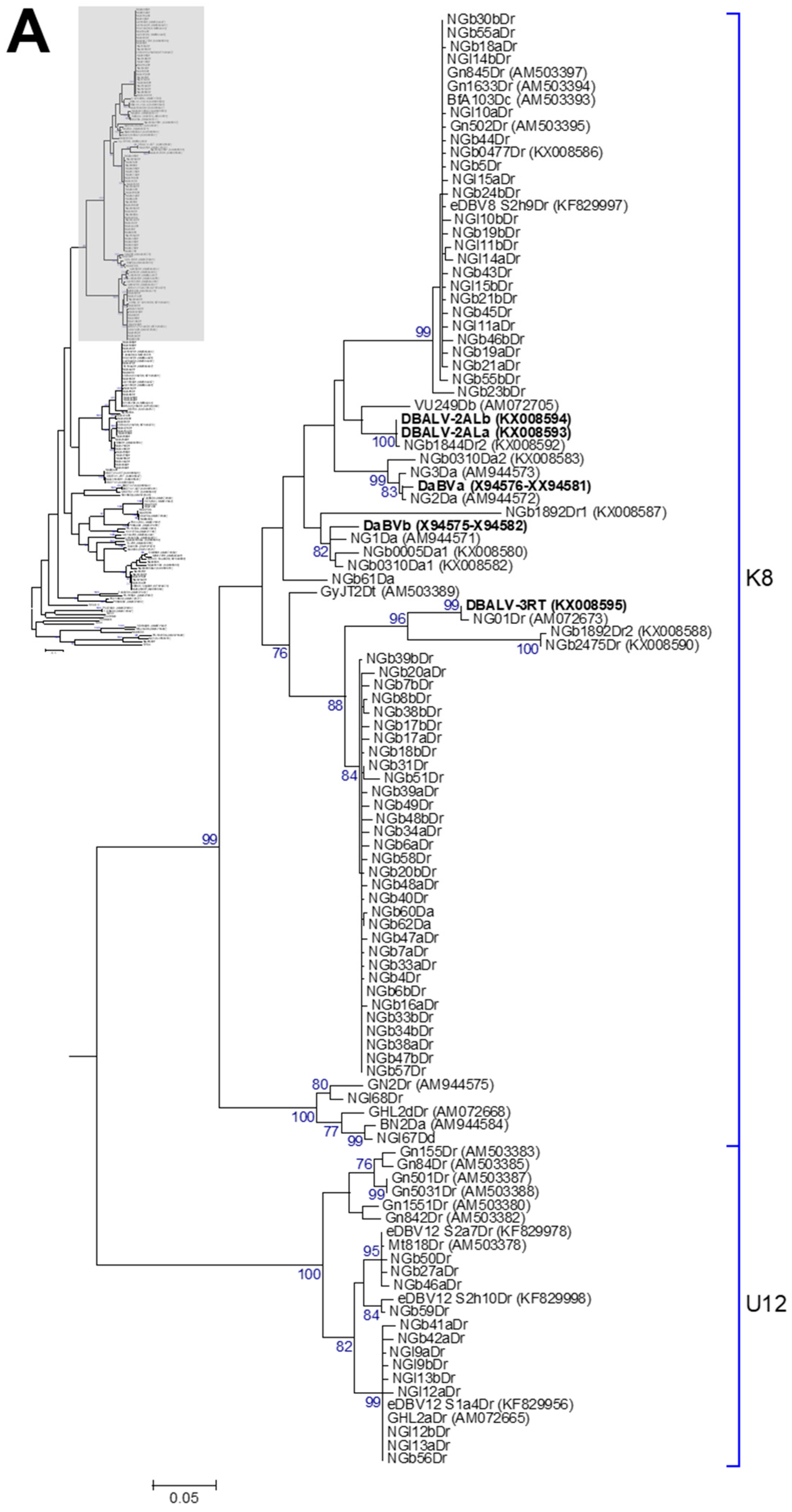
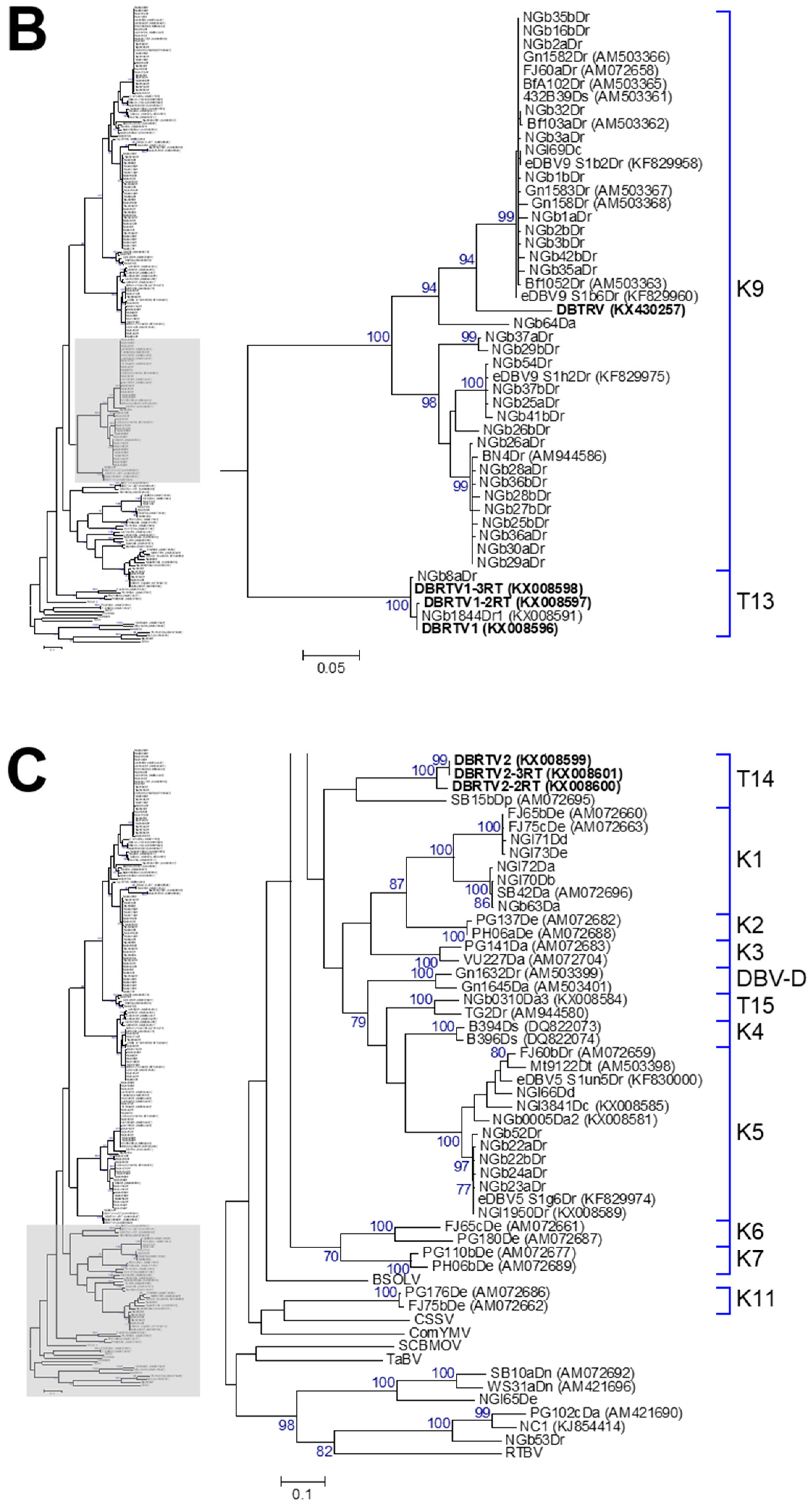
3.2. Phylogenetic Diversity of Dioscorea Badnavirus Sequences
3.3. Monophyletic Group Assignment of Sequences Identified in This Study
3.4. Conservation of Amino Acid Motifs in Partial RT-RNaseH Badnavirus Sequences
4. Discussion
4.1. Potential of DGGE in Badnavirus Diversity Studies and Identification of Potential Integrated Sequences
4.2. DGGE-Captured Badnavirus Diversity
4.3. Endogenous Badnavirus Partial RT-RNaseH Sequences
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BF | Badna-forward primer |
| BLAST | basic local alignment search tool |
| BR | Badna-reverse primer |
| BSOLV | Banana streak OL virus |
| BSV | banana streak virus |
| ComYMV | Commelina yellow mottle virus |
| CSSV | Cacao swollen shoot virus |
| CTAB | cetyltrimethylammonium bromide |
| DBALV | Dioscorea bacilliform alata virus |
| DBRTV | Dioscorea bacilliform rotundata (RT) virus |
| DBV | Dioscorea bacilliform virus |
| DGGE | denaturing gradient gel electrophoresis |
| DOAJ | Directory of open access journals |
| eDBV | endogenous Dioscorea bacilliform virus |
| GC | GC clamp |
| ICTV | International Committee on Taxonomy of Viruses |
| IITA | International Institute of Tropical Agriculture |
| ISEM | immunosorbent electron microscopy |
| LD | linear dichroism |
| MAFFT | Multiple Alignment using Fast Fourier Transform |
| NCBI | National Centre for Biotechnology Information |
| NG | Nigeria |
| NGS | next-generation sequencing |
| NRI | Natural Resources Institute |
| o.p. | open pollinated |
| ORF | open reading frame |
| PCR | polymerase chain reaction |
| RCA | rolling circle amplification |
| RFLP | restriction fragment length polymorphism |
| RNaseH | ribonuclease H |
| RT | reverse transcriptase |
| RTBV | Rice tungro bacilliform virus |
| SCBMOV | Sugarcane bacilliform MO virus |
| TaBV | Taro bacilliform virus |
| TAE | Tris-acetate-EDTA |
| TBE | Tris-Boric acid-EDTA |
| TDa | Dioscorea alata accession |
| TDb | Dioscorea bulbifera accession |
| TDc | Dioscorea cayenensis accession |
| TDe | Dioscorea esculenta accession |
| TDr | Dioscorea rotundata accession |
Appendix A
References
- Borah, B.K.; Sharma, S.; Kant, R.; Johnson, A.M.A.; Saigopal, D.V.R.; Dasgupta, I. Bacilliform DNA-containing plant viruses in the tropics: Commonalities within a genetically diverse group. Mol. Plant Pathol. 2013, 14, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Harper, G.; Hart, D.; Moult, S.; Hull, R.; Geering, A.; Thomas, J. The diversity of Banana streak virus isolates in Uganda. Arch. Virol. 2005, 150, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, L.; Lebas, B.S.M.; Seal, S.E. Yams (Dioscorea spp.) from the South Pacific Islands contain many novel badnaviruses: Implications for international movement of yam germplasm. Arch. Virol. 2008, 153, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, B.E.L. Purification and serology of a bacilliform virus associated with Banana streak disease. Phytopathology 1986, 80, 995–999. [Google Scholar] [CrossRef]
- Muller, E.; Dupuy, V.; Blondin, L.; Bauffe, F.; Daugrois, J.H.; Laboureau, N.; Iskra-Caruana, M.L. High molecular variability of sugarcane bacilliform viruses in Guadeloupe implying the existence of at least three new species. Virus Res. 2011, 160, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Harper, G.; Dahal, G.; Thottappilly, G.; Hull, R. Detection of episomal banana streak badnavirus by IC-PCR. J. Virol. Methods 1999, 79, 1–8. [Google Scholar] [CrossRef]
- Ndowora, T.; Dahal, G.; LaFleur, D.; Harper, G.; Hull, R.; Olszewski, N.E.; Lockhart, B. Evidence that badnavirus infection in Musa can originate from integrated pararetroviral sequences. Virology 1999, 255, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Le Provost, G.; Iskra-Caruana, M.L.; Acina, I.; Teycheney, P.Y. Improved detection of episomal Banana streak viruses by multiplex immunocapture PCR. J. Virol. Methods 2006, 137, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.I.; Hohn, T.; Selvarajan, R. Badnaviruses: The Current Global Scenario. Viruses 2016, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.W.; Maumus, F.; Copetti, D.; Choisne, N.; Zwickl, D.J.; Zytnicki, M.; McTaggart, A.R.; Scalabrin, S.; Vezzulli, S.; Wing, R.A.; et al. Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat. Commun. 2014, 5, 5269. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.W.; Scharaschkin, T.; Teycheney, P.Y. The classification and nomenclature of endogenous viruses of the family Caulimoviridae. Arch. Virol. 2010, 155, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Mette, M.F.; Kanno, T.; Aufsatz, W.; Jakowitsch, J.; van der Winden, J.; Matzke, M.A.; Matzke, A.J.M. Endogenous viral sequences and their potential contribution to heritable virus resistance in plants. EMBO J. 2002, 21, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Staginnus, C.; Iskra-Caruana, M.L.; Lockhart, B.; Hohn, T.; Richert-Pöggeler, K.R. Suggestions for a nomenclature of endogenous pararetroviral sequences in plants. Arch. Virol. 2009, 154, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, D.A.; Lockhart, B.E.L.; Olszewski, N.E. Portions of the banana streak badnavirus genome are integrated in the genome of its host Musa sp. Phytopathology 1996, 86, S100. [Google Scholar]
- Richert-Pöggeler, K.R.; Shepherd, R.J. Petunia vein-clearing virus: A plant pararetrovirus with the core sequences for an integrase function. Virology 1997, 236, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Jakowitsch, J.; Mette, M.F.; van Der Winden, J.; Matzke, M.A.; Matzke, A.J. Integrated pararetroviral sequences define a unique class of dispersed repetitive DNA in plants. Proc. Natl. Acad. Sci. USA 1999, 96, 13241–13246. [Google Scholar] [CrossRef] [PubMed]
- Kunii, M.; Kanda, M.; Nagano, H.; Uyeda, I.; Kishima, Y.; Sano, Y. Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: Implications for integration and evolution. BMC Genom. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.N.; Harper, G.; Heslop-Harrison, J.S. Characterisation of pararetrovirus-like sequences in the genome of potato (Solanum tuberosum). Cytogenet. Genome Res. 2005, 110, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Staginnus, C.; Richert-Pöggeler, K.R. Endogenous pararetroviruses: Two-faced travelers in the plant genome. Trends Plant Sci. 2006, 11, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Chabannes, M.; Baurens, F.C.; Duroy, P.O.; Bocs, S.; Vernerey, M.S.; Rodier-Goud, M.; Barbe, V.; Gayral, P.; Iskra-Caruana, M.L. Three infectious viral species lying in wait in the banana genome. J. Virol. 2013, 87, 8624–8637. [Google Scholar] [CrossRef] [PubMed]
- Gayral, P.; Noa-Carrazana, J.C.; Lescot, M.; Lheureux, F.; Lockhart, B.E.; Matsumoto, T.; Piffanelli, P.; Iskra-Caruana, M.L. A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J. Virol. 2008, 82, 6697–6710. [Google Scholar] [CrossRef] [PubMed]
- Richert-Pöggeler, K.R.; Noreen, F.; Schwarzacher, T.; Harper, G.; Hohn, T. Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J. 2003, 22, 4836–4845. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, B.E.; Menke, J.; Dahal, G.; Olszewski, N.E. Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol. 2000, 81, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.; McMichael, L.A.; Dietzgen, R.G.; Thomas, J.E. Genetic Diversity Among Banana streak virus Isolates from Australia. Phytopathology 2000, 90, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Iskra-Caruana, M.L.; Baurens, F.C.; Gayral, P.; Chabannes, M. A four-partner plant-virus interaction: Enemies can also come from within. Mol. Plant Microbe Interact. 2010, 23, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Iskra-Caruana, M.L.; Chabannes, M.; Duroy, P.O.; Muller, E. A possible scenario for the evolution of Banana streak virus in banana. Virus Res. 2014, 186, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Côte, F.X.; Galzi, S.; Folliot, M.; LamagnÈre, Y.; Teycheney, P.Y.; Iskra-Caruana, M.L. Micropropagation by tissue culture triggers differential expression of infectious endogenous Banana streak virus sequences (eBSV) present in the B genome of natural and synthetic interspecific banana plantains. Mol. Plant Pathol. 2010, 11, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Dallot, S.; Acuna, P.; Rivera, C.; Ramirez, P.; Cote, F.; Lockhart, B.E.L.; Caruana, M.L. Evidence that the proliferation stage of micropropagation procedure is determinant in the expression of Banana streak virus integrated into the genome of the FHIA 21 hybrid (Musa AAAB). Arch. Virol. 2001, 146, 2179–2190. [Google Scholar] [CrossRef] [PubMed]
- Harper, G.; Hull, R.; Lockhart, B.; Olszewski, N. Viral sequences integrated into plant genomes. Annu. Rev. Phytopathol. 2002, 40, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Chabannes, M.; Iskra-Caruana, M.L. Endogenous pararetroviruses—A reservoir of virus infection in plants. Curr. Opin. Virol. 2013, 3, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, R.; Sartie, A. Crops that feed the World 1. Yams. Food Secur. 2010, 2, 305–315. [Google Scholar] [CrossRef]
- Ekanayake, I.J.; Asiedu, R. Problems and Perspectives of Yam-Based Cropping Systems in Africa. J. Crop Prod. 2003, 9, 531–558. [Google Scholar] [CrossRef]
- Bömer, M.; Turaki, A.; Silva, G.; Kumar, P.; Seal, S. A Sequence-Independent Strategy for Amplification and Characterisation of Episomal Badnavirus Sequences Reveals Three Previously Uncharacterised Yam Badnaviruses. Viruses 2016, 8, 188. [Google Scholar] [CrossRef] [PubMed]
- Bousalem, M.; Durand, O.; Scarcelli, N.; Lebas, B.S.M.; Kenyon, L.; Marchand, J.L.; Lefort, F.; Seal, S.E. Dilemmas caused by endogenous pararetroviruses regarding the taxonomy and diagnosis of yam (Dioscorea spp.) badnaviruses: Analyses to support safe germplasm movement. Arch. Virol. 2009, 154, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Eni, A.O.; Hughes, J.D.; Asiedu, R.; Rey, M.E.C. Sequence diversity among badnavirus isolates infecting yam (Dioscorea spp.) in Ghana, Togo, Benin and Nigeria. Arch. Virol. 2008, 153, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Galzi, S.; Scutt, R.; Prophète, P.; Roumagnac, P.; Filloux, D. Assessment and Characterization of the Genetic Diversity of Viruses Infecting Cultivated Yams (Dioscorea spp.) in Haïti. In Rencontres de Virologie Végétale; Marais Armelle, R.F., Ed.; SFP INRA: Paris, France, 2013; p. 70. [Google Scholar]
- Lima, J.S.; Lima, G.S.A.; Micheref, S.J. Variabilidade genética de isolados de badnavírus infectando inhame (Dioscorea spp.) no nordeste do Brasil. Trop. Plant Pathol. 2013, 38, 349–353. [Google Scholar] [CrossRef]
- Seal, S.; Turaki, A.; Muller, E.; Kumar, P.L.; Kenyon, L.; Filloux, D.; Galzi, S.; Lopez-Montes, A.; Iskra-Caruana, M.L. The prevalence of badnaviruses in West African yams (Dioscorea cayenensis-rotundata) and evidence of endogenous pararetrovirus sequences in their genomes. Virus Res. 2014, 186, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Umber, M.; Filloux, D.; Muller, E.; Laboureau, N.; Galzi, S.; Roumagnac, P.; Iskra-Caruana, M.L.; Pavis, C.; Teycheney, P.Y.; Seal, S.E. The genome of African yam (Dioscorea cayenensis-rotundata complex) hosts endogenous sequences from four distinct badnavirus species. Mol. Plant Pathol. 2014, 15, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Gayral, P.; Iskra-Caruana, M.L. Phylogeny of banana streak virus reveals recent and repetitive endogenization in the genome of its banana host (Musa sp.). J. Mol. Evol. 2009, 69, 65–80. [Google Scholar] [CrossRef] [PubMed]
- James, A.P.; Geijskes, R.J.; Dale, J.L.; Harding, R.M. Molecular characterisation of six badnavirus species associated with leaf streak disease of banana in East Africa. Ann. Appl. Biol. 2011, 158, 346–353. [Google Scholar] [CrossRef]
- Jaufeerally-Fakim, Y.; Khorugdharry, A.; Harper, G. Genetic variants of Banana streak virus in Mauritius. Virus Res. 2006, 115, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.G.; Lerman, L.S. Length-independent separation of DNA restriction fragments in two-dimensional gel electrophoresis. Cell 1979, 16, 191–200. [Google Scholar] [CrossRef]
- Green, S.J.; Leigh, M.B.; Neufeld, J.D. Denaturing Gradient Gel Electrophoresis (DGGE) for Microbial Community Analysis. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 4137–4158. ISBN 978-3-540-77587-4. [Google Scholar]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [PubMed]
- Sheffield, V.C.; Cox, D.R.; Lerman, L.S.; Myers, R.M. Attachment of a 40-base-pair G + C-rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chain reaction results in improved detection of single-base changes. Proc. Natl. Acad. Sci. USA 1989, 86, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Suttle, C.A. Sequence Analysis of Marine Virus Communities Reveals that Groups of Related Algal Viruses Are Widely Distributed in Nature. Appl. Environ. Microbiol. 2002, 68, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Top, B. A simple method to attach a universal 50-bp GC-clamp to PCR fragments used for mutation analysis by DGGE. Genome Res. 1992, 2, 83–85. [Google Scholar] [CrossRef]
- Ercolini, D. PCR-DGGE fingerprinting: Novel strategies for detection of microbes in food. J. Microbiol. Methods 2004, 56, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, H.; Lu, H.; Qian, G.; Lv, L.; Zhang, C.; Guo, J.; Jiang, H.; Zheng, B.; Yang, F.; et al. The Effect of Probiotic Treatment on Patients Infected with the H7N9 Influenza Virus. PLoS ONE 2016, 11, e0151976. [Google Scholar] [CrossRef] [PubMed]
- Valaskova, V.; Baldrian, P. Denaturing gradient gel electrophoresis as a fingerprinting method for the analysis of soil microbial communities. Plant Soil Environ. 2009, 55, 413–423. [Google Scholar]
- Michaelsen, A.; Pinzari, F.; Ripka, K.; Lubitz, W.; Piñar, G. Application of molecular techniques for identification of fungal communities colonising paper material. Int. Biodeterior. Biodegrad. 2006, 58, 133–141. [Google Scholar] [CrossRef]
- Carmona, M.; Sepúlveda, D.; Cárdenas, C.; Nilo, L.; Marshall, S.H. Denaturing gradient gel electrophoresis (DGGE) as a powerful novel alternative for differentiation of epizootic ISA virus variants. PLoS ONE 2012, 7, e37353. [Google Scholar] [CrossRef] [PubMed]
- Lyttle, D.J.; Orlovich, D.A.; Guy, P.L. Detection and analysis of endogenous badnaviruses in the New Zealand flora. AoB Plants 2011, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Suttle, C.A. Use of the polymerase chain reaction and denaturing gradient gel electrophoresis to study diversity in natural virus communities. Hydrobiologia 1999, 401, 19–32. [Google Scholar] [CrossRef]
- Lerner, A.; Shor, Y.; Vinokurov, A.; Okon, Y.; Jurkevitch, E. Can denaturing gradient gel electrophoresis (DGGE) analysis of amplified 16s rDNA of soil bacterial populations be used in forensic investigations? Soil Biol. Biochem. 2006, 38, 1188–1192. [Google Scholar] [CrossRef]
- Liu, B.; Lowes, F. Multiple Divergent ITS1 Copies Were Identified in Single Tomato Genome Using DGGE Analysis. Plant Mol. Biol. Report. 2013, 31, 272–279. [Google Scholar] [CrossRef]
- Riedel, G.E.; Swanberg, S.L.; Kuranda, K.D.; Marquette, K.; LaPan, P.; Bledsoe, P.; Kennedy, A.; Lin, B.Y. Denaturing gradient gel electrophoresis identifies genomic DNA polymorphism with high frequency in maize. Theor. Appl. Genet. 1990, 80, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.C.; Hafner, G.J.; Revill, P.A.; Dale, J.L.; Harding, R.M. Sequence diversity of South Pacific isolates of Taro bacilliform virus and the development of a PCR-based diagnostic test. Arch. Virol. 2003, 148, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Lebas, B.S.M. Diversity of Viruses Infecting Dioscorea Species in the South Pacific. Ph.D. Thesis, University of Greenwich, Chatham Maritime, UK, 2002. [Google Scholar]
- Mumford, R.A.; Seal, S.E. Rapid single-tube immunocapture RT-PCR for the detection of two yam potyviruses. J. Virol. Methods 1997, 69, 73–79. [Google Scholar] [CrossRef]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Family—Caulimoviridae. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 429–443. ISBN 978-0-12-384684-6. [Google Scholar]
- Silva, G.; Bömer, M.; Nkere, C.; Lava Kumar, P.; Seal, S.E. Rapid and specific detection of Yam mosaic virus by reverse-transcription recombinase polymerase amplification. J. Virol. Methods 2015, 222, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- National Centre for Biotechnology Information (NCBI) GenBank. Available online: https://www.ncbi.nlm.nih.gov/genbank/ (accessed on 10 June 2017).
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Multiple Alignment Using Fast Fourier Transform (MAFFT). Available online: http://www.ebi.ac.uk/Tools/msa/mafft/ (accessed on 10 June 2017).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using CLUSTAL OMEGA. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Clustal Omega. Available online: http://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 10 June 2017).
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Umber, M.; Gomez, R.-M.; Gélabale, S.; Bonheur, L.; Pavis, C.; Teycheney, P.-Y. The genome sequence of Dioscorea bacilliform TR virus, a member of the genus Badnavirus infecting Dioscorea spp., sheds light on the possible function of endogenous Dioscorea bacilliform viruses. Arch. Virol. 2017, 162, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Harper, G.; Hull, R. Cloning and sequence analysis of Banana streak virus. Virus Genes 1998, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Briddon, R.W.; Phillips, S.; Brunt, A.; Hull, R. Analysis of the sequence of Dioscorea alata bacilliform virus; comparison to other members of the badnavirus group. Virus Genes 1999, 18, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Tijdens, M.; Hoogveld, H.L.; Kamst-Van Agterveld, M.P.; Simis, S.G.H.; Baudoux, A.C.; Laanbroek, H.J.; Gons, H.J. Population dynamics and diversity of viruses, bacteria and phytoplankton in a shallow eutrophic lake. Microb. Ecol. 2008, 56, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.W.; Olszewski, N.E.; Harper, G.; Lockhart, B.E.L.; Hull, R.; Thomas, J.E. Banana contains a diverse array of endogenous badnaviruses. J. Gen. Virol. 2005, 86, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Ferris, M.J.; Muyzer, G.; Ward, D.M. Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl. Environ. Microbiol. 1996, 62, 340–346. [Google Scholar] [PubMed]
- Bouhida, M.; Lockhartz, B.E.L.; Olszewski, N.E. An analysis of the complete sequence of a sugarcane bacilliform virus genome infectious to banana and rice. J. Gen. Virol. 1993, 74, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Medberry, S.L.; Lockhart, B.E.L.; Olszewski, N.L. Properties of Commelina yellow mottle virus’s complete DNA sequence, genomic discontinuities and transcript suggest that it is a pararetrovirus. Nucleic Acids Res. 1990, 18, 5505–5513. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.; Muller, E. Molecular analysis of a full-length sequence of a new yam badnavirus from Dioscorea sansibarensis. Arch. Virol. 2007, 152, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.W.; Parry, J.N.; Thomas, J.E. Complete genome sequence of a novel badnavirus, banana streak IM virus. Arch. Virol. 2011, 156, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Sackey, S. Molecular variability analysis of five new complete cacao swollen shoot virus genomic sequences. Arch. Virol. 2005, 150, 53–66. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Accession a | DGGE Sequence b | Accession | Primers c | Size (bp) | NCBI Nearest Match | Identity (%) | Species Group d |
|---|---|---|---|---|---|---|---|
| TDr 89/02475 | NGb1aDr | KY555456 | BF-GC + BR | 528 | eDBV9_S1h6Dr (KF829977) | 99 | K9 |
| TDr 89/02475 | NGb1bDr | KY555457 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 89/02475 | NGb2aDr | KY555458 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 100 | K9 |
| TDr 89/02475 | NGb2bDr | KY555459 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 89/02475 | NGb3aDr | KY555460 | BF-GC + BR | 527 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 89/02475 | NGb3bDr | KY555461 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 89/02475 | NGb4Dr | KY555462 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 89/02475 | NGb5Dr | KY555463 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 89/02475 | NGb6aDr | KY555464 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb6bDr | KY555465 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 89/02475 | NGb7aDr | KY555466 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb7bDr | KY555467 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb8aDr | KY555468 | BF-GC + BR | 528 | DBRTV1-[3RT] (KX008598) | 99 | T13 |
| TDr 89/02475 | NGb8bDr | KY555469 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| Pona | NGl9aDr | KY555470 | BF-GC + BR | 528 | eDBV12_S1a4Dr (KF829956) | 99 | U12 |
| Pona | NGl9bDr | KY555471 | BF-GC + BR | 528 | eDBV12_S1a4Dr (KF829956) | 99 | U12 |
| Pona | NGl10aDr | KY555472 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| Pona | NGl10bDr | KY555473 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| Pona | NGl11aDr | KY555474 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| Pona | NGl11bDr | KY555475 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| Pona | NGl12aDr | KY555476 | BF + BR-GC | 528 | eDBV12_S1a4Dr (KF829956) | 99 | U12 |
| Pona | NGl12bDr | KY555477 | BF + BR-GC | 528 | eDBV12_S1a4Dr (KF829956) | 100 | U12 |
| Pona | NGl13aDr | KY555478 | BF + BR-GC | 528 | eDBV12_S1a4Dr (KF829956) | 100 | U12 |
| Pona | NGl13bDr | KY555479 | BF + BR-GC | 528 | eDBV12_S1a4Dr (KF829956) | 99 | U12 |
| Pona | NGl14aDr | KY555480 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| Pona | NGl14bDr | KY555481 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 100 | K8 |
| Pona | NGl15aDr | KY555482 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| Pona | NGl15bDr | KY555483 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 89/02475 | NGb16aDr | KY555484 | BF + BR-GC | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb16bDr | KY555485 | BF + BR-GC | 527 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 89/02475 | NGb17aDr | KY555486 | BF + BR-GC | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb17bDr | KY555487 | BF + BR-GC | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb18aDr | KY555488 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 89/02475 | NGb18bDr | KY555489 | BF + BR-GC | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 89/02475 | NGb19aDr | KY555490 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 89/02475 | NGb19bDr | KY555491 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 89/02475 | NGb20aDr | KY555492 | BF + BR-GC | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02475 | NGb20bDr | KY555493 | BF + BR-GC | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 95/18544 | NGb21aDr | KY555494 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 95/18544 | NGb21bDr | KY555495 | BF + BR-GC | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 95/18544 (o.p.) | NGb22aDr | KY555496 | BF-GC + BR | 528 | eDBV5_S1g6Dr (KF829974) | 99 | K5 |
| TDr 95/18544 (o.p.) | NGb22bDr | KY555497 | BF-GC + BR | 528 | eDBV5_S1g6Dr (KF829974) | 99 | K5 |
| TDr 95/18544 (o.p.) | NGb23aDr | KY555498 | BF-GC + BR | 528 | eDBV5_S1g6Dr (KF829974) | 99 | K5 |
| TDr 95/18544 (o.p.) | NGb23bDr | KY555499 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 97 | K8 |
| TDr 95/18544 (o.p.) | NGb24aDr | KY555500 | BF-GC + BR | 528 | eDBV5_S1g6Dr (KF829974) | 99 | K5 |
| TDr 95/18544 (o.p.) | NGb24bDr | KY555501 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 97/00917 × TDr 99/02607 | NGb25aDr | KY555502 | BF-GC + BR | 528 | eDBV9_S1h2Dr (KF829975) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb25bDr | KY555503 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb26aDr | KY555504 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb26bDr | KY555505 | BF-GC + BR | 528 | eDBV9_G1Dr (KF830002) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb27aDr | KY555506 | BF-GC + BR | 528 | eDBV12_S2a7Dr (KF829978) | 99 | U12 |
| TDr 97/00917 × TDr 99/02607 | NGb27bDr | KY555507 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb28aDr | KY555508 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb28bDr | KY555509 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb29aDr | KY555510 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb29bDr | KY555511 | BF-GC + BR | 528 | eDBV9_S2f8Dr (KF829993) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb30aDr | KY555512 | BF-GC + BR | 527 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb30bDr | KY555513 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 100 | K8 |
| TDr 99/02793 × TDr 1892 | NGb31Dr | KY555514 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 99/02793 × TDr 1892 | NGb32Dr | KY555515 | BF-GC + BR | 528 | eDBV9_S1e3Dr (KF829969) | 100 | K9 |
| TDr 04/219 × TDr 98/02677 | NGb33aDr | KY555516 | BF-GC + BR | 527 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 04/219 × TDr 98/02677 | NGb33bDr | KY555517 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 97/00917 (o.p.) | NGb34aDr | KY555518 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 97/00917 (o.p.) | NGb34bDr | KY555519 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 96/00629 × TDr 99/02607 | NGb35aDr | KY555520 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 96/00629 × TDr 99/02607 | NGb35bDr | KY555521 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 100 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb36aDr | KY555522 | BF-GC + BR | 528 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb36bDr | KY555523 | BF-GC + BR | 527 | BN4Dr (AM944586) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb37aDr | KY555524 | BF-GC + BR | 528 | eDBV9_S2f8Dr (KF829993) | 99 | K9 |
| TDr 97/00917 × TDr 99/02607 | NGb37bDr | KY555525 | BF-GC + BR | 528 | eDBV9_S1h2Dr (KF829975) | 99 | K9 |
| TDr 04/219 × TDr 04/219 | NGb38aDr | KY555526 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 04/219 × TDr 04/219 | NGb38bDr | KY555527 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 04/219 × TDr 04/219 | NGb39aDr | KY555528 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 04/219 × TDr 04/219 | NGb39bDr | KY555529 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 04/219 × TDr 04/219 | NGb40Dr | KY555530 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 97/00205 ×TDr 1892 | NGb41aDr | KY555531 | BF-GC + BR | 528 | eDBV12_S1a4Dr (KF829956) | 99 | U12 |
| TDr 97/00205 ×TDr 1892 | NGb41bDr | KY555532 | BF-GC + BR | 528 | eDBV9_S1h2Dr (KF829975) | 99 | K9 |
| TDr 96/00629 × TDr 99/02607 | NGb42aDr | KY555533 | BF-GC + BR | 528 | eDBV12_S1a4Dr (KF829956) | 99 | U12 |
| TDr 96/00629 × TDr 99/02607 | NGb42bDr | KY555534 | BF-GC + BR | 528 | 432B39Ds (AM503361) | 99 | K9 |
| TDr 95/18544 | NGb43Dr | KY555535 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 95/18544 | NGb44Dr | KY555536 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 95/18544 | NGb45Dr | KY555537 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 96/00629 | NGb46aDr | KY555538 | BF-GC + BR | 528 | eDBV12_S2a7Dr (KF829978) | 99 | U12 |
| TDr 96/00629 | NGb46bDr | KY555539 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 89/02677 | NGb47aDr | KY555540 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02677 | NGb47bDr | KY555541 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 89/02677 | NGb48aDr | KY555542 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02677 | NGb48bDr | KY555543 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 89/02677 | NGb49Dr | KY555544 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 96/00629 | NGb50Dr | KY555545 | BF-GC + BR | 528 | eDBV12_S2a7Dr (KF829978) | 99 | U12 |
| TDr 89/02677 | NGb51Dr | KY555546 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 91 | K8 |
| TDr 97/00917 (o.p.) | NGb52Dr | KY555547 | BF-GC + BR | 528 | eDBV5_S1g6Dr (KF829974) | 98 | K5 |
| TDr 97/00917 (o.p.) | NGb53Dr | KY555548 | BF-GC + BR | 528 | NC1 (KJ854414) | 79 | K13 |
| TDr 97/00917 × TDr 99/02607 | NGb54Dr | KY555549 | BF-GC + BR | 528 | eDBV9_S1h2Dr (KF829975) | 99 | K9 |
| TDr 99/02793 (o.p.) | NGb55aDr | KY555550 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 100 | K8 |
| TDr 99/02793 (o.p.) | NGb55bDr | KY555551 | BF-GC + BR | 528 | BfA103Dc (AM503393) | 99 | K8 |
| TDr 96/00621 (o.p.) | NGb56Dr | KY555552 | BF-GC + BR | 528 | eDBV12_S1a4Dr (KF829956) | 100 | U12 |
| TDr 97/00917 × TDr 97/00777 | NGb57Dr | KY555553 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDr 96/00629 × TDr 99/02607 | NGb58Dr | KY555554 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDr 97/00917 × TDr 99/02607 | NGb59Dr | KY555555 | BF-GC + BR | 528 | eDBV12_S2h10Dr (KF829998) | 99 | U12 |
| TDa 99/00240 × TDa 95/00310 | NGb60Da | KY555556 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 92 | K8 |
| TDa 99/00240 × TDa 01/00012 | NGb61Da | KY555557 | BF-GC + BR | 528 | NG1Da (AM944571) | 93 | K8 |
| TDa 01/00081 × TDa 02/00012 | NGb62Da | KY555558 | BF-GC + BR | 528 | GyJT2Dt (AM503389) | 93 | K8 |
| TDa 1/00081×TDa 98/00150 | NGb63Da | KY555559 | BF-GC + BR | 528 | SB42Da (AM072696) | 99 | K1 |
| TDa 00/00194 × TDa 98/00150 | NGb64Da | KY555560 | BF-GC + BR | 528 | eDBV9_S1b6Dr (KF829960) | 89 | K9 |
| TDe 3049A | NGl65De | KY555561 | BF-GC + BR | 528 | WS31aDn (AM421696) | 73 | K12 |
| TDd 4118B | NGl66Dd | KY555562 | BF-GC + BR | 528 | eDBV5_S1un5Dr (KF830000) | 93 | K5 |
| TDd 4118B | NGl67Dd | KY555563 | BF-GC + BR | 528 | BN2Da (AM944584) | 98 | K8 |
| TDr 1950B | NGl68Dr | KY555564 | BF-GC + BR | 528 | GN2Dr (AM944575) | 96 | K8 |
| TDc 3808C | NGl69Dc | KY555565 | BF-GC + BR | 528 | eDBV9_S1b2Dr (KF829958) | 100 | K9 |
| TDb 3045B | NGl70Db | KY555566 | BF-GC + BR | 528 | SB42Da (AM072696) | 99 | K1 |
| TDd 3778B | NGl71Dd | KY555567 | BF-GC + BR | 528 | FJ65bDe (AM072660) | 99 | K1 |
| TDa 1013C | NGl72Da | KY555568 | BF-GC + BR | 528 | SB42Da (AM072696) | 99 | K1 |
| TDe 3028A | NGl73De | KY555569 | BF-GC + BR | 528 | FJ65bDe (AM072660) | 99 | K1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turaki, A.A.; Bömer, M.; Silva, G.; Kumar, P.L.; Seal, S.E. PCR-DGGE Analysis: Unravelling Complex Mixtures of Badnavirus Sequences Present in Yam Germplasm. Viruses 2017, 9, 181. https://doi.org/10.3390/v9070181
Turaki AA, Bömer M, Silva G, Kumar PL, Seal SE. PCR-DGGE Analysis: Unravelling Complex Mixtures of Badnavirus Sequences Present in Yam Germplasm. Viruses. 2017; 9(7):181. https://doi.org/10.3390/v9070181
Chicago/Turabian StyleTuraki, Aliyu A., Moritz Bömer, Gonçalo Silva, P. Lava Kumar, and Susan E. Seal. 2017. "PCR-DGGE Analysis: Unravelling Complex Mixtures of Badnavirus Sequences Present in Yam Germplasm" Viruses 9, no. 7: 181. https://doi.org/10.3390/v9070181
APA StyleTuraki, A. A., Bömer, M., Silva, G., Kumar, P. L., & Seal, S. E. (2017). PCR-DGGE Analysis: Unravelling Complex Mixtures of Badnavirus Sequences Present in Yam Germplasm. Viruses, 9(7), 181. https://doi.org/10.3390/v9070181