
A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea



Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Purification of Phages
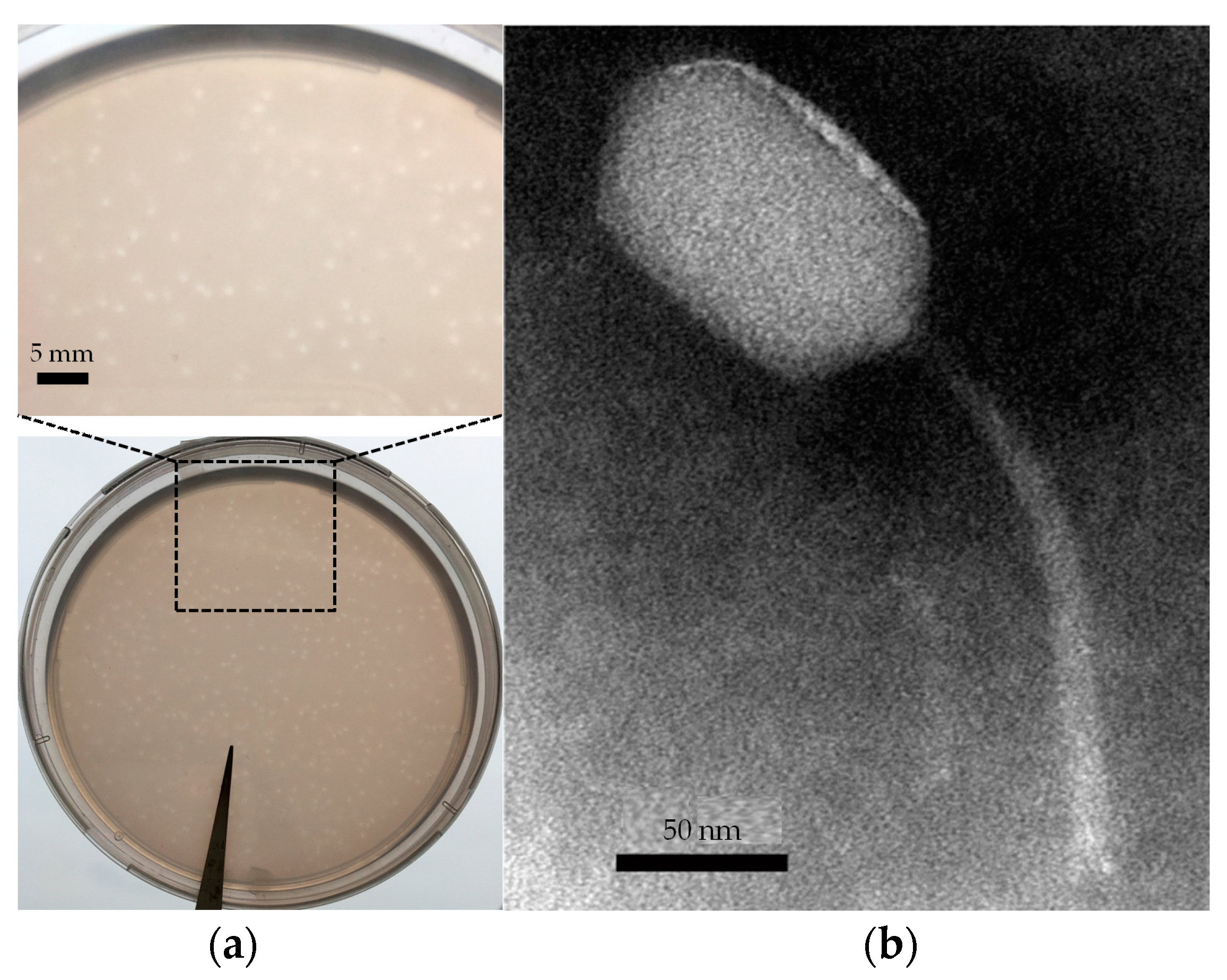
2.2. Transmission Electron Microscopy (TEM)
2.3. Host Range
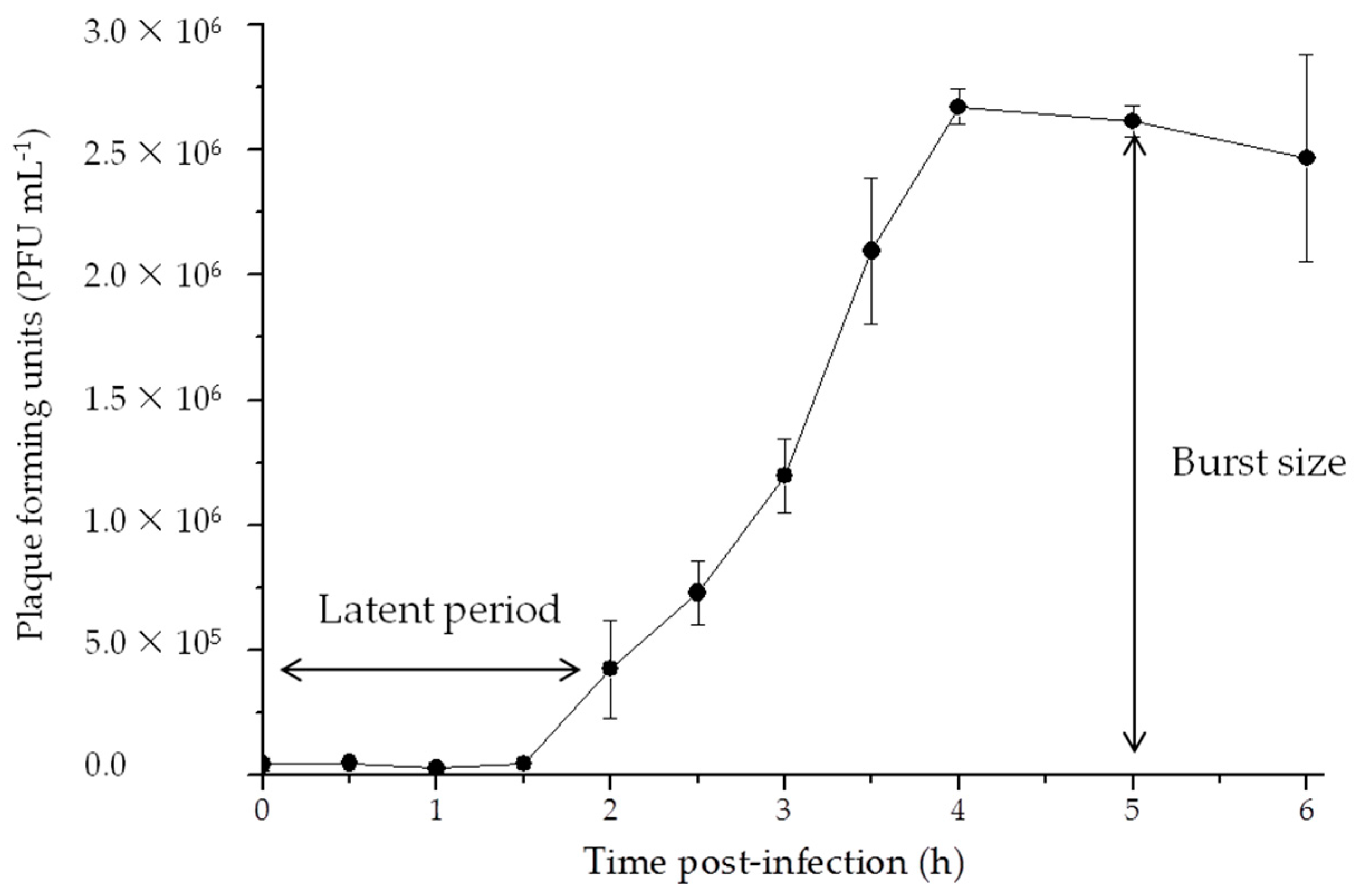
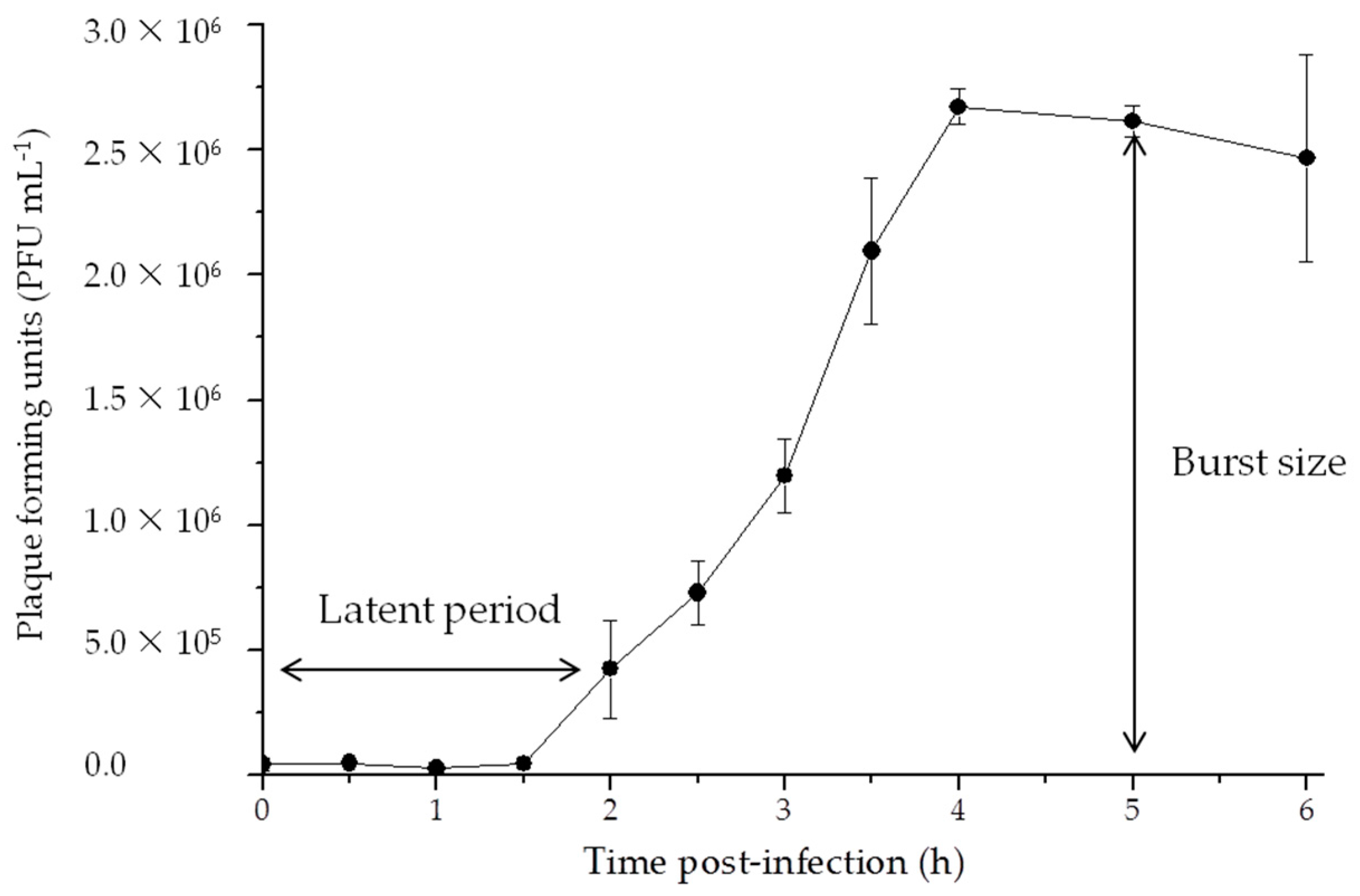
2.4. One-Step Growth Curve
2.5. Lipid in the Viral Capsid
2.6. DNA Extraction
2.7. Genome Sequencing and Analysis, and Phylogenetic Analyses
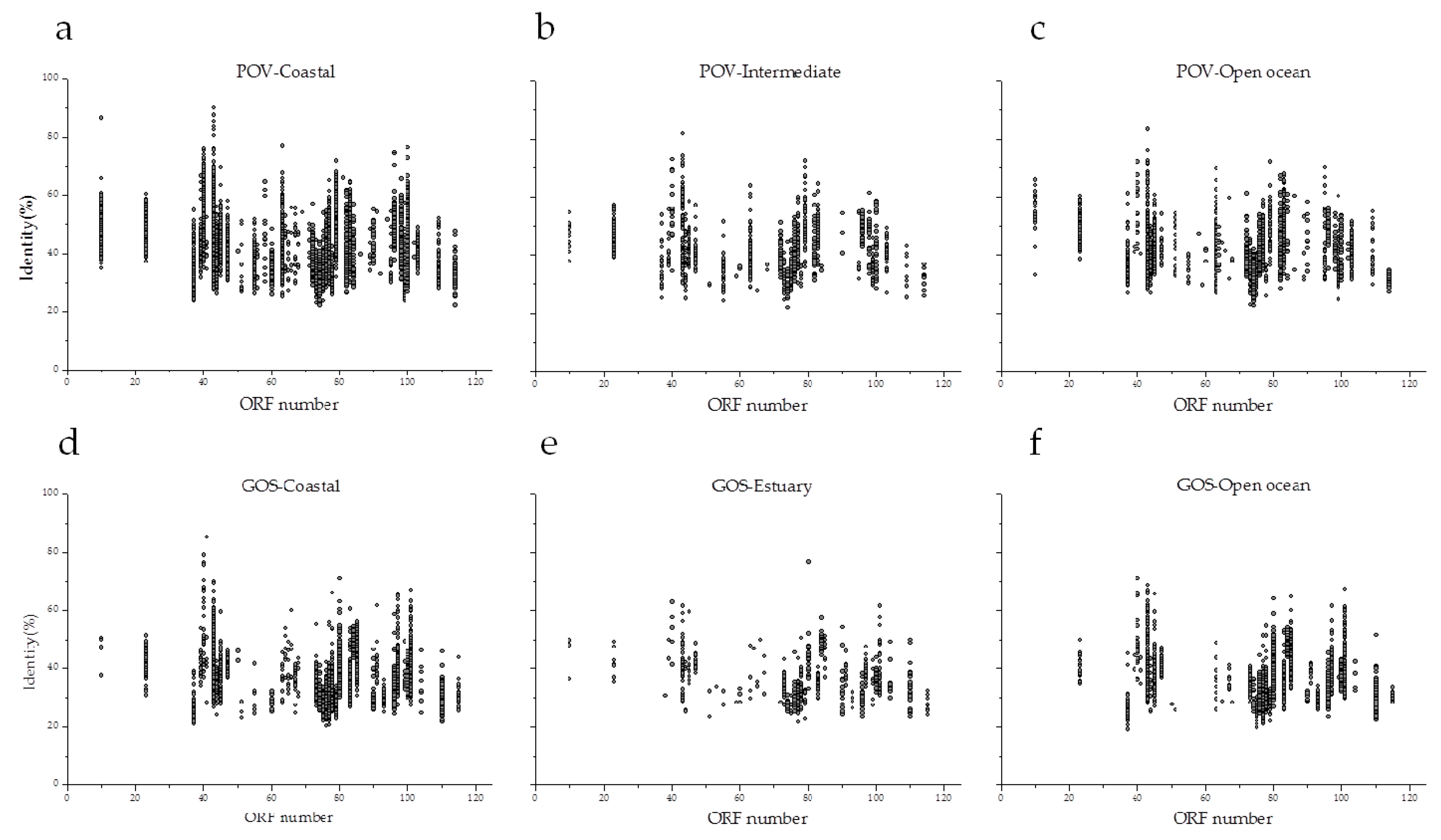
2.8. Recruitment of Metagenomic Data
3. Results and Discussion
3.1. Isolation and Characterization of Phage R5C
3.2. Life Cycle
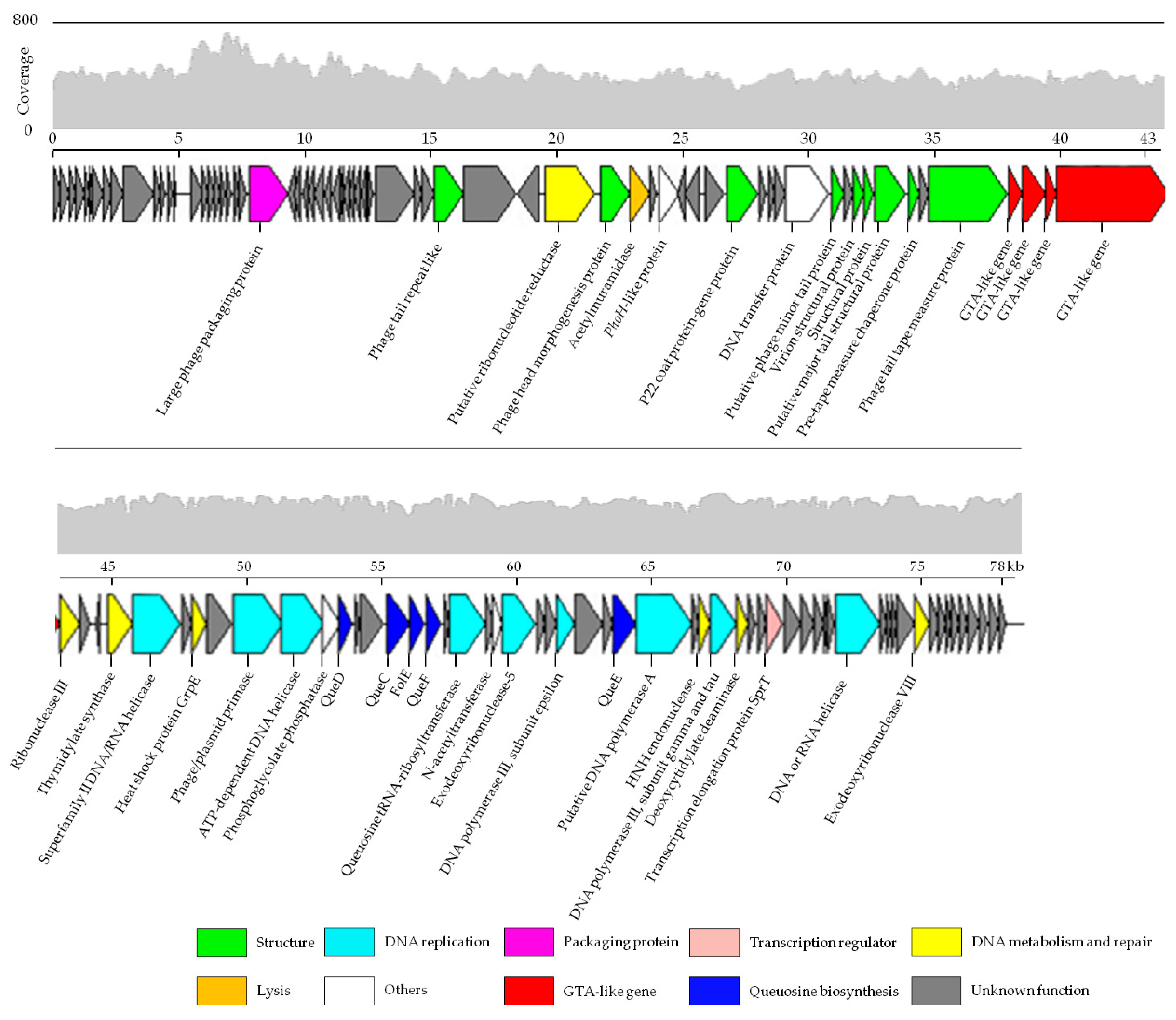
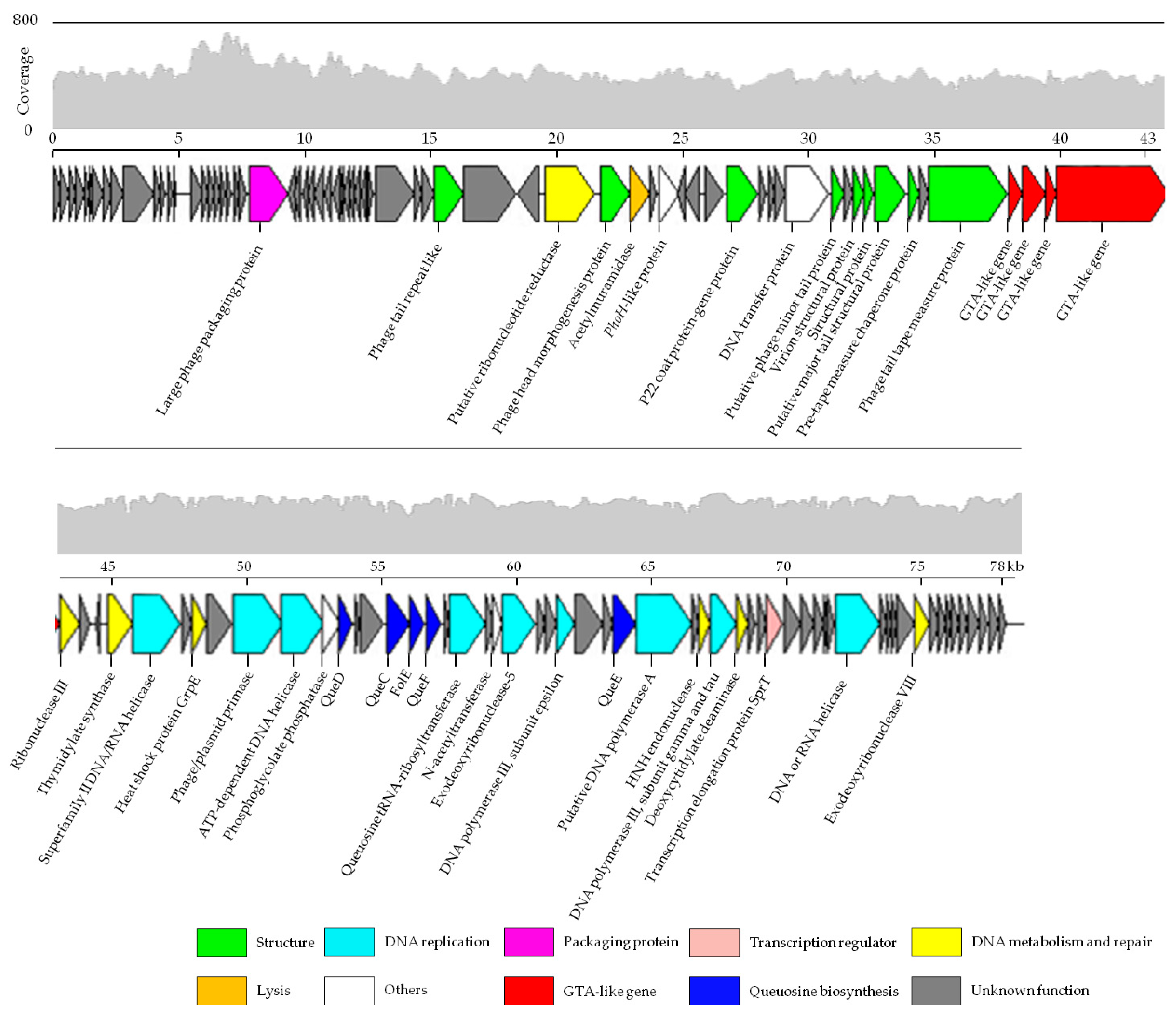
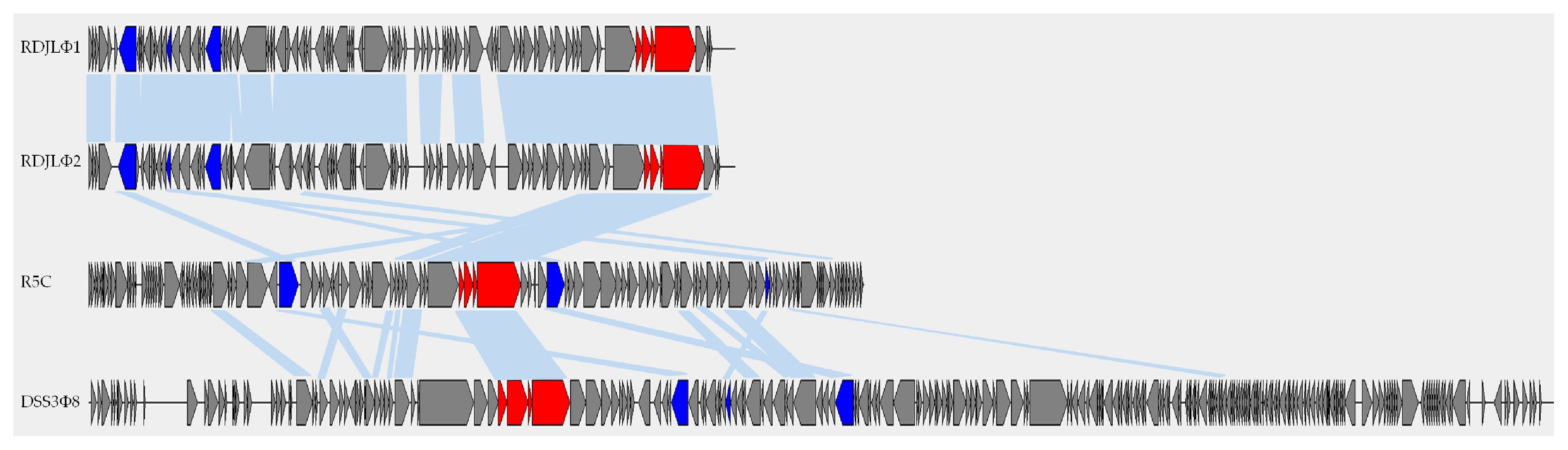
3.3. Genome Features
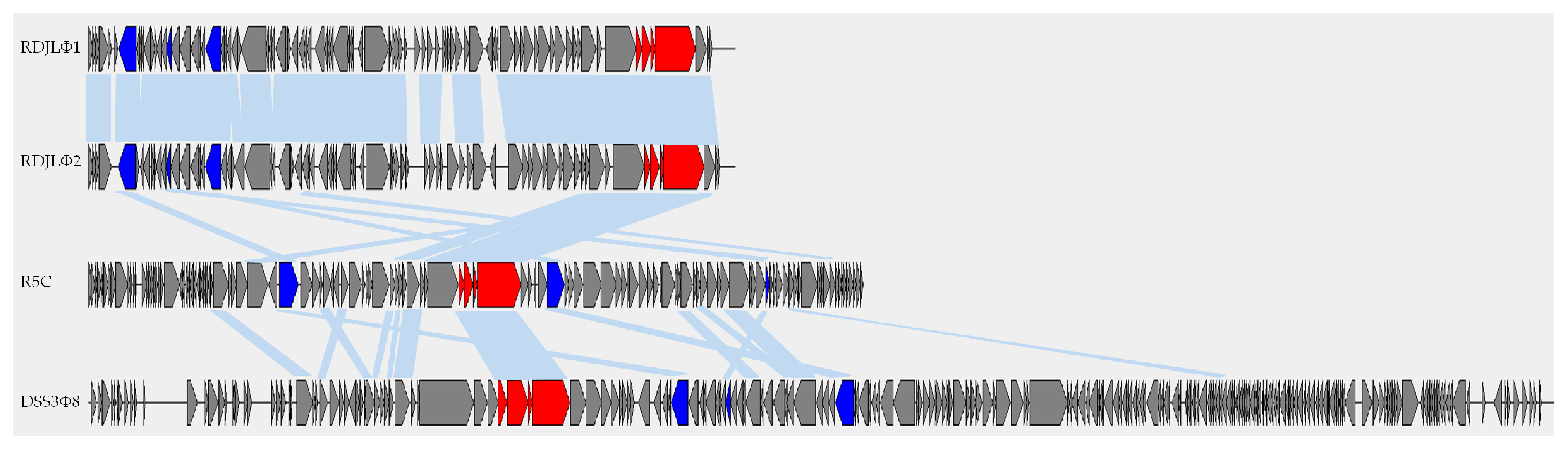
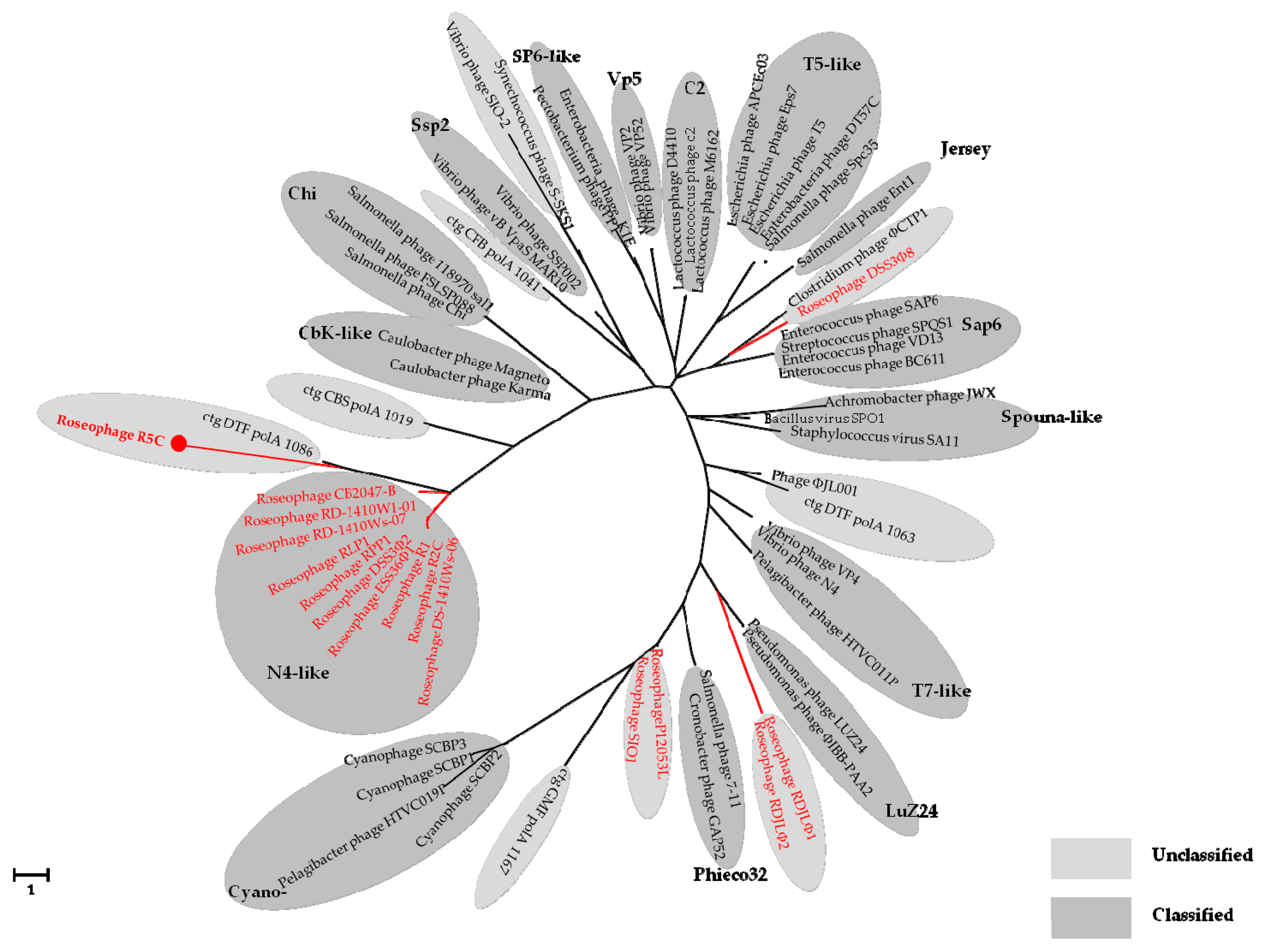
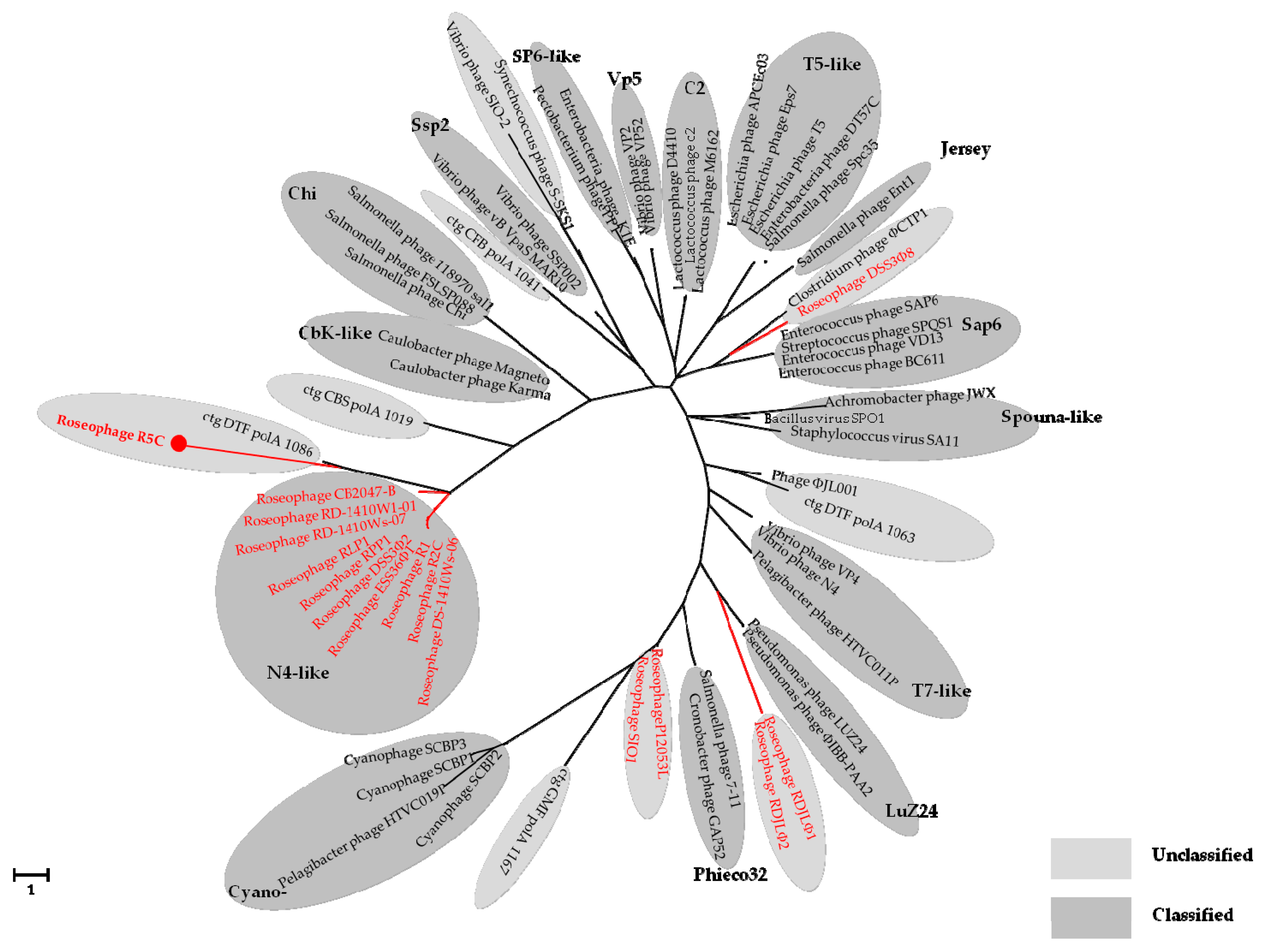
3.4. Phylogenetic Analyses
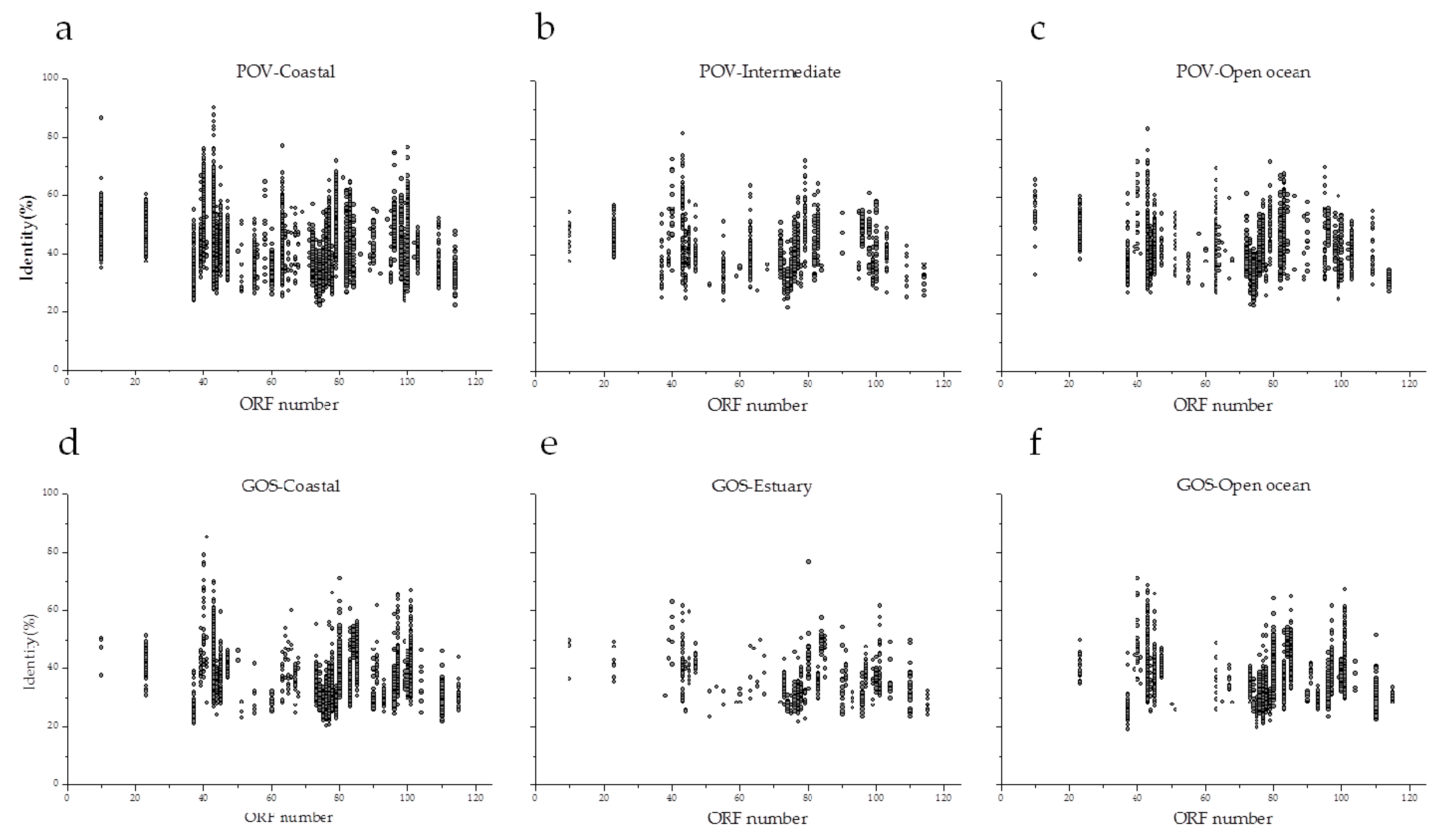
3.5. Environmental Distribution
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; Vargas, C.D.; Gasol, J.M.; et al. Patterns and ecological drivers of ocean viral communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Frois-Moniz, K.; Osburne, M.S.; Kelly, L.; Roggensack, S.E.; Sullivan, M.B.; Gearin, G.; Zeng, Q.; Fitzgerald, M.; Henn, M.R.; et al. Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ. Microbiol. 2013, 15, 1356–1376. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, A.C.; Hendrix, R.W.; Lander, G.C.; Bailly, X.; Podell, S.; Paillard, C.; Johnson, J.E.; Potter, C.S.; Carragher, B.; Azam, F. Genomic and functional analysis of Vibrio phage SIO-2 reveals novel insights into ecology and evolution of marine siphoviruses. Environ. Microbiol. 2012, 14, 2071–2086. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Temperton, B.; Thrash, J.C.; Schwalbach, M.S.; Vergin, K.L.; Landry, Z.C.; Ellisman, M.; Deerinck, T.; Sullivan, M.B.; Giovannoni, S.J. Abundant SAR11 viruses in the ocean. Nature 2013, 494, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Coleman, M.L.; Quinlivan, V.; Rosenkrantz, J.E.; Defrancesco, A.S.; Tan, G.; Fu, R.; Lee, J.A.; Waterbury, J.B.; Bielawski, J.P. Portal protein diversity and phage ecology. Environ. Microbiol. 2008, 10, 2810–2823. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.H.; Clokie, M.R.; Millard, A.; Cook, A.; Wilson, W.H.; Wheatley, P.J.; Letarov, A.; Krisch, H.M. The genome of S-PM2, a “photosynthetic” T4-type bacteriophage that infects marine Synechococcus strains. J. Bacteriol. 2005, 187, 3188–3200. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, L.I.; Martiny, J.B.; Marston, M.F. Selection and characterization of cyanophage resistance in marine Synechococcus strains. Appl. Environ. Microbiol. 2007, 73, 5516–5522. [Google Scholar] [CrossRef] [PubMed]
- Avrani, S.; Wurtzel, O.; Sharon, I.; Sorek, R.; Lindell, D. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature 2011, 474, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Brinkhoff, T.; Giebel, H.A.; Simon, M. Diversity, ecology, and genomics of the Roseobacter clade: A short overview. Arch. Microbiol. 2008, 189, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, Y.; Chen, F.; Jiao, N. Complete genome sequence of a marine roseophage provides evidence into the evolution of gene transfer agents in alphaproteobacteria. J. Virol. 2011, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Huang, S.; Voget, S.; Simon, M.; Chen, F. A novel roseobacter phage possesses features of podoviruses, siphoviruses, prophages and gene transfer agents. Sci. Rep. 2016, 6, 30372. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Yang, Y.; Jiao, N.; Zhang, R. Complete genome sequence of vB_DshP-R2C, a N4-like lytic roseophage. Mar. Genom. 2015, 22, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Budinof, C.R. Diversity and Activity of Roseobacters and Roseophage. Ph.D. Thesis, University of Tennessee, Knoxville, TS, USA, 2012. Available online: http://trace.tennessee.edu/utk_graddiss/1276 (accessed on 18 April 2017).
- Zhao, Y.; Wang, K.; Jiao, N.; Chen, F. Genome sequences of two novel phages infecting marine roseobacters. Environ. Microbiol. 2009, 11, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Buchan, A.; Gonzalez, J.M.; Moran, M.A. Overview of the marine roseobacter lineage. Appl. Environ. Microbiol. 2005, 71, 5665–5677. [Google Scholar] [CrossRef] [PubMed]
- Christie-Oleza, J.A.; Armengaud, J. Proteomics of the Roseobacter clade, a window to the marine microbiology landscape. Proteomics 2015, 15, 3928–3942. [Google Scholar] [CrossRef] [PubMed]
- Biebl, H.; Allgaier, M.; Tindall, B.J.; Koblizek, M.; Lunsdorf, H.; Pukall, R.; Wagner-Dobler, I. Dinoroseobacter shibae gen. nov., sp. nov., a new aerobic phototrophic bacterium isolated from dinoflagellates. Int. J. Syst. Evol. Microbiol. 2005, 55, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Zhang, Y.; Zeng, Y.; Hong, N.; Liu, R.; Chen, F.; Wang, P. Distinct distribution pattern of abundance and diversity of aerobic anoxygenic phototrophic bacteria in the global ocean. Environ. Microbiol. 2007, 9, 3091–3099. [Google Scholar] [CrossRef] [PubMed]
- Kolber, Z.S.; Plumley, F.G.; Lang, A.S.; Beatty, J.T.; Blankenship, R.E.; VanDover, C.L.; Vetriani, C.; Koblizek, M.; Rathgeber, C.; Falkowski, P.G. Contribution of aerobic photoheterotrophic bacteria to the carbon cycle in the ocean. Science 2001, 292, 2492–2495. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Dobler, I.; Biebl, H. Environmental biology of the marine Roseobacter lineage. Annu. Rev. Microbiol. 2006, 60, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Allgaier, M.; Uphoff, H.; Felske, A.; Wagner-Dobler, I. Aerobic anoxygenic photosynthesis in Roseobacter clade bacteria from diverse marine habitats. Appl. Environ. Microbiol. 2003, 69, 5051–5059. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Dobler, I.; Ballhausen, B.; Berger, M.; Brinkhoff, T.; Buchholz, I.; Bunk, B.; Cypionka, H.; Daniel, R.; Drepper, T.; Gerdts, G.; et al. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: A hitchhiker’s guide to life in the sea. ISME J. 2010, 4, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Jang, H.; Oh, H.-M.; Cho, J.-C. Complete genome sequence of Celeribacter bacteriophage P12053L. J. Virol. 2012, 86, 8339–8340. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Segall, A.; Steward, G.; Seguritan, V.; Breitbart, M.; Wolven, F.; Azam, F. The complete genomic sequence of the marine phage Roseophage SIO1 shares homology with nonmarine phages. Limnol. Oceanogy 2000, 45, 408–418. [Google Scholar] [CrossRef]
- Ankrah, N.Y.D.; Budinoff, C.R.; Wilson, W.H.; Wilhelm, S.W.; Buchan, A. Genome sequence of the Sulfitobacter sp. strain 2047-infecting lytic phage ΦCB2047-B. Genome Announc. 2014, 2, e00945-13. [Google Scholar] [CrossRef] [PubMed]
- Ankrah, N.Y.D.; Budinoff, C.R.; Wilson, W.H.; Wilhelm, S.W.; Buchana, A. Genome sequences of two temperate phages, ΦCB2047-A and ΦCB2047-C, infecting Sulfitobacter sp strain 2047. Genome Announc. 2014, 2, e00108-14. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Z.; Millard, A.D.; Mann, N.H.; Schafer, H. Comparative genomics defines the core genome of the growing N4-like phage genus and identifies N4-like Roseophage specific genes. Front. Microbiol. 2014, 5, 506. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zhang, R.; Jiao, N. Complete genome sequence of roseophage vB_DshP-R1, which infects Dinoroseobacter shibae DFL12. Stand. Genomic. Sci. 2015, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, Y.; Zhou, C.; Chen, Z.; Yang, S.; Yan, C.; Jiao, N. Complete genome sequence of the siphovirus roseophage RDJLФ2 infecting Roseobacter denitrificans OCh114. Mar. Genom. 2016, 25, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, S.; Long, L.; Huang, S. Characterization and complete genome sequences of three N4-like roseobacter phages isolated from the South China Sea. Curr. Microbiol. 2016, 73, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.; Youle, M.; Nosrat, B.; Srinagesh, S.; Rodriguez-Brito, B.; McNairnie, P.; Deyanat-Yazdi, G.; Breitbart, M.; Rohwer, F. Genomic analysis of multiple Roseophage SIO1 strains. Environ. Microbiol. 2009, 11, 2863–2873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, N. Roseophage RDJLФ1, infecting the aerobic anoxygenic phototrophic bacterium Roseobacter denitrificans OCh114. Appl. Environ. Microbiol. 2009, 75, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Middelboe, M.; Chan, A.M.; Bertelsen, S.K. Isolation and life cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria. In Manual of Aquatic Viral Ecology; Wilhelm, S.W., Weinbauer, M.G., Suttle, C.A., Eds.; American Society of Limnology and Oceanography: Waco, TX, USA, 2010; pp. 118–133. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Li, S.; An, X.; Pei, G.; Huang, Y.; Fan, H.; Mi, Z.; Zhang, Z.; Wang, W.; et al. A novel termini analysis theory using HTS data alone for the identification of Enterococcus phage EF4-like genome termini. BMC Genom. 2015, 16, 414. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A vector for high-throughput gene identificatio. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2014, 42, D7–D17. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007, 5, e77. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Chen, I.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA viruses and retroviruses. Nucleic Acids Res. 2017, 45, D457–D465. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Watai, H.; Honda, T.; Mihara, T.; Omae, K.; Roux, S.; Blanc-Mathieu, R.; Yamamoto, K.; Hingamp, P.; Sako, Y.; et al. Environmental viral genomes shed new light on virus-host interactions in the ocean. mSphere 2017, 2, e00359-16. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonte, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef] [PubMed]
- Parada, V.; Herndl, G.J.; Weinbauer, M.G. Viral burst size of heterotrophic prokaryotes in aquatic systems. J. Mar. Biol. Assos. UK 2006, 86, 613–621. [Google Scholar] [CrossRef]
- Hwang, C.Y.; Cho, B.C. Virus-infected bacteria in oligotrophic open waters of the East Sea, Korea. Aquat. Microb. Ecol. 2002, 30, 1–9. [Google Scholar] [CrossRef]
- Rocha, P.C.E.; Danchin, A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002, 18, 291–294. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, J. Ribonucleotide reductases: The link between an RNA and a DNA world? Curr. Opin. Struct. Biol. 2000, 10, 731–736. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Coleman, M.L.; Weigele, P.; Rohwer, F.; Chisholm, S.W. Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLoS Biol. 2005, 3, e144. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Lu, J. Genomic sequence and evolution of marine cyanophage P60: A new insight on lytic and lysogenic phages. Appl. Environ. Microbiol. 2002, 68, 2589–2594. [Google Scholar] [CrossRef] [PubMed]
- Matson, S.W.; Bean, D.W.; George, J.W. DNA helicases: Enzymes with essential roles in all aspects of DNA metabolism. BioEssays 1993, 16, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Tuteja, R. Unraveling DNA helicases. Motif, structure, mechanism and function. Eur. J. Biochem. 2004, 271, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.T.; Silversmith, R.E.; Maley, G.F.; Maley, F. T4-phage deoxycytidylate deaminase is a metalloprotein containing two zinc atoms per subunit. J. Biol. Chem. 1993, 268, 2288–2291. [Google Scholar] [PubMed]
- Weigele, P.R.; Pope, W.H.; Pedulla, M.L.; Houtz, J.M.; Smith, A.L.; Conway, J.F.; King, J.; Hatfull, G.F.; Lawrence, J.G.; Hendrix, R.W. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ. Microbiol. 2007, 9, 1675–1695. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.S.; Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Durkin, A.S.; Ciecko, A.; Feldblyum, T.V.; White, O.; Paulsen, I.T.; Nierman, W.C.; et al. Complete genome sequence of the broad-host-range vibriophage KVP40: Comparative genomics of a T4-related bacteriophage. J. Bacteriol. 2003, 185, 5220–5233. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.E.; Vassieva, O.; Gelfand, M.S.; Osterman, A.; Overbeek, R. Bioinformatics classification and functional analysis of phoH homologs. In Silico Biol. 2003, 3, 3–15. [Google Scholar] [PubMed]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, J.B.; Saltik, M.; Chiaocca, S.; Michou, A.-I.; Moseley, P.; Cotten, M. Activation of heat-shock response by an adenovirus is essential for virus replication. Nature 2000, 407, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.; Walter, W.; Gross, C.A. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma 32. Genes Dev. 1990, 4, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Liberek, K.; Marszalek, J.; Ang, D.; Georgopoulos, C.; Zylicz, M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. USA 1991, 88, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, M.; Pathak, C. Queuosine modification of tRNA: Its divergent role in cellular machinery. Biosci. Rep. 2009, 30, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.; Hauser, R.; Ouellette, M.; Liu, J.; Dehbi, M.; Moeck, G.; Garcia, E.; Titz, B.; Uetz, P.; Moineau, S. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J. Bacteriol. 2011, 193, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, E.E.; Golomidova, A.K.; Letarova, M.A.; Kostryukova, E.S.; Zelenin, A.S.; Prokhorov, N.S.; Letarov, A.V. Genomic sequencing and biological characteristics of a novel Escherichia coli bacteriophage 9g, a putative representative of a new Siphoviridae genus. Viruses 2014, 6, 5077–5092. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Ravet, V.; Pereira, O.; Sullivan, M.B. Genomic characteristics and environmental distributions of the uncultivated Far-T4 phages. Front. Microbiol. 2015, 6, 199. [Google Scholar] [CrossRef] [PubMed]
- Loh, E.; Loeb, L.A. Mutability of DNA polymerase I: Implications for the creation of mutant DNA polymerases. DNA Repair 2005, 4, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.F.; Sakowski, E.G.; Williamson, S.J.; Polson, S.W.; Wommack, K.E. Shotgun metagenomics indicates novel family A DNA polymerases predominate within marine virioplankton. ISME J. 2014, 8, 103–114. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | R5C | DSS3Φ8 | RDJLФ1 | RLP1 | RPP1 | DSS3Φ2 | ESS36Φ1 | DS-1410Ws-06 | RD-1410W1-01 | RD-1410Ws-07 |
|---|---|---|---|---|---|---|---|---|---|---|
| D. shibae DFL12T | + | + | + | + | ||||||
| Acinetobacter sp. JL1404 | − | |||||||||
| Alcanivorax sp. JL1378 | − | |||||||||
| Alteromonas sp. JL1357 | − | |||||||||
| Antarctobacter sp. JL351 | − | |||||||||
| Citromicrobium sp. JL1363 | − | |||||||||
| Citromicrobium sp. JL1351 | − | − | ||||||||
| Citromicrobium sp. JL2201 | − | |||||||||
| Citromicrobium sp. JL354 | − | − | ||||||||
| Citromicrobium sp. WPS32 | − | |||||||||
| Cytophaga sp. JL1362 | − | |||||||||
| Dinoroseobacter sp. JL1447 | − | − | ||||||||
| Erythrobacter litoralis DMS 8509 | − | − | ||||||||
| Erythrobacter longus DSM 6997 | − | − | ||||||||
| Erythrobacter sp. JL1350 | − | |||||||||
| Erythrobacter sp. JL359 | − | |||||||||
| Erythrobacter sp. JL475. | − | − | ||||||||
| Furvibacter sp. JL1383 | − | |||||||||
| Hoeflea phototrophica DFL-43T | − | |||||||||
| Leisingera methylohalidivorans MB2 | − | − | ||||||||
| Marinovum algicola ATCC 51440T | − | − | ||||||||
| Micrococcus sp. JL1389 | − | |||||||||
| Nocardioides sp. JL1369 | − | |||||||||
| Paenibacillus sp. JL1210 | − | |||||||||
| Phaeobacter sp. 27–4 | − | − | ||||||||
| Pseudoalteromonas sp. JL1391 | − | |||||||||
| Rhodobacteraceae sp. 176 | − | − | ||||||||
| Roseobacter denitrificans ATCC 33942 | − | − | ||||||||
| Roseobacter denitrificans OCh 114 | − | − | + | + | + | + | ||||
| Roseobacter litoralis ATCC 49566 | − | − | ||||||||
| Roseobacter litoralis DMS 6996 | − | |||||||||
| Roseobacter litoralis Och149 | − | + | + | |||||||
| Roseobacter sp. JL1336 | − | |||||||||
| Roseobacter sp. TM1038 | − | − | ||||||||
| Roseobacter sp. TM1039 | − | − | ||||||||
| Roseomonas sp. JL2290 | − | |||||||||
| Roseomonas sp. JL2293 | − | |||||||||
| Roseovarius sp.TM1042 | − | − | ||||||||
| Roseovarius nubinhibens ISMT | − | − | + | − | − | − | − | − | ||
| Roseovarius sp. 217 | + | − | ||||||||
| Roseovarius sp. JL2434 | − | |||||||||
| Roseovarius crassostreae CV919–312T | − | − | ||||||||
| Roseovarius mucosus DFL–24T | − | − | ||||||||
| Ruegeria atlantica AMA–03 | − | − | ||||||||
| Ruegeria sp. 198 | − | − | ||||||||
| Ruegeria sp. JL126 | − | |||||||||
| Sagittula stellata E–37 | − | − | ||||||||
| Silicibacter pomeroyi DSS3 | − | + | − | − | + | + | + | + | ||
| Silicibacter sp. TM1040 | − | − | − | |||||||
| Sphingobium sp. JL1088 | − | |||||||||
| Stappia sp. JL1358 | − | |||||||||
| Sulfitobacter sp. 1921 | − | − | ||||||||
| Sulfitobacter pseudonitzschiae H3 | − | − | − | |||||||
| Sulfitobacter sp. EE–36 | − | − | + | − | − | − | ||||
| Sulfitobacter sp. JL1353 | − | |||||||||
| Sulfitobacter sp CBB406 | − | − | ||||||||
| Sulfitobacter sp. CHSB4 | − | − | ||||||||
| Sulfitobacter sp. JL351 | − | − | ||||||||
| Sulfitobacter sp. T11 | − | − |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Cai, L.; Ma, R.; Xu, Y.; Tong, Y.; Huang, Y.; Jiao, N.; Zhang, R. A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea. Viruses 2017, 9, 109. https://doi.org/10.3390/v9050109
Yang Y, Cai L, Ma R, Xu Y, Tong Y, Huang Y, Jiao N, Zhang R. A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea. Viruses. 2017; 9(5):109. https://doi.org/10.3390/v9050109
Chicago/Turabian StyleYang, Yunlan, Lanlan Cai, Ruijie Ma, Yongle Xu, Yigang Tong, Yong Huang, Nianzhi Jiao, and Rui Zhang. 2017. "A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea" Viruses 9, no. 5: 109. https://doi.org/10.3390/v9050109
APA StyleYang, Y., Cai, L., Ma, R., Xu, Y., Tong, Y., Huang, Y., Jiao, N., & Zhang, R. (2017). A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea. Viruses, 9(5), 109. https://doi.org/10.3390/v9050109