
The Role of cccDNA in HBV Maintenance

{kind=link}
Abstract
:1. Introduction
2. HBV Infection and cccDNA Establishment
3. CccDNA Activity and Pool Size
4. HBV Treatment and Impact on the cccDNA
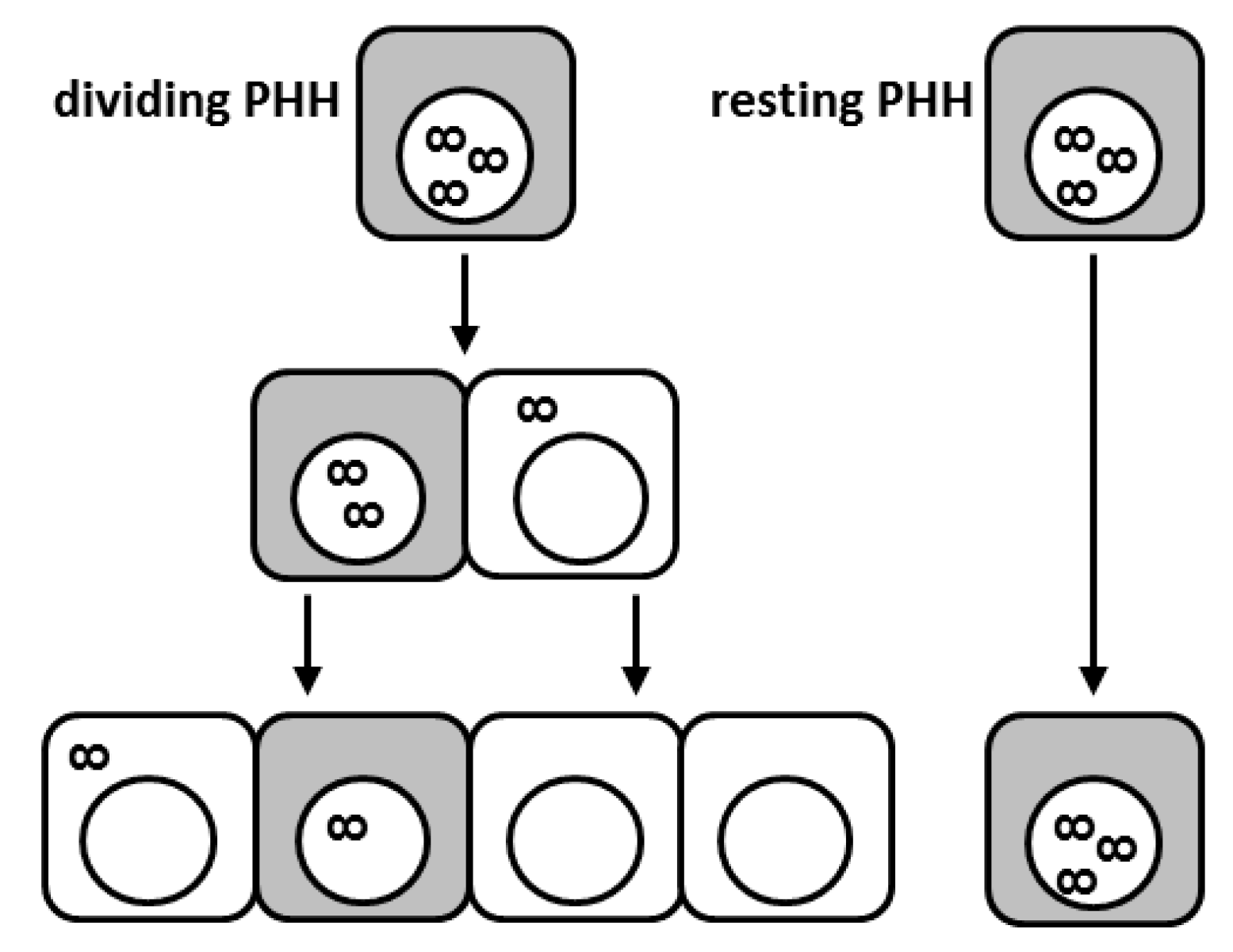
5. The Contribution of Cell Division to HBV Resolution
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zeisel, M.B.; Lucifora, J.; Mason, W.S.; Sureau, C.; Beck, J.; Levrero, M.; Kann, M.; Knolle, P.A.; Benkirane, M.; Durantel, D.; et al. Towards an HBV cure: State-of-the-art and unresolved questions—Report of the anrs workshop on HBV cure. Gut 2015, 64, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.; Glebe, D.; Kann, M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J. Virol. 2006, 80, 5465–5473. [Google Scholar] [CrossRef] [PubMed]
- Kann, M.; Schmitz, A.; Rabe, B. Intracellular transport of hepatitis B virus. World J. Gastroenterol. 2007, 13, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xu, C.; Zhou, T.; Block, T.M.; Guo, J.T. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS ONE 2012, 7, e43270. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Nassal, M. A role for the host DNA damage response in hepatitis B virus cccDNA formation-and beyond? Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [PubMed]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA—Repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Cortes Ledesma, F.; Caldecott, K.W.; Seeger, C.; Hu, J. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [PubMed]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Quasdorff, M.; Protzer, U. Control of hepatitis B virus at the level of transcription. J. Viral. Hepat. 2010, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus x protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B virus X protein promotes degradation of Smc5/6 to enhance HBV replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Lebosse, F.; Testoni, B.; Fresquet, J.; Facchetti, F.; Galmozzi, E.; Fournier, M.; Hervieu, V.; Berthillon, P.; Berby, F.; Bordes, I.; et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 2017, 66, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, T.; Huang, X.; Kumar, G.R.; Chen, X.; Zeng, Z.; Zhang, R.; Chen, R.; Li, T.; Zhang, T.; et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J. Hepatol. 2016, 65, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Allweiss, L.; Volz, T.; Dandri, M.; Lutgehetmann, M. Serum HBV pgRNA as a clinical marker for cccDNA activity. J. Hepatol. 2017, 66, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Van Bommel, F.; Bartens, A.; Mysickova, A.; Hofmann, J.; Kruger, D.H.; Berg, T.; Edelmann, A. Serum hepatitis B virus RNA levels as an early predictor of hepatitis B envelope antigen seroconversion during treatment with polymerase inhibitors. Hepatology 2015, 61, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef] [PubMed]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [PubMed]
- Hantz, O.; Parent, R.; Durantel, D.; Gripon, P.; Guguen-Guillouzo, C.; Zoulim, F. Persistence of the hepatitis B virus covalently closed circular DNA in heparg human hepatocyte-like cells. J. Gen. Virol. 2009, 90, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kock, J.; Rosler, C.; Zhang, J.J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef] [PubMed]
- Laras, A.; Koskinas, J.; Dimou, E.; Kostamena, A.; Hadziyannis, S.J. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 2006, 44, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Lutgehetmann, M.; Wachtler, P.; Jacob, A.; Quaas, A.; Murray, J.M.; Dandri, M.; Petersen, J. Impaired intrahepatic hepatitis B virus productivity contributes to low viremia in most HBeAg-negative patients. Gastroenterology 2007, 133, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E., 4th; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Volz, T.; Lutgehetmann, M.; Giersch, K.; Bornscheuer, T.; Lohse, A.W.; Petersen, J.; Ma, H.; Klumpp, K.; Fletcher, S.P.; et al. Immune cell responses are not required to induce substantial hepatitis B virus antigen decline during pegylated interferon-α administration. J. Hepatol. 2014, 60, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Lutgehetmann, M.; Bornscheuer, T.; Volz, T.; Allweiss, L.; Bockmann, J.H.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Dandri, M. Hepatitis B virus limits response of human hepatocytes to interferon-α in chimeric mice. Gastroenterology 2011, 140, 2074–2083.e2. [Google Scholar] [CrossRef] [PubMed]
- Lutgehetmann, M.; Mancke, L.V.; Volz, T.; Helbig, M.; Allweiss, L.; Bornscheuer, T.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Urban, S.; et al. Humanized chimeric uPAa mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012, 55, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccDNA—The holy grail to hepatitis B cure. J. Hepatol. 2016, 64, S41–S48. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Will, H.; Petersen, J. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology 2000, 32, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Wursthorn, K.; Buggisch, P.; Lutgehetmann, M.; Zollner, B.; Petersen, J. Temporary HBV resolution in an HIV-coinfected patient during HBV-directed combination therapy followed by relapse of HBV. Antivir. Ther. 2006, 11, 647–652. [Google Scholar] [PubMed]
- Lutgehetmann, M.; Volzt, T.; Quaas, A.; Zankel, M.; Fischer, C.; Dandri, M.; Petersen, J. Sequential combination therapy leads to biochemical and histological improvement despite low ongoing intrahepatic hepatitis B virus replication. Antivir. Ther. 2008, 13, 57–66. [Google Scholar] [PubMed]
- Bowden, S.; Locarnini, S.; Chang, T.T.; Chao, Y.C.; Han, K.H.; Gish, R.G.; de Man, R.A.; Yu, M.; Llamoso, C.; Tang, H. Covalently closed-circular hepatitis B virus DNA reduction with entecavir or lamivudine. World J. Gastroenterol. 2015, 21, 4644–4651. [Google Scholar] [PubMed]
- Sung, J.J.; Wong, M.L.; Bowden, S.; Liew, C.T.; Hui, A.Y.; Wong, V.W.; Leung, N.W.; Locarnini, S.; Chan, H.L. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology 2005, 128, 1890–1897. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.K.; Yuen, M.F.; Ngai, V.W.; Fung, J.; Lai, C.L. One-year entecavir or lamivudine therapy results in reduction of hepatitis B virus intrahepatic covalently closed circular DNA levels. Antivir. Ther. 2006, 11, 909–916. [Google Scholar] [PubMed]
- Zheng, Q.; Zhu, Y.Y.; Chen, J.; Liu, Y.R.; You, J.; Dong, J.; Zeng, D.W.; Gao, L.Y.; Chen, L.H.; Jiang, J.J. Decline in intrahepatic cccDNA and increase in immune cell reactivity after 12 weeks of antiviral treatment were associated with HBeAg loss. J. Viral. Hepat. 2014, 21, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Lacombe, K.; Lavocat, F.; Maylin, S.; Miailhes, P.; Lascoux-Combe, C.; Delaugerre, C.; Girard, P.M.; Zoulim, F. Decay of ccc-DNA marks persistence of intrahepatic viral DNA synthesis under tenofovir in HIV-HBV co-infected patients. J. Hepatol. 2016, 65, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Sevastianos, V.; Rapti, I.; Vassilopoulos, D.; Hadziyannis, E. Sustained responses and loss of hbsag in HBeAg-negative patients with chronic hepatitis B who stop long-term treatment with adefovir. Gastroenterology 2012, 143, 629–636.e1. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L.; Liaw, Y.F.; Hadziyannis, S.J. Systematic review: Cessation of long-term nucleos(t)ide analogue therapy in patients with hepatitis B e antigen-negative chronic hepatitis B. Aliment. Pharmacol. Ther. 2015, 42, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V. Cytotoxic T cells and viral hepatitis. J. Clin. Investig. 1997, 99, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999, 284, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Litwin, S.; Xu, C.; Jilbert, A.R. Hepatocyte turnover in transient and chronic hepadnavirus infections. J. Viral. Hepat. 2007, 14 (Suppl. 1), 22–28. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hosel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-γ and tumor necrosis factor-α produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Hosel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, G.A.; Scisciani, C.; Pediconi, N.; Lupacchini, L.; Alfalate, D.; Guerrieri, F.; Calvo, L.; Salerno, D.; Di Cocco, S.; Levrero, M.; et al. IL6 inhibits HBV transcription by targeting the epigenetic control of the nuclear cccDNA minichromosome. PLoS ONE 2015, 10, e0142599. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.J.; Block, T.M.; McMahon, B.J.; Ghany, M.G.; Urban, S.; Guo, J.T.; Locarnini, S.; Zoulim, F.; Chang, K.M.; Lok, A.S. Present and future therapies of hepatitis B: From discovery to cure. Hepatology 2015, 62, 1893–1908. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Guidotti, L.G.; Chisari, F.V. Intrahepatic induction of α/β interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J. Virol. 2000, 74, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Eustaquio, A.; Whitten-Bauer, C.; Boyd, B.; Chisari, F.V. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc. Natl. Acad. Sci. USA 2005, 102, 9913–9917. [Google Scholar] [CrossRef] [PubMed]
- Uprichard, S.L.; Wieland, S.F.; Althage, A.; Chisari, F.V. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc. Natl. Acad. Sci. USA 2003, 100, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Guo, H.; Pan, X.B.; Mao, R.; Yu, W.; Xu, X.; Wei, L.; Chang, J.; Block, T.M.; Guo, J.T. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J. Virol. 2010, 84, 9332–9340. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 2001, 19, 65–91. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Gut 2012, 61, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.R.; Walters, K.A.; Wong, W.W.; Wilson, J.S.; Madej, D.; Jewell, L.D.; Tyrrell, D.L. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J. Virol. 2002, 76, 6356–6363. [Google Scholar] [CrossRef] [PubMed]
- Reaiche-Miller, G.Y.; Thorpe, M.; Low, H.C.; Qiao, Q.; Scougall, C.A.; Mason, W.S.; Litwin, S.; Jilbert, A.R. Duck hepatitis B virus covalently closed circular DNA appears to survive hepatocyte mitosis in the growing liver. Virology 2013, 446, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal expansion of normal-appearing human hepatocytes during chronic hepatitis B virus infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [PubMed]
- Lechardeur, D.; Sohn, K.J.; Haardt, M.; Joshi, P.B.; Monck, M.; Graham, R.W.; Beatty, B.; Squire, J.; O’Brodovich, H.; Lukacs, G.L. Metabolic instability of plasmid DNA in the cytosol: A potential barrier to gene transfer. Gene Ther. 1999, 6, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Le, N.; Denoth-Lippuner, A.; Barral, Y.; Kroschewski, R. Asymmetric partitioning of transfected DNA during mammalian cell division. Proc. Natl. Acad. Sci. USA 2016, 113, 7177–7182. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Volz, T.; Giersch, K.; Kah, J.; Raffa, G.; Petersen, J.; Lohse, A.W.; Beninati, C.; Pollicino, T.; Urban, S.; et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.K.; Li, C.C.; Chen, H.J.; Chang, J.L.; Jeng, K.S.; Chou, C.K.; Hsu, M.T.; Tsai, T.F. Blocking of g1/s transition and cell death in the regenerating liver of hepatitis B virus X protein transgenic mice. Biochem. Biophys. Res. Commun. 2006, 340, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Quetier, I.; Brezillon, N.; Duriez, M.; Massinet, H.; Giang, E.; Ahodantin, J.; Lamant, C.; Brunelle, M.N.; Soussan, P.; Kremsdorf, D. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J. Hepatol. 2013, 59, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; O’Connor, T.; Protzer, U.; Heikenwalder, M. The direct and indirect roles of HBV in liver cancer: Prospective markers for hcc screening and potential therapeutic targets. J. Pathol. 2015, 235, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.; Bertoletti, A. Circumventing failed antiviral immunity in chronic hepatitis B virus infection: Triggering virus-specific or innate-like t cell response? Med. Microbiol. Immunol. 2015, 204, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Honer Zu Siederdissen, C.; Rinker, F.; Maasoumy, B.; Wiegand, S.B.; Filmann, N.; Falk, C.S.; Deterding, K.; Port, K.; Mix, C.; Manns, M.P.; et al. Viral and host responses after stopping long-term nucleos(t)ide analogue therapy in HBeAg-negative chronic hepatitis B. J. Infect. Dis. 2016, 214, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. https://doi.org/10.3390/v9060156
Allweiss L, Dandri M. The Role of cccDNA in HBV Maintenance. Viruses. 2017; 9(6):156. https://doi.org/10.3390/v9060156
Chicago/Turabian StyleAllweiss, Lena, and Maura Dandri. 2017. "The Role of cccDNA in HBV Maintenance" Viruses 9, no. 6: 156. https://doi.org/10.3390/v9060156
APA StyleAllweiss, L., & Dandri, M. (2017). The Role of cccDNA in HBV Maintenance. Viruses, 9(6), 156. https://doi.org/10.3390/v9060156