
Modulation of Human β-Defensin-1 Production by Viruses



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Human β-Defensin-1
3. hBD-1 and hBD-2 Induction by Lipopolysaccharide
4. hBD-1 Induction by Viruses In Vitro
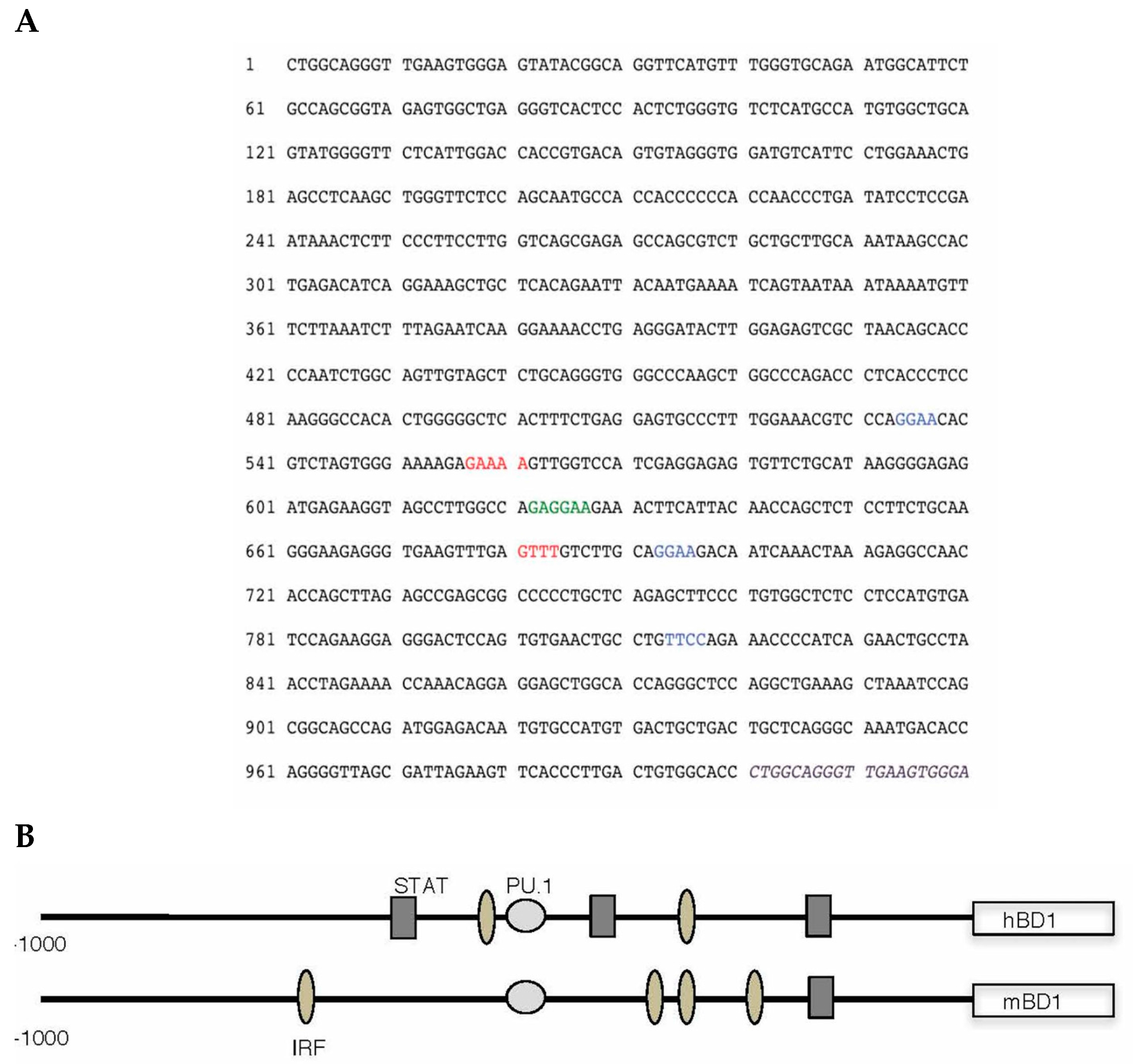
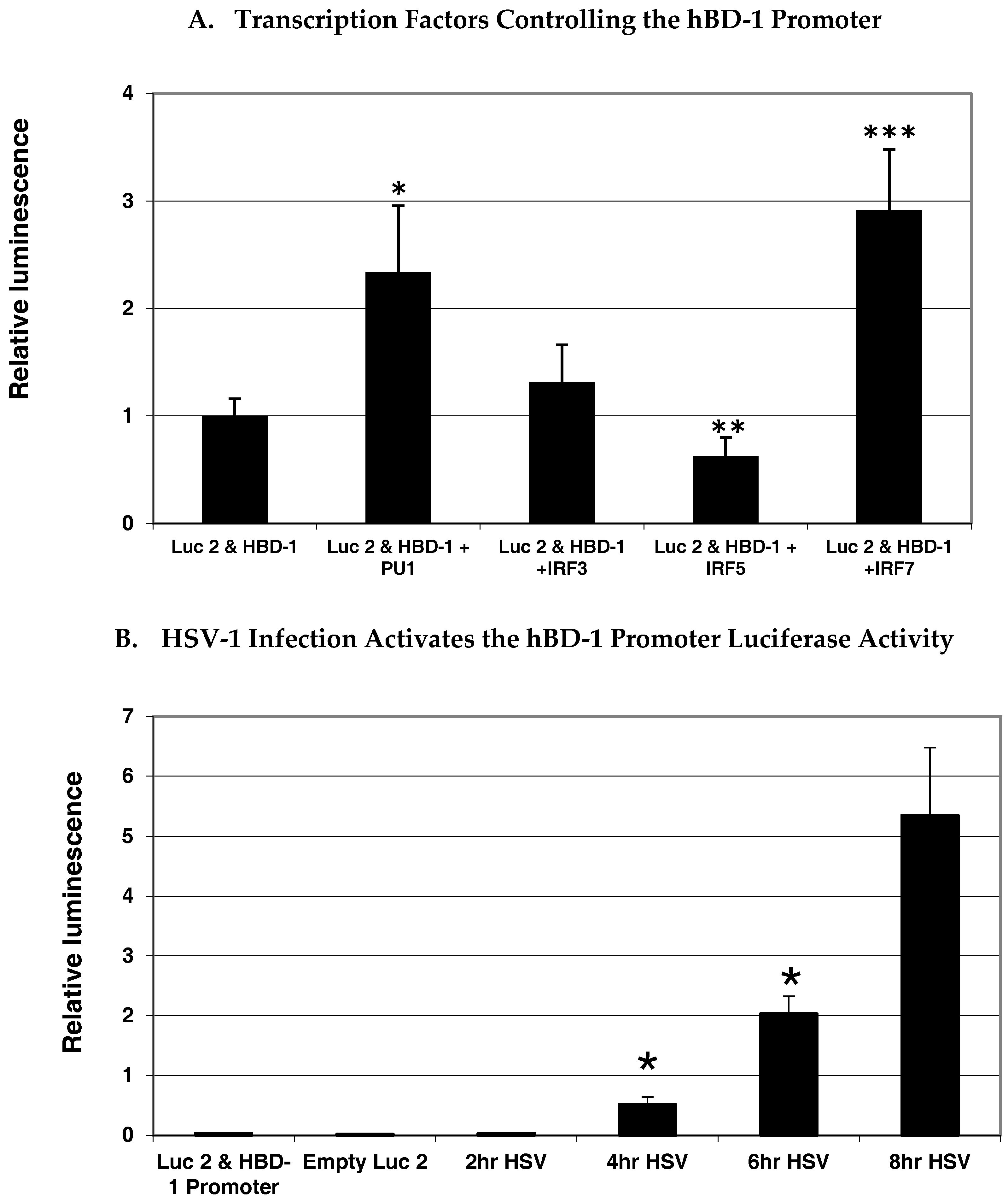
5. Transcriptional Control of hBD-1
6. In Vivo Induction of hBD-1 by Virus in Humans
7. In Vivo Induction of hBD-1 in Animal Models of Viral Infection
8. Viral Innate Immune Escape
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lehrer, R.I.; Lu, W. alpha-Defensins in human innate immunity. Immunol. Rev. 2012, 245, 84–112. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Wiens, M.E.; Holly, M.K.; Smith, J.D. Defensins at the Mucosal Surface: Latest Insights into Defensin-Virus Interactions. J. Virol. 2016, 90, 5216–5218. [Google Scholar] [CrossRef] [PubMed]
- Semple, C.A.; Gautier, P.; Taylor, K.; Dorin, J.R. The changing of the guard: Molecular diversity and rapid evolution of beta-defensins. Mol. Divers. 2006, 10, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gao, B. Evolutionary origin of beta-defensins. Dev. Comp. Immunol. 2013, 39, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bensch, K.W.; Raida, M.; Magert, H.-J.; Schulz-Knappe, P.; Forssmann, W.-G. hBD-1: A novel beta-defensin from human plasma. FEBS Lett. 1995, 368, 331–335. [Google Scholar] [CrossRef]
- Valore, E.V.; Park, C.H.; Quayle, A.J.; Wiles, K.R.; McCray, P.B.; Ganz, T. Human b-defensin-1: An antimicrobial peptide of urogenital tissues. J. Clin. Investig. 1998, 101, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, C.; Heng, H.H.Q.; Ganz, T. The human b-defensin-1 and a-defensins are encoded by adjacent genes: Two peptide families with differing disulfide topology share a common ancestry. Genomics 1997, 43, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 2011, 469, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, I.; Lehrer, R.I. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS Lett. 1996, 396, 319–322. [Google Scholar] [CrossRef]
- McCray, P.B.; Bentley, L. Human airway epithelia express a beta-defensin. Am. J. Respir. Cell. Mol. Biol. 1997, 16, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.-M. A peptide antibiotic from human skin. Nature 1997, 387, 861. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human b-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef]
- Mehlotra, R.K.; Zimmerman, P.A.; Weinberg, A.; Jurevic, R.J. Variation in human beta-defensin genes: New insights from a multi-population study. Int. J. Immunogenet. 2013, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Jones, D.E.; Bevins, C.L. Airway epithelial cells are the site of expression of a mammalian antimicrobial peptide gene. Proc. Nat. Acad. Sci. USA 1993, 90, 4596–4600. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Kaiser, V.; Rhodes, J.; Russell, J.P.; Bevins, C.L. Transcriptional Regulation of b-defensin Gene Expression in Tracheal Epithelial Cells. Infect. Immun. 2000, 68, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Zasloff, M.; Eck, H.; Brasseur, M.; Maloy, W.L.; Bevins, C.L. Tracheal antimicrobial peptide, a novel cysteine-rich peptide from mammalian tracheal mucosa: peptide isolation and cloning of a cDNA. Proc. Natl. Acad. Sci. USA 1991, 88, 3952–3956. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Siebert, R.; Zhang, Y.; Matthiesen, P.; Christophers, E.; Schlegelberger, B.; Schroder, J.M. Mapping of the gene encoding human beta-defensin-2 (DEFB2) to chromosome region 8p22-p23.1. Genomics 1997, 46, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, R.N.; Mahida, Y.R. Expression and regulation of antimicrobial peptides in the gastrointestinal tract. J. Leukoc. Biol. 2004, 75, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.N.; Diamond, G.; Verghese, M.W.; Randell, S.H. CD14-dependent lipopolysaccharide-induced beta-defensin-2 expression in human tracheobronchial epithelium. J. Biol. Chem. 2000, 275, 29731–29736. [Google Scholar] [CrossRef] [PubMed]
- Duits, L.A.; Ravensbergen, B.; Rademaker, M.; Hiemstra, P.S.; Nibbering, P.H. Expression of beta-defensin 1 and 2 mRNA by human monocytes, macrophages and dendritic cells. Immunology 2002, 106, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.M.; Shu, Q.; Chen, Q.X.; Book, M.; Sahl, H.G.; Hoeft, A.; Stuber, F. Differential expression of alpha- and beta-defensins in human peripheral blood. Eur. J. Clin. Investig. 2003, 33, 82–87. [Google Scholar] [CrossRef]
- Ryan, L.K.; Diamond, G.; Amrute, S.; Feng, Z.; Weinberg, A.; Fitzgerald-Bocarsly, P. Detection of HBD1 peptide in peripheral blood mononuclear cell subpopulations by intracellular flow cytometry. Peptides 2003, 24, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Ryan, L.K.; Dai, J.; Yin, Z.; Megjugorac, N.; Uhlhorn, V.; Yim, S.; Schwartz, K.D.; Abrahams, J.M.; Diamond, G.; Fitzgerald-Bocarsly, P. Modulation of human {beta}-defensin-1 (hBD-1) in plasmacytoid dendritic cells (PDC), monocytes, and epithelial cells by influenza virus, Herpes simplex virus, and Sendai virus and its possible role in innate immunity. J. Leukoc. Biol. 2011, 90, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Ryan, L.K.; Dai, J.; Fitzgerald-Bocarsly, P.; Schwartz, K.; Diamond, G. Induction of human beta-defensin-1 gene expression is dependent on IRF-7 and PU.1. J. Immun. 2013, 190, 571. [Google Scholar]
- Corleis, B.; Lisanti, A.C.; Korner, C.; Schiff, A.E.; Rosenberg, E.S.; Allen, T.M.; Altfeld, M.; Kwon, D.S. Early type I Interferon response induces upregulation of human beta-defensin 1 during acute HIV-1 infection. PLoS ONE 2017, 12, e0173161. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Wei, R.; Sun, C.; Gombart, A.F.; Koeffler, H.P.; Diamond, G.; Christakos, S. C/EBPalpha and the Vitamin D Receptor Cooperate in the Regulation of Cathelicidin in Lung Epithelial Cells. J. Cell Physiol. 2015, 230, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Megjugorac, N.J.; Amrute, S.B.; Fitzgerald-Bocarsly, P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J. Immunol. 2004, 173, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Braida, L.; Boniotto, M.; Pontillo, A.; Tovo, P.A.; Amoroso, A.; Crovella, S. A single-nucleotide polymorphism in the human beta-defensin 1 gene is associated with HIV-1 infection in Italian children. Aids 2004, 18, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Segat, L.; Milanese, M.; Boniotto, M.; Crovella, S.; Bernardon, M.; Costantini, M.; Alberico, S. DEFB-1 genetic polymorphism screening in HIV-1 positive pregnant women and their children. J. Matern. Fetal. Neonatal. Med. 2006, 19, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Segat, L.; Zupin, L.; Moura, R.R.; Coelho, A.V.; Chagas, B.S.; de Freitas, A.C.; Crovella, S. DEFB1 polymorphisms are involved in susceptibility to human papillomavirus infection in Brazilian gynaecological patients. Mem. Inst. Oswaldo Cruz 2014, 109, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Zapata, W.; Rodriguez, B.; Weber, J.; Estrada, H.; Quinones-Mateu, M.E.; Zimermman, P.A.; Lederman, M.M.; Rugeles, M.T. Increased levels of human beta-defensins mRNA in sexually HIV-1 exposed but uninfected individuals. Curr. HIV Res. 2008, 6, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, I.; Hasegawa, K.; Nakata, K.; Yasuda, K.; Tokunaga, K.; Keicho, N. Genetic variants of human beta-defensin-1 and chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2002, 291, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Jurevic, R.J.; Bai, M.; Chadwick, R.B.; White, T.C.; Dale, B.A. Single-Nucleotide Polymorphisms (SNPs) in Human beta-Defensin 1: High-Throughput SNP Assays and Association with Candida Carriage in Type I Diabetics and Nondiabetic Controls. J. Clin. Microbiol. 2003, 41, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schroeder, J.M.; Wang, J.M.; Howard, O.M.; et al. beta-Defensins: Linking Innate and Adaptive Immunity Through Dendritic and T Cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Biragyn, A.; Belyakov, I.M.; Chow, Y.H.; Dimitrov, D.S.; Berzofsky, J.A.; Kwak, L.W. DNA vaccines encoding human immunodeficiency virus-1 glycoprotein 120 fusions with proinflammatory chemoattractants induce systemic and mucosal immune responses. Blood 2002, 100, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Prado Montes de Oca, E. Antimicrobial peptide elicitors: New hope for the post-antibiotic era. Innate Immun. 2013, 19, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.H.; Casanova, V.; Stevens, C.; Barlow, P.G. Antiviral host defence peptides. In Host Defense Peptides and Their Potential as Therapeutic Agents; Epand, R.M., Ed.; Springer: Switzerland, 2016; pp. 57–94. [Google Scholar]
- Wiens, M.E.; Wilson, S.S.; Lucero, C.M.; Smith, J.G. Defensins and viral infection: Dispelling common misconceptions. PLoS Pathog. 2014, 10, e1004186. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef] [PubMed]
- Tolfvenstam, T.; Lindblom, A.; Schreiber, M.J.; Ling, L.; Chow, A.; Ooi, E.E.; Hibberd, M.L. Characterization of early host responses in adults with dengue disease. BMC Infect. Dis. 2011, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Chong, K.T.; Xiang, L.; Wang, X.; Jun, E.L.; Xi, L.F.; Schweinfurth, J.M. High level expression of human epithelial beta-defensins (hBD-1, 2 and 3) in papillomavirus induced lesions. Virol. J. 2006, 3, 75. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dauletbaev, N.; Gropp, R.; Frye, M.; Loitsch, S.; Wagner, T.O.; Bargon, J. Expression of human beta defensin (HBD-1 and HBD-2) mRNA in nasal epithelia of adult cystic fibrosis patients, healthy individuals, and individuals with acute cold. Respiration 2002, 69, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Grubor, B.; Gallup, J.M.; Meyerholz, D.K.; Crouch, E.C.; Evans, R.B.; Brogden, K.A.; Lehmkuhl, H.D.; Ackermann, M.R. Enhanced surfactant protein and defensin mRNA levels and reduced viral replication during parainfluenza virus type 3 pneumonia in neonatal lambs. Clin. Diagn. Lab. Immunol. 2004, 11, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Goldman, M.J.; Wilson, J.M. Mouse b-defensin 1 is a salt-sensitive antimicrobial peptide present in epithelia of the lung and urogenital tract. Infect. Immun. 1998, 66, 1225–1232. [Google Scholar] [PubMed]
- Morrison, G.M.; Davidson, D.J.; Kilanowski, F.M.; Borthwick, D.W.; Crook, K.; Maxwell, A.I.; Govan, J.R.W.; Dorin, J.R. Mouse beta defensin-1 is a functional homolog of human beta defensin-1. Mamm. Genome 1998, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Chong, K.T.; Thangavel, R.R.; Tang, X. Enhanced expression of murine beta-defensins (MBD-1, -2, -3, and -4) in upper and lower airway mucosa of influenza virus infected mice. Virology 2008, 380, 136–143. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryan, L.K.; Diamond, G. Modulation of Human β-Defensin-1 Production by Viruses. Viruses 2017, 9, 153. https://doi.org/10.3390/v9060153
Ryan LK, Diamond G. Modulation of Human β-Defensin-1 Production by Viruses. Viruses. 2017; 9(6):153. https://doi.org/10.3390/v9060153
Chicago/Turabian StyleRyan, Lisa Kathleen, and Gill Diamond. 2017. "Modulation of Human β-Defensin-1 Production by Viruses" Viruses 9, no. 6: 153. https://doi.org/10.3390/v9060153
APA StyleRyan, L. K., & Diamond, G. (2017). Modulation of Human β-Defensin-1 Production by Viruses. Viruses, 9(6), 153. https://doi.org/10.3390/v9060153