
Distinct Contributions of Autophagy Receptors in Measles Virus Replication

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
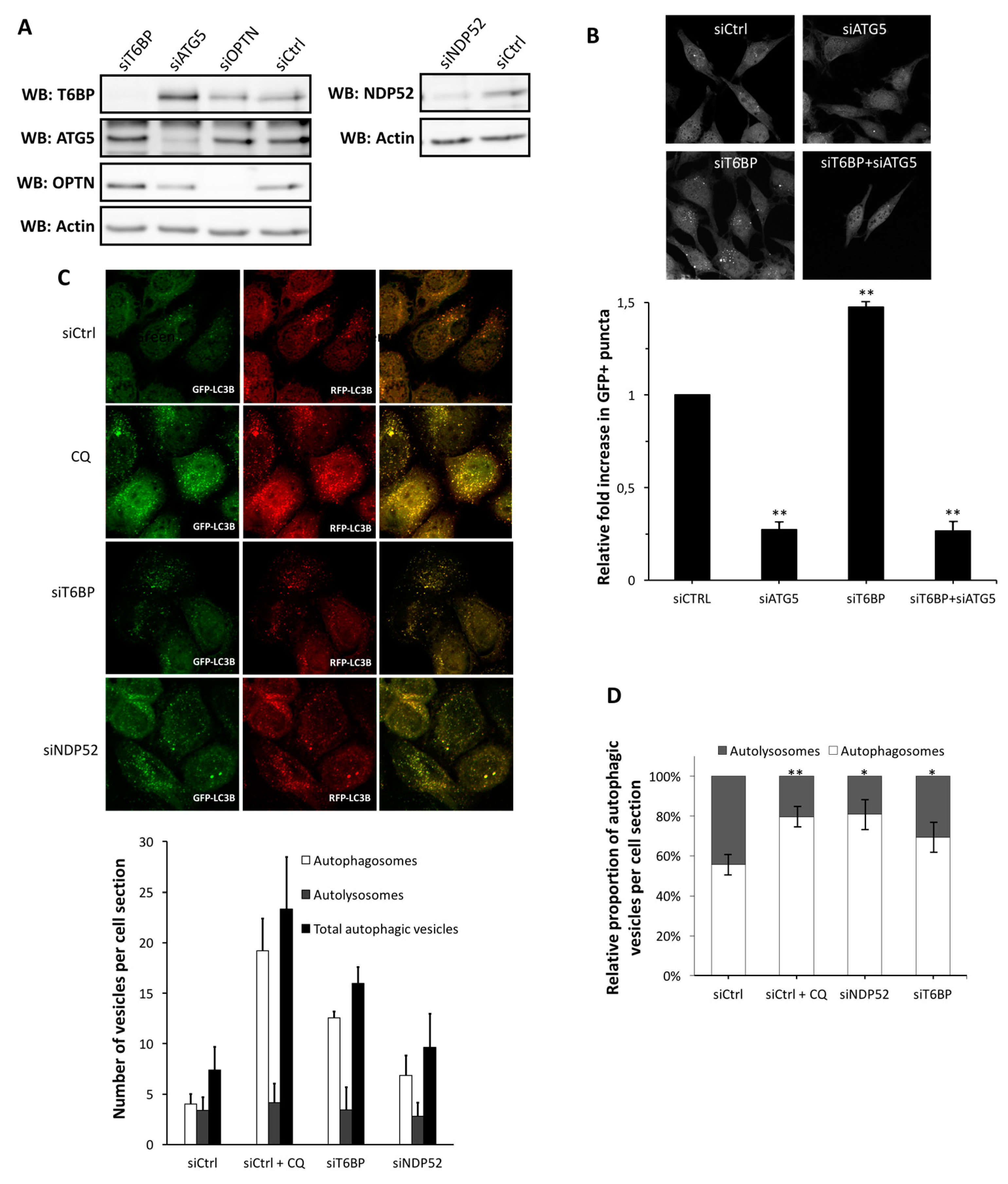
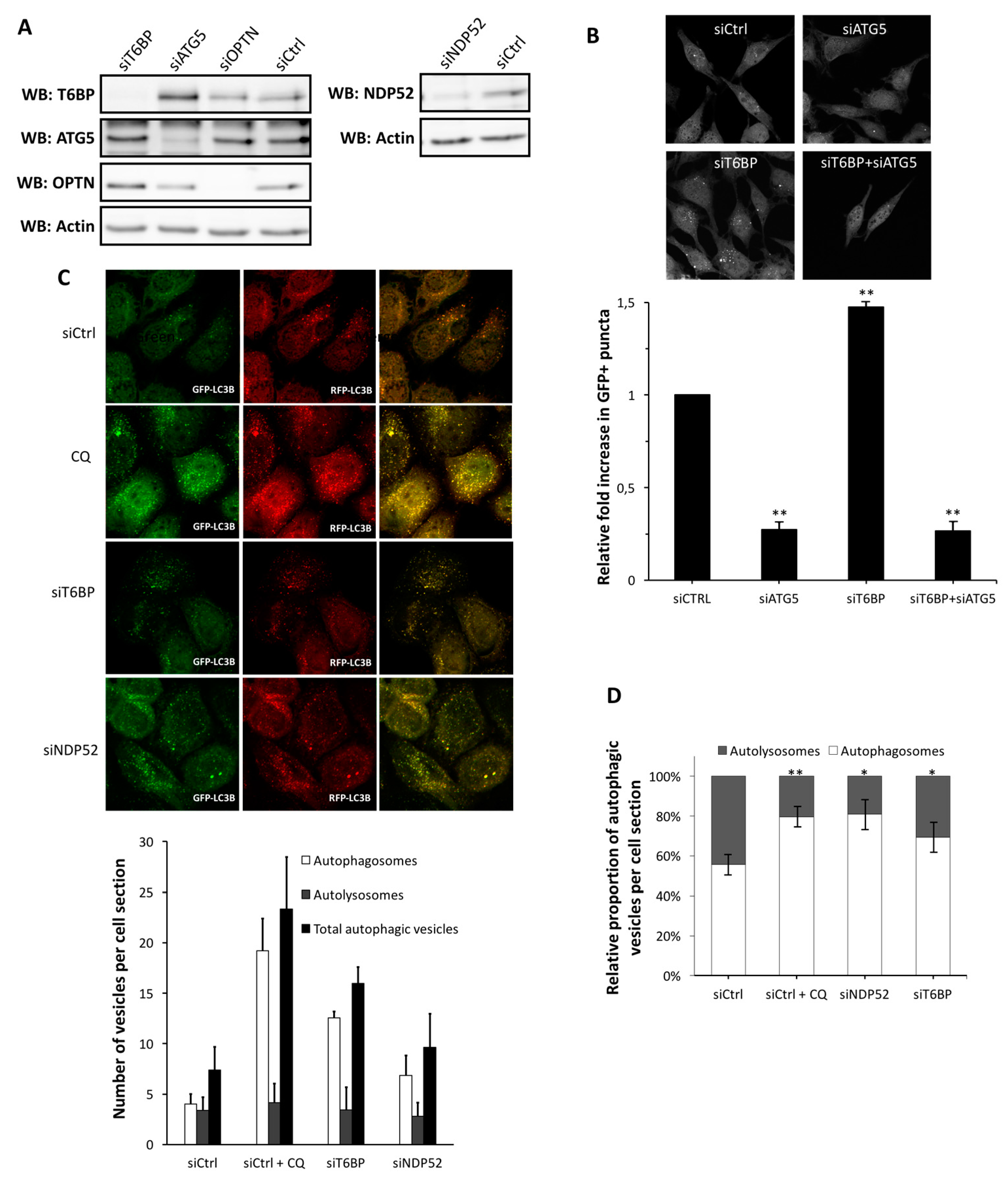
2.1. T6BP Promotes Autophagosome Maturation
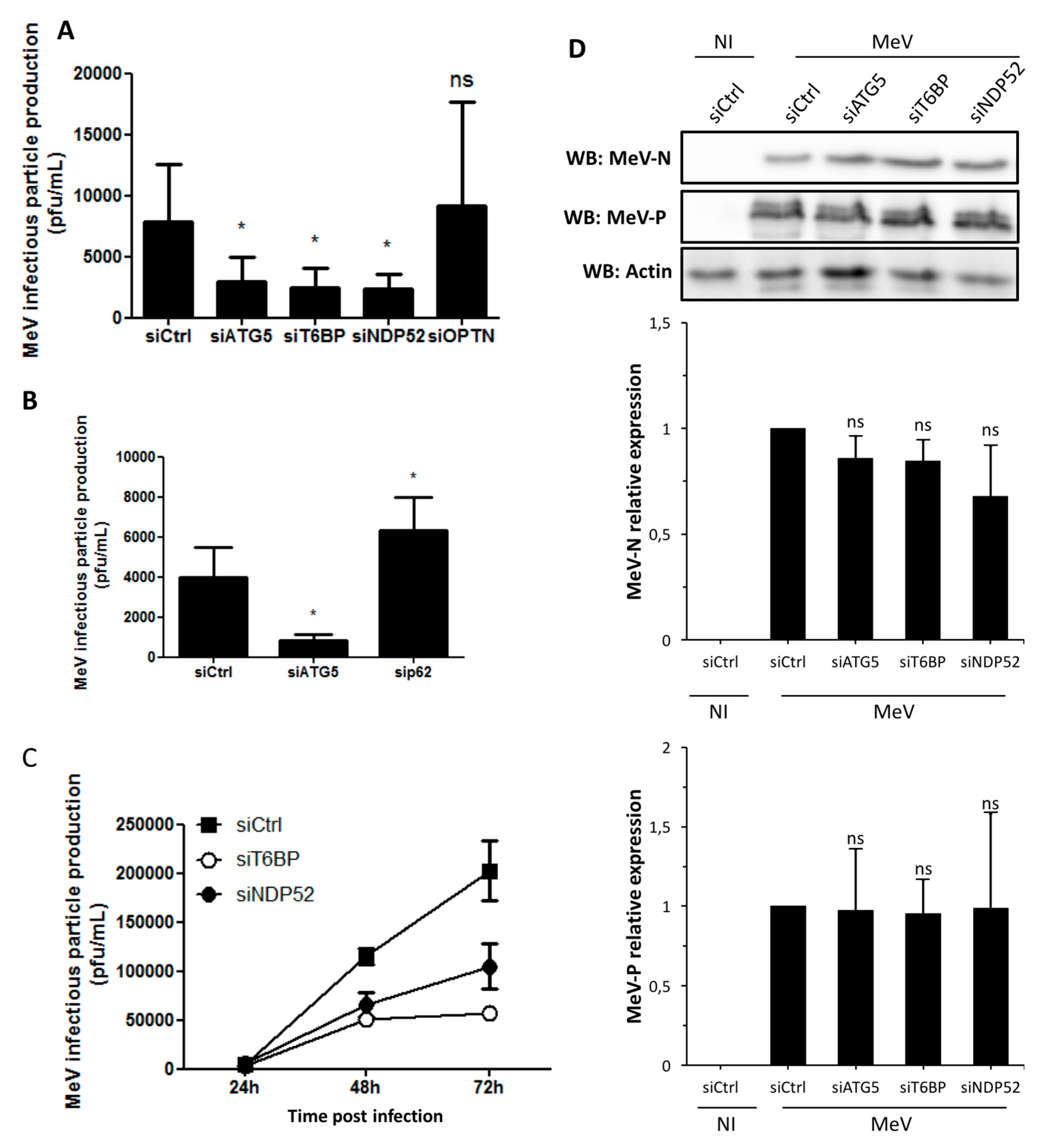
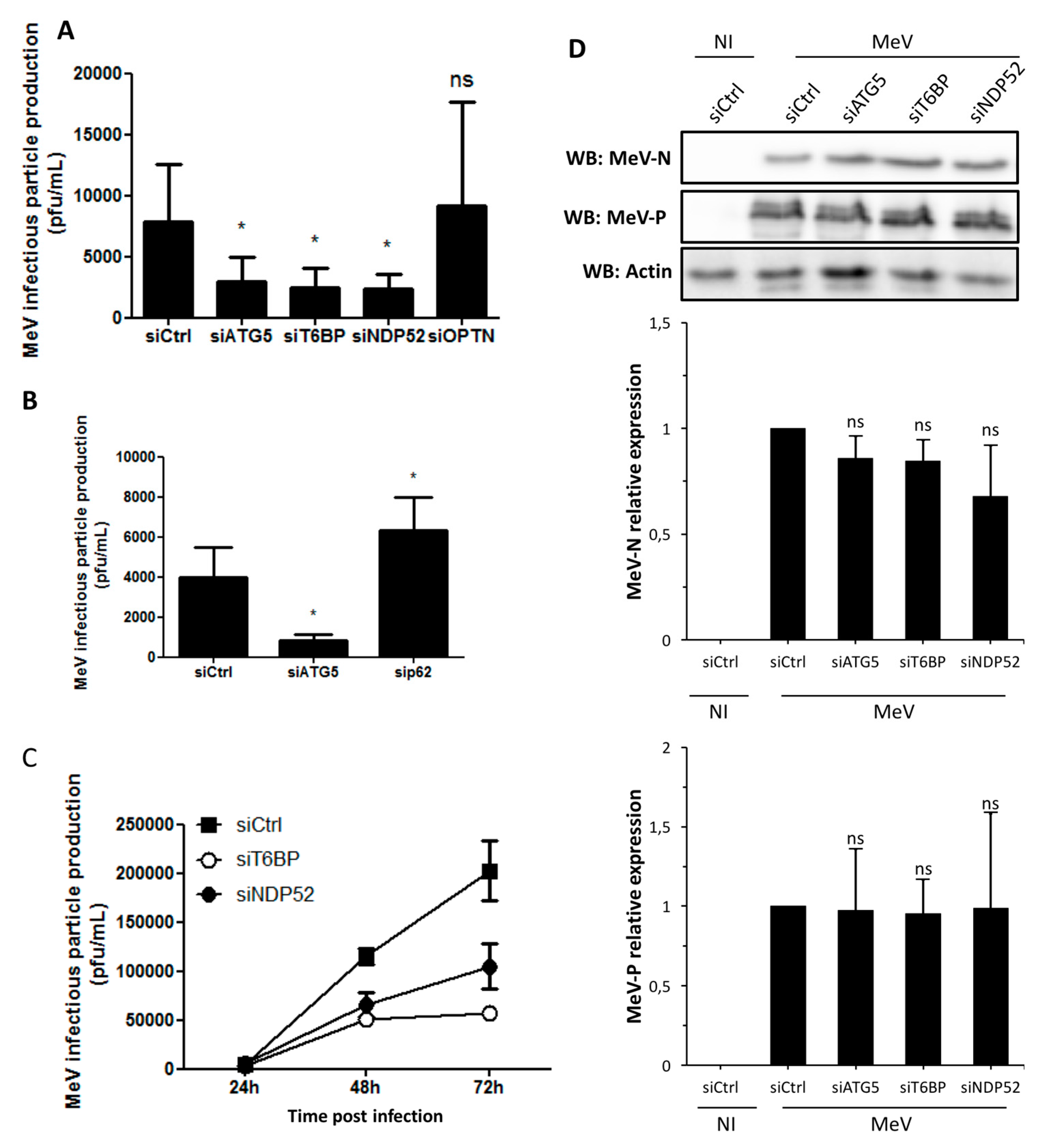
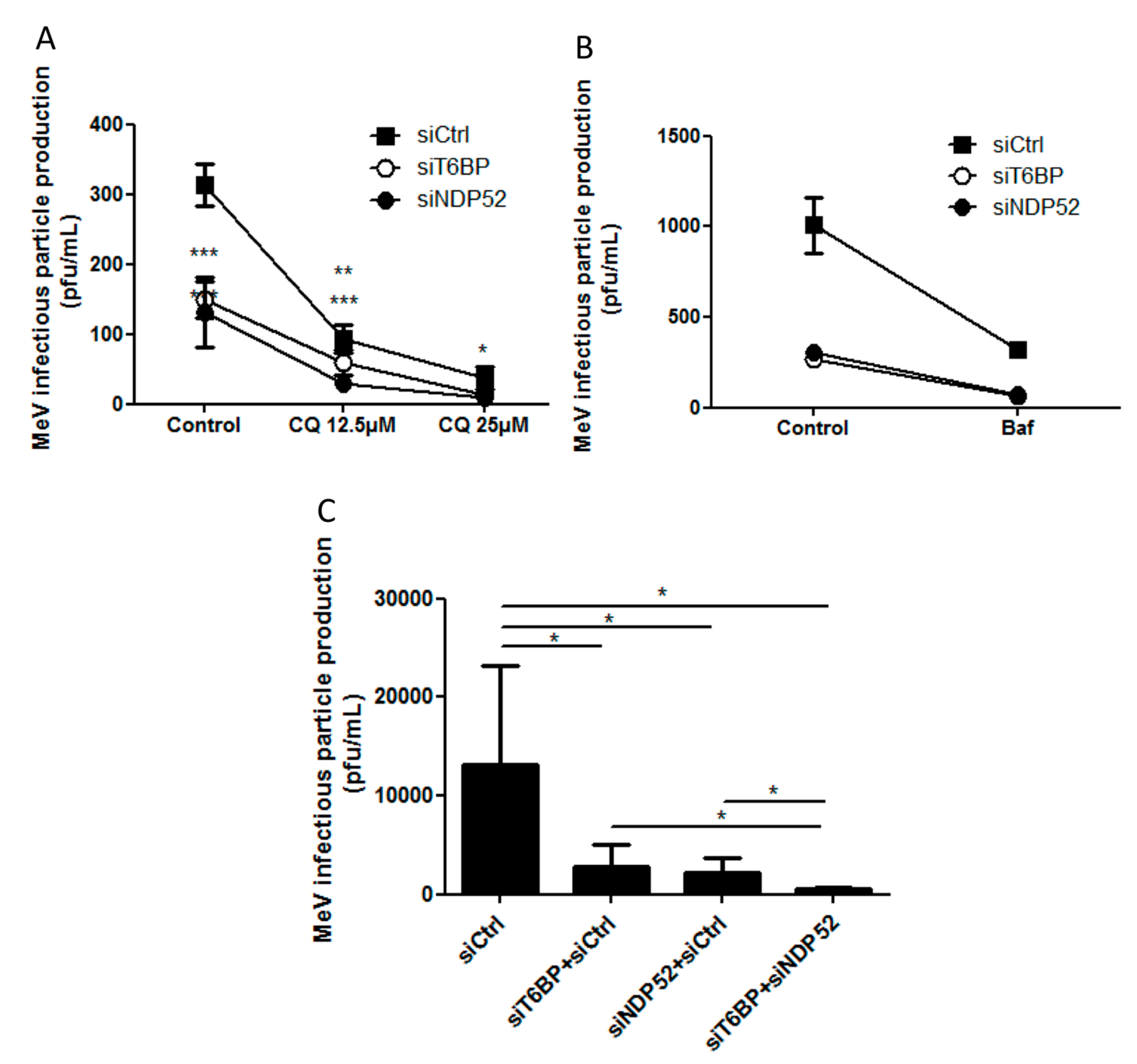
2.2. T6BP and NDP52, but Not OPTN, Are Required for MeV Replication
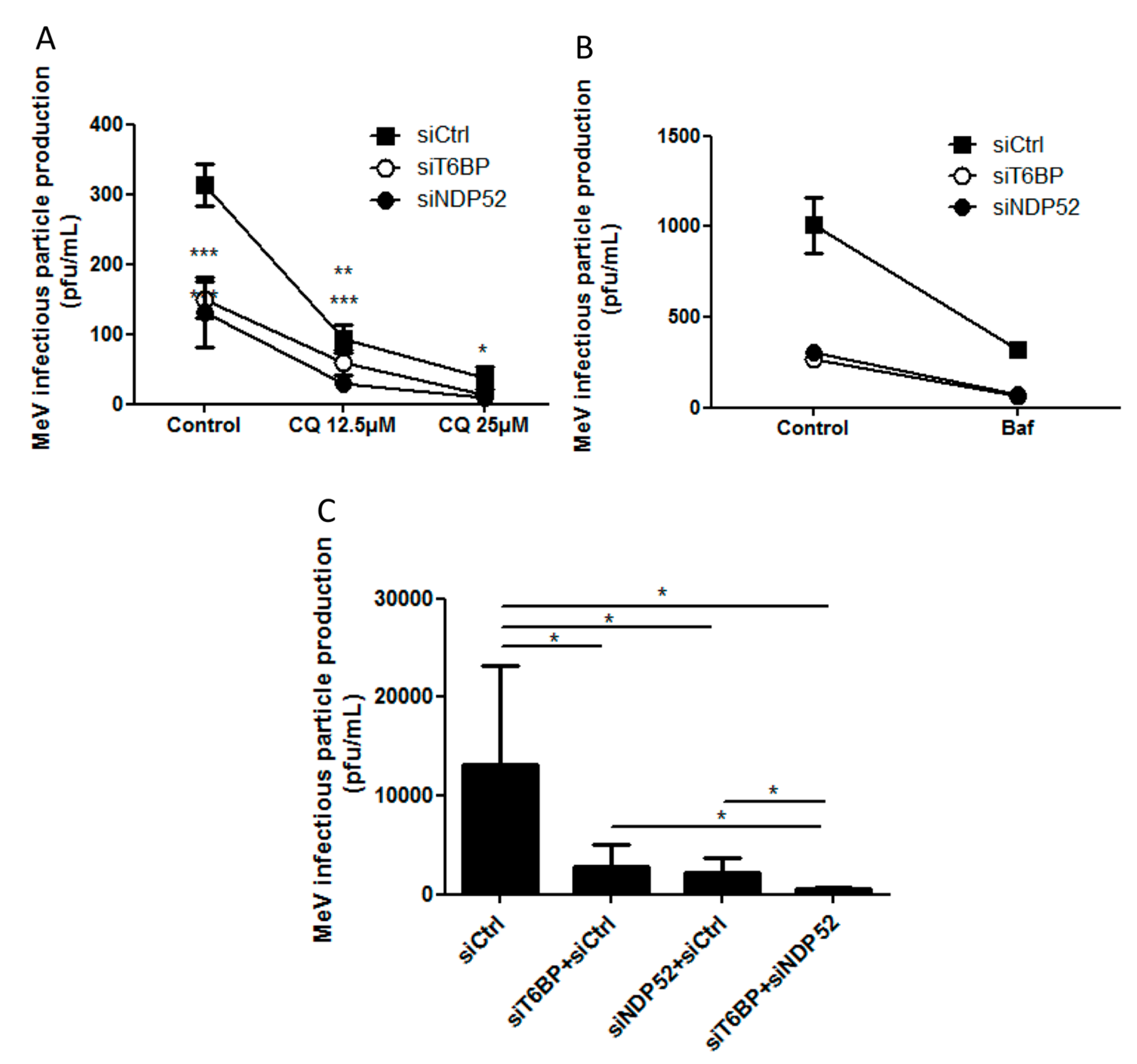
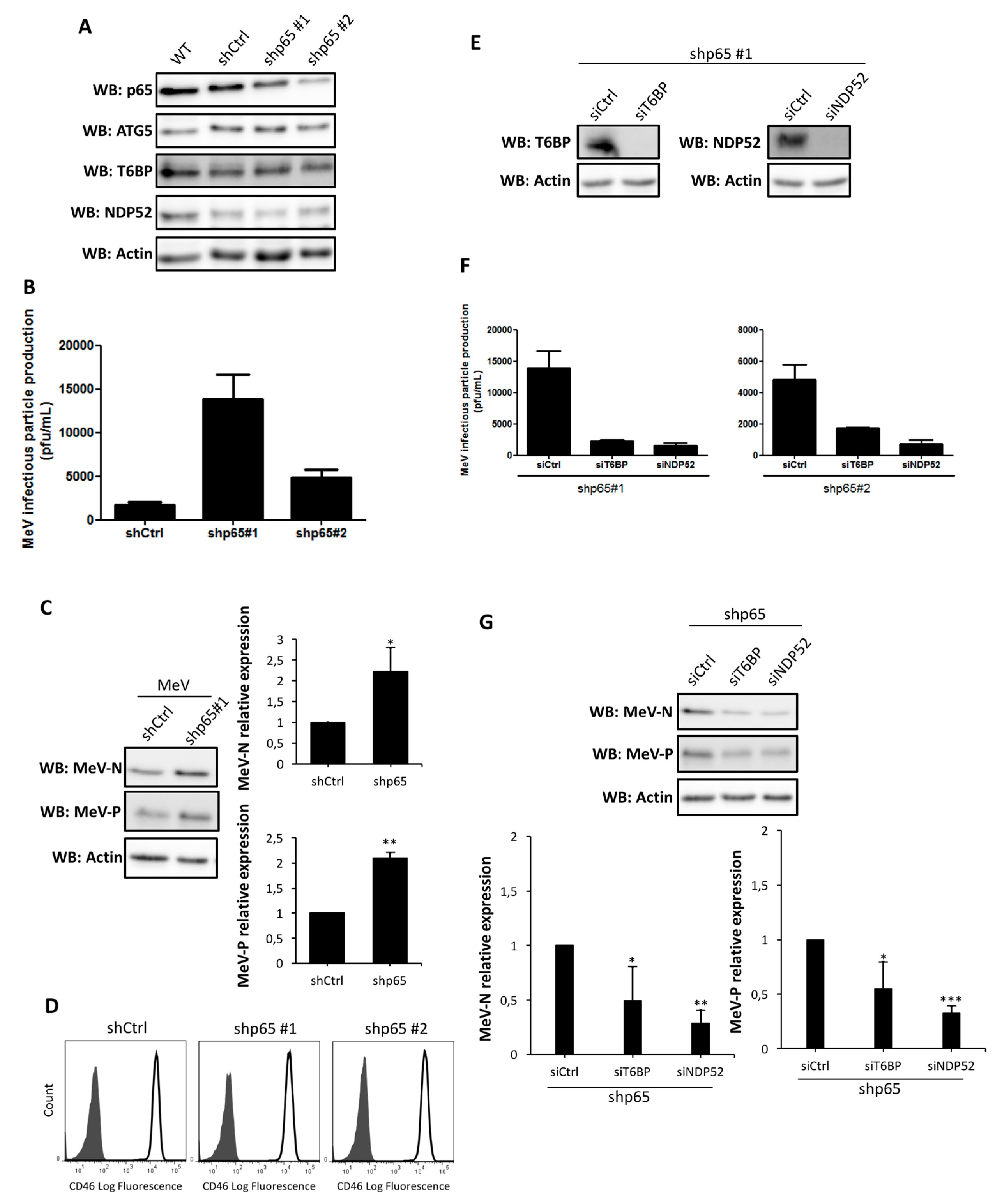
2.3. NF-κB Independent role of T6BP and NDP52 in MeV Replication
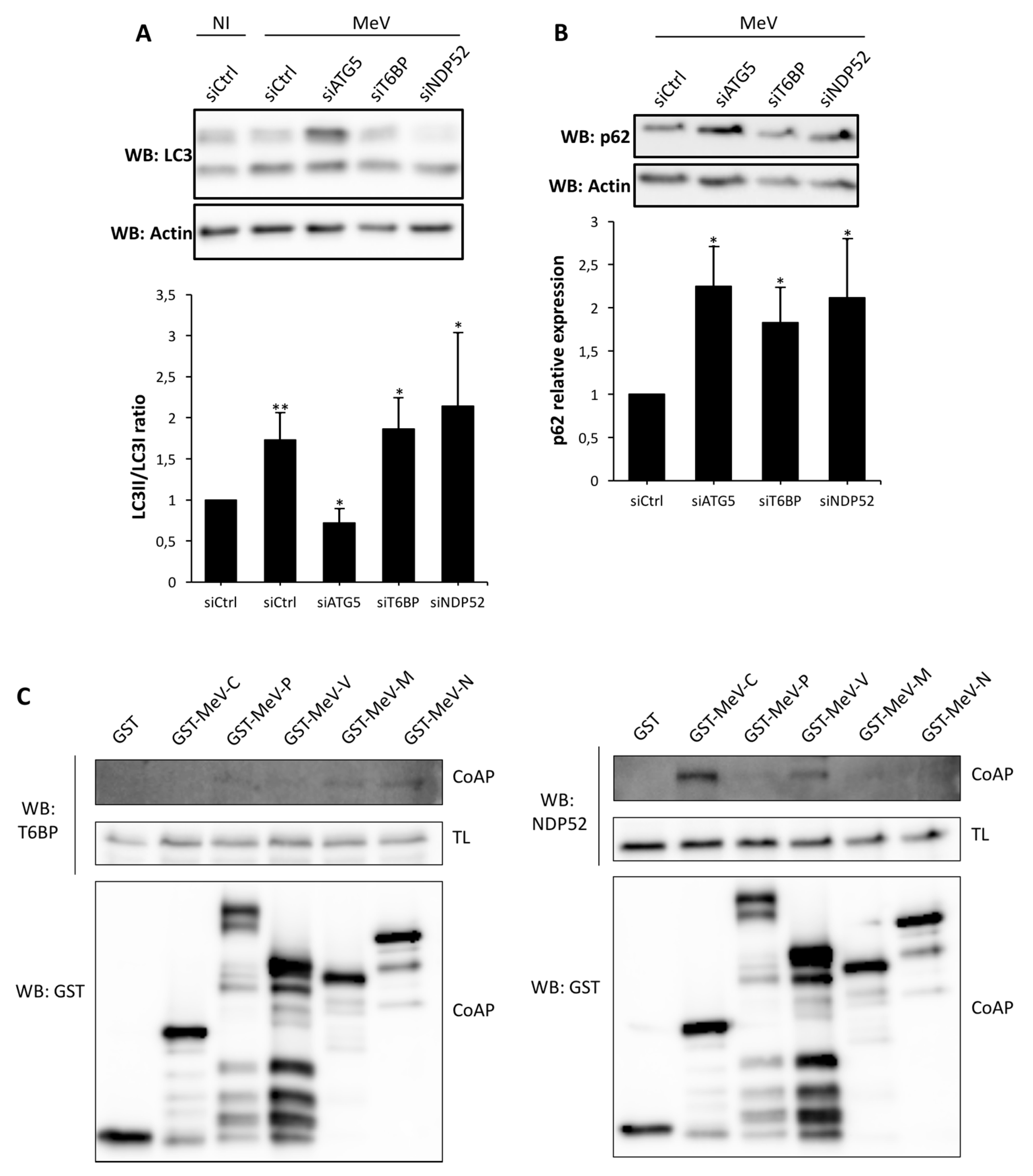
2.4. T6BP and NDP52 Can both Interact with MeV Proteins
2.5. Independent Contribution of T6BP and NDP52 in MeV Replication
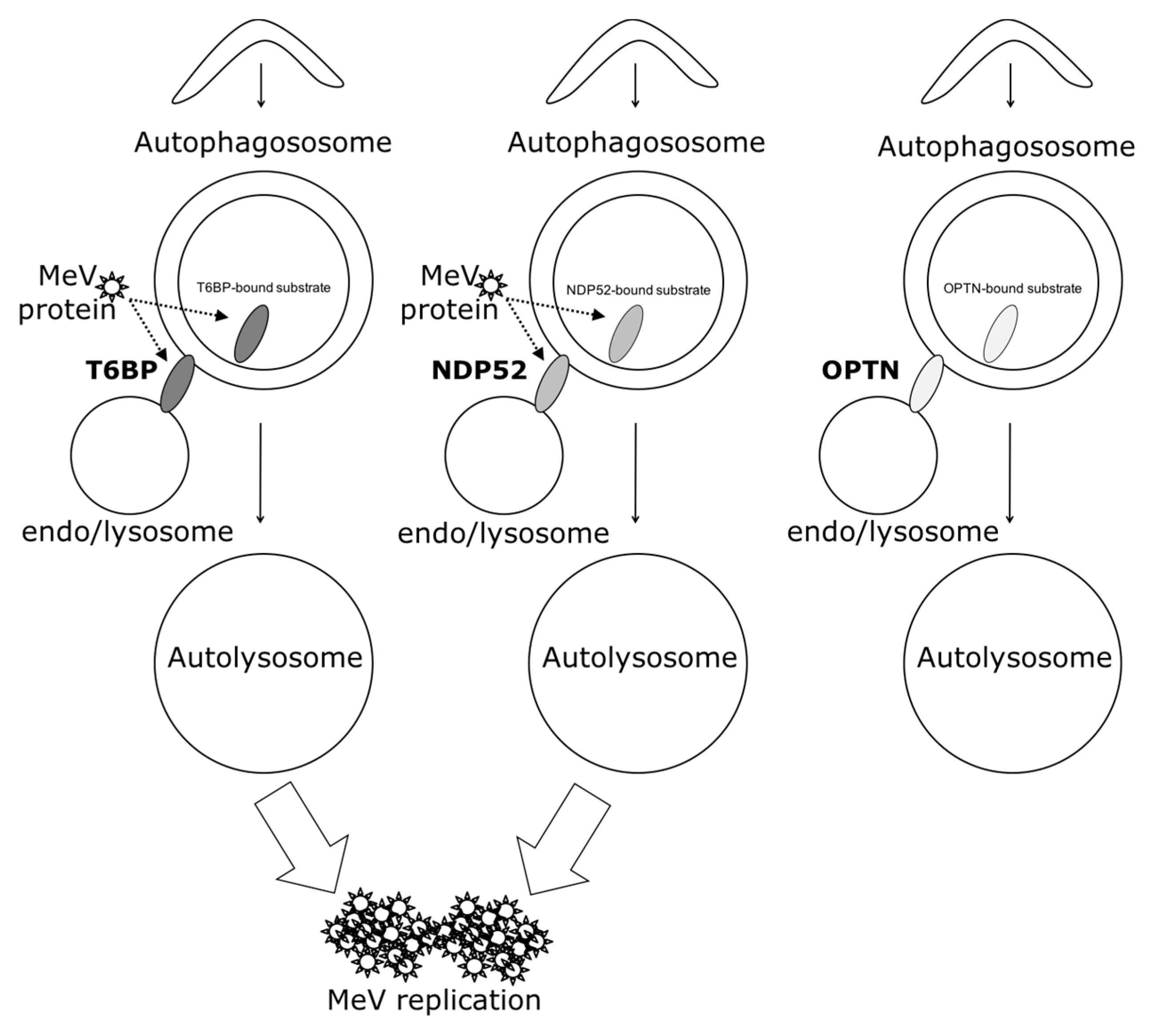
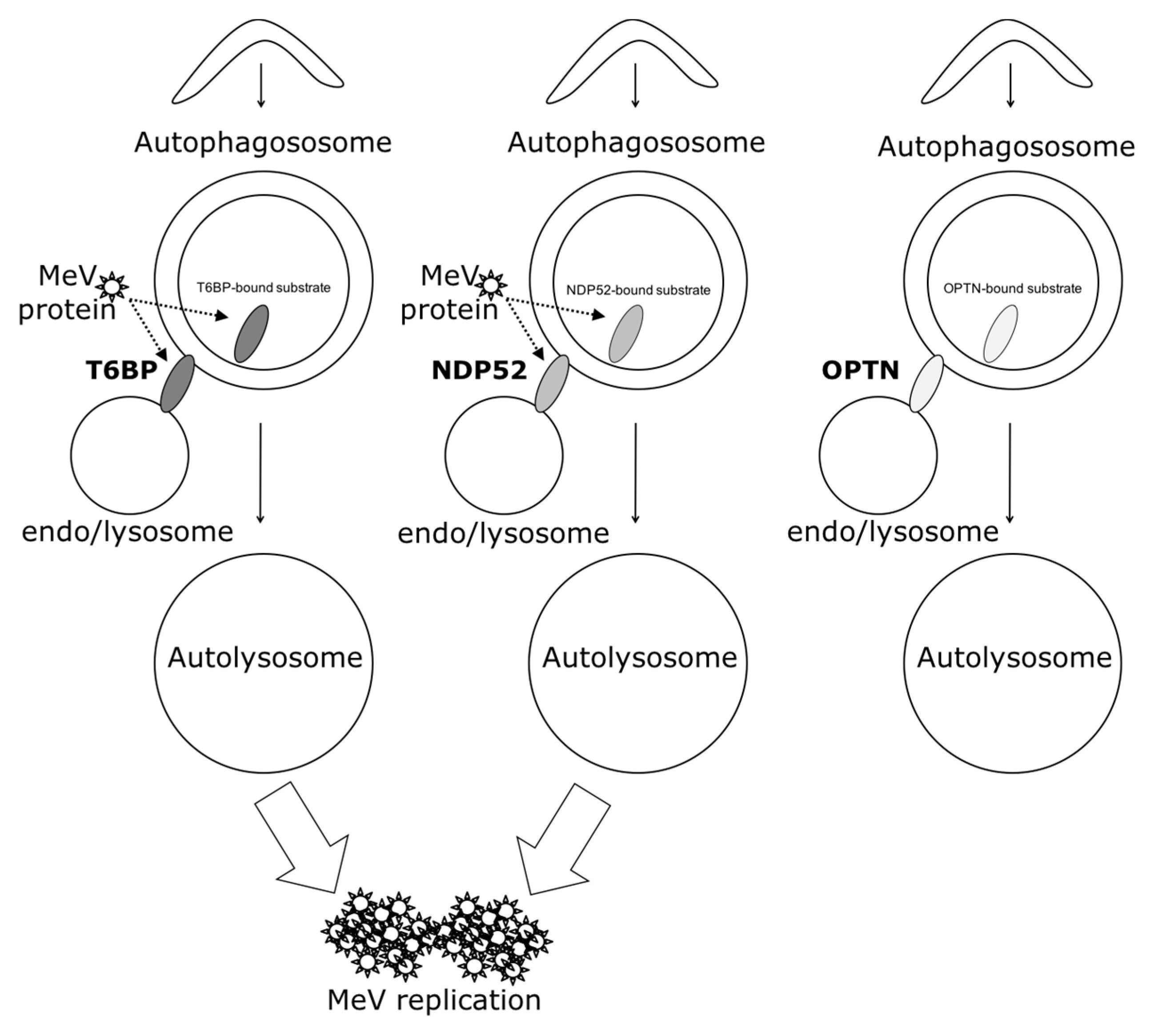
3. Discussion
4. Experimental Procedures
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. siRNA Transfection
4.4. MeV Strains and Titration by Plaque Assay
4.5. Molecular Cloning
4.6. GST Co-Affinity Purification Assays
4.7. Confocal Microscopy
4.8. Immunofluorescence-coupled Flow Cytometry
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Grégoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef] [PubMed]
- Moss, W.J.; Griffin, D.E. Measles. Lancet 2012, 379, 153–164. [Google Scholar] [CrossRef]
- Griffin, D.E. Measles virus-induced suppression of immune responses. Immunol. Rev. 2010, 236, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Moss, W.J.; Griffin, D.E. Global measles elimination. Nat. Rev. Microbiol. 2006, 4, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, I.P.; Rabourdin-Combe, C.; Faure, M. Autophagy and RNA virus interactomes reveal IRGM as a common target. Autophagy 2012, 8, 1136–1137. [Google Scholar] [CrossRef] [PubMed]
- Petkova, D.S.; Viret, C.; Faure, M. IRGM in autophagy and viral infections. Front Immunol. 2013, 17, 426. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Meiffren, G.; Grégoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Meiffren, G.; Joubert, P.E.; Grégoire, I.P.; Codogno, P.; Rabourdin-Combe, C.; Faure, M. Pathogen recognition by the cell surface receptor CD46 induces autophagy. Autophagy 2010, 6, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Naniche, D.; Varior-Krishnan, G.; Cervoni, F.; Wild, T.F.; Rossi, B.; Rabourdin-Combe, C.; Gerlier, D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993, 67, 6025–6032. [Google Scholar] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Grégoire, I.P.; Azocar, O.; Petkova, D.S.; Baguet, J.; Viret, C.; Faure, M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 2015, 17, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Viret, C.; Faure, M. Dual function of CALCOCO2/NDP52 during xenophagy. Autophagy 2015, 11, 965–966. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Manna, P.T.; Allen, M.; Bycroft, M.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 2015, 11, e1005174. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovitch, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Pujari, R.; Harhaj, N.S.; Abbott, D.W.; Harhaj, E.W. The kinase IKKalpha inhibits activation of the transcription factor NF-kappaB by phosphorylating the regulatory molecule TAX1BP1. Nat. Immunol. 2011, 12, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Inomata, M.; Niida, S.; Shibata, K.; Into, T. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell Mol. Life Sci. 2012, 69, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, K.M.; Pfaller, C.K.; Conzelmann, K.K. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 2011, 85, 3162–3171. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Abou-Samra, M.; Richet, E.; Guyot, B.; Arrigo, A.P.; Kretz-Remy, C. NF-kappaB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex. J. Cell Sci. 2012, 125, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Nivon, M.; Richet, E.; Cogogno, P.; Arrigo, A.P.; Kretz-Remy, C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 2009, 5, 766–783. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Rabourdin-Combe, C. Innate immunity modulation in virus entry. Curr. Opin. Virol. 2011, 1, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; McPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prévost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Faure, M. Autophagy in antiviral innate immunity. Cell Microbiol. 2013, 15, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Lafont, F. Pathogen-induced autophagy signaling in innate immunity. J. Innate Immun. 2013, 5, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; Youle, R.J. Self and Nonself: How Autophagy Targets Mitochondria and Bacteria. Cell Host Microbe 2014, 15, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Morriswood, B.; Ryzhakov, G.; Puri, C.; Arden, S.D.; Roberts, R.; Dendrou, C.; Kendrick-Jones, J.; Buss, F. T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. J. Cell Sci. 2007, 120, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.C.; Scholefield, C.L.; Kemp, A.J.; Newman, M.; McIver, E.G.; Kamal, A.; Wilkinson, S. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-kappaB signalling. PLoS ONE 2012, 7, e50672. [Google Scholar] [CrossRef] [PubMed]
- Kachaner, D.; Genic, P.; Laplantine, E.; Weil, R. Toward an integrative view of Optineurin functions. Cell Cycle 2012, 11, 2808–2818. [Google Scholar] [CrossRef] [PubMed]
- Journo, C.; Filipe, J.; About, F.; Chevalier, S.A.; Afonso, P.V.; Brady, J.N.; Flynn, D.; Tangy, F.; Israël, A.; Vidalain, P.O.; et al. NRP/Optineurin Cooperates with TAX1BP1 to potentiate the activation of NF-kappaB by human T-lymphotropic virus type 1 tax protein. PLoS Pathog. 2009, 5, e1000521. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkova, D.S.; Verlhac, P.; Rozières, A.; Baguet, J.; Claviere, M.; Kretz-Remy, C.; Mahieux, R.; Viret, C.; Faure, M. Distinct Contributions of Autophagy Receptors in Measles Virus Replication. Viruses 2017, 9, 123. https://doi.org/10.3390/v9050123
Petkova DS, Verlhac P, Rozières A, Baguet J, Claviere M, Kretz-Remy C, Mahieux R, Viret C, Faure M. Distinct Contributions of Autophagy Receptors in Measles Virus Replication. Viruses. 2017; 9(5):123. https://doi.org/10.3390/v9050123
Chicago/Turabian StylePetkova, Denitsa S., Pauline Verlhac, Aurore Rozières, Joël Baguet, Mathieu Claviere, Carole Kretz-Remy, Renaud Mahieux, Christophe Viret, and Mathias Faure. 2017. "Distinct Contributions of Autophagy Receptors in Measles Virus Replication" Viruses 9, no. 5: 123. https://doi.org/10.3390/v9050123
APA StylePetkova, D. S., Verlhac, P., Rozières, A., Baguet, J., Claviere, M., Kretz-Remy, C., Mahieux, R., Viret, C., & Faure, M. (2017). Distinct Contributions of Autophagy Receptors in Measles Virus Replication. Viruses, 9(5), 123. https://doi.org/10.3390/v9050123