Deciphering the Multifactorial Susceptibility of Mucosal Junction Cells to HPV Infection and Related Carcinogenesis


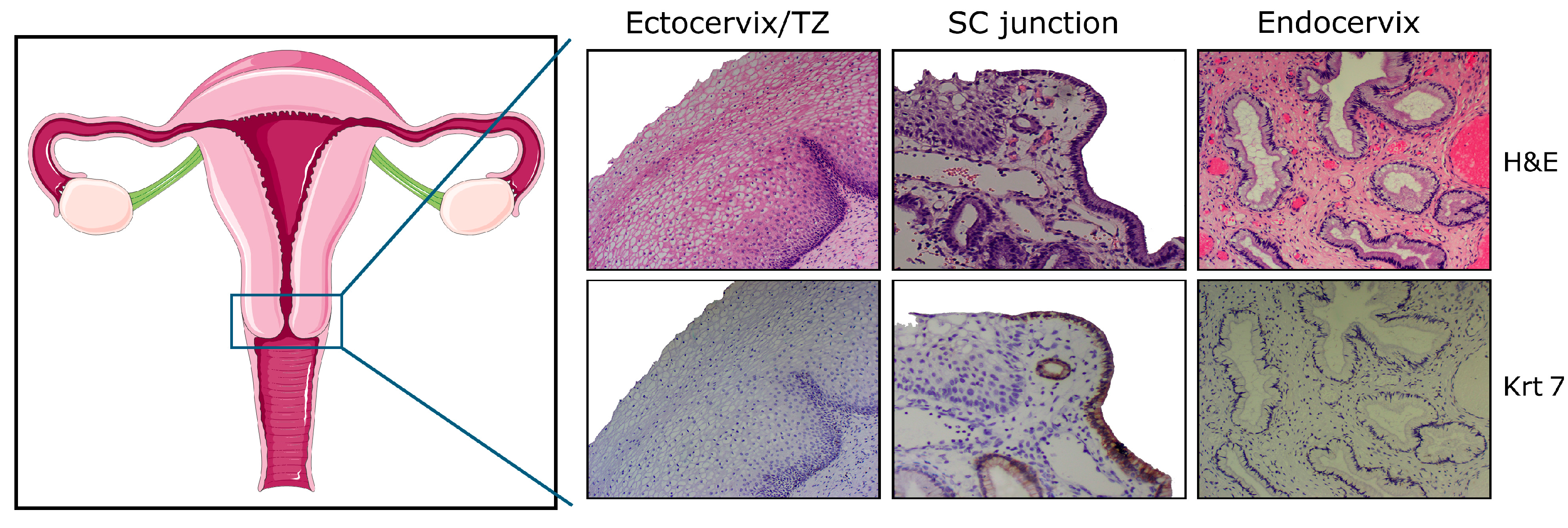
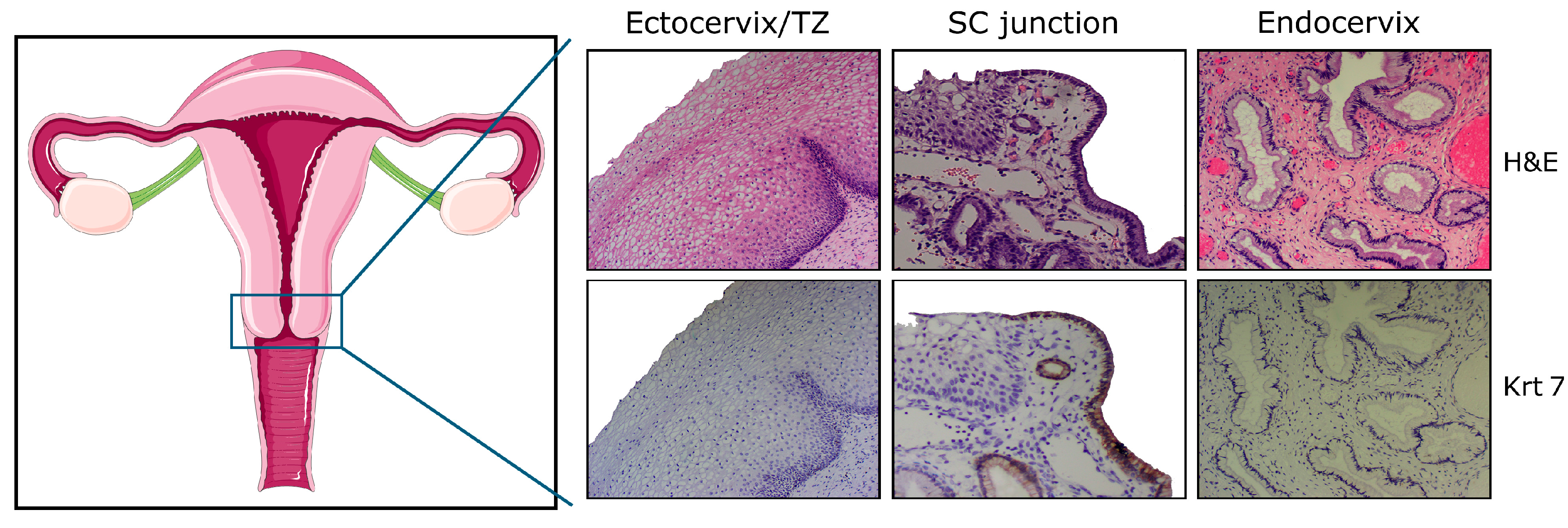{kind=link}
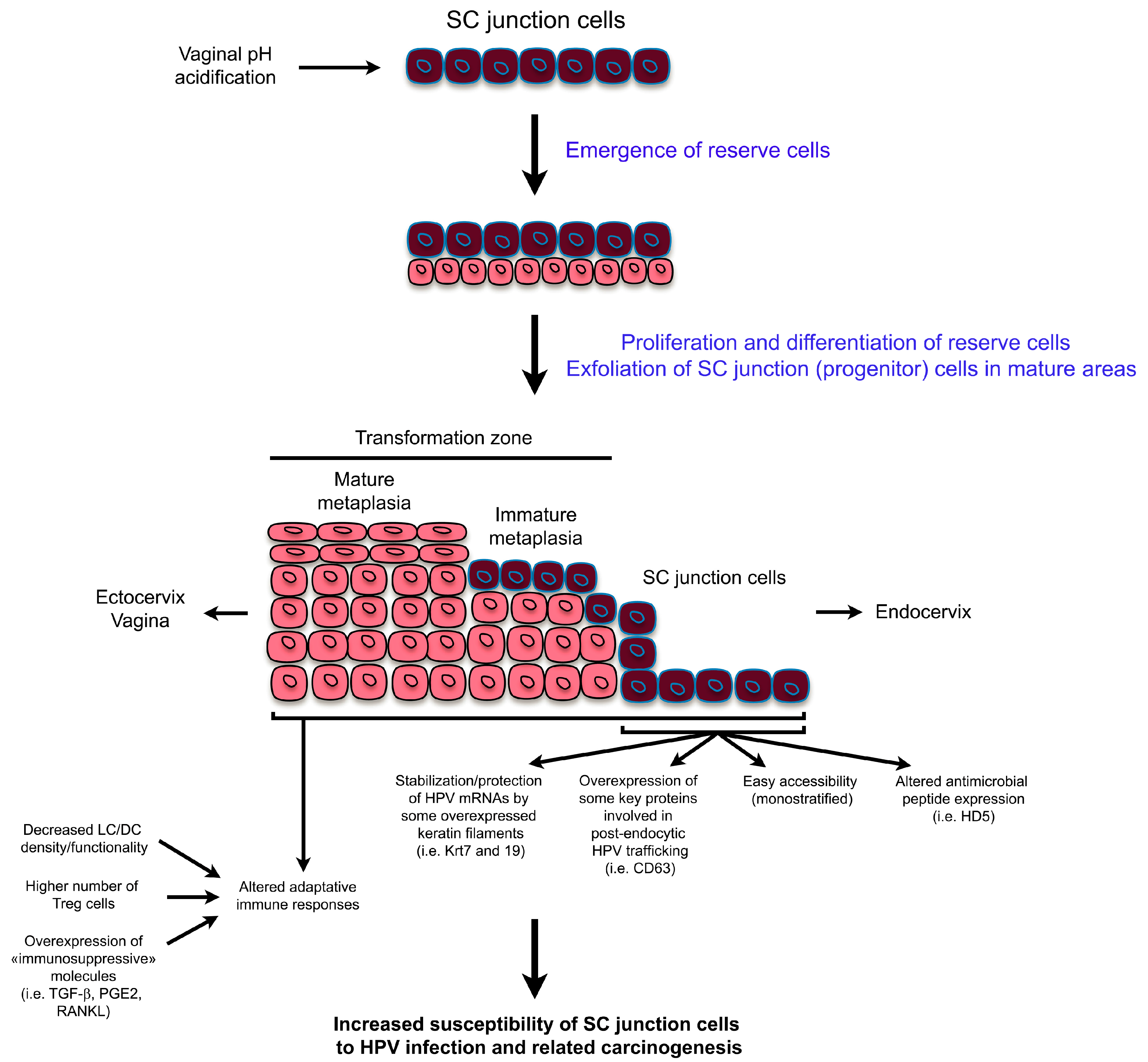{kind=link}
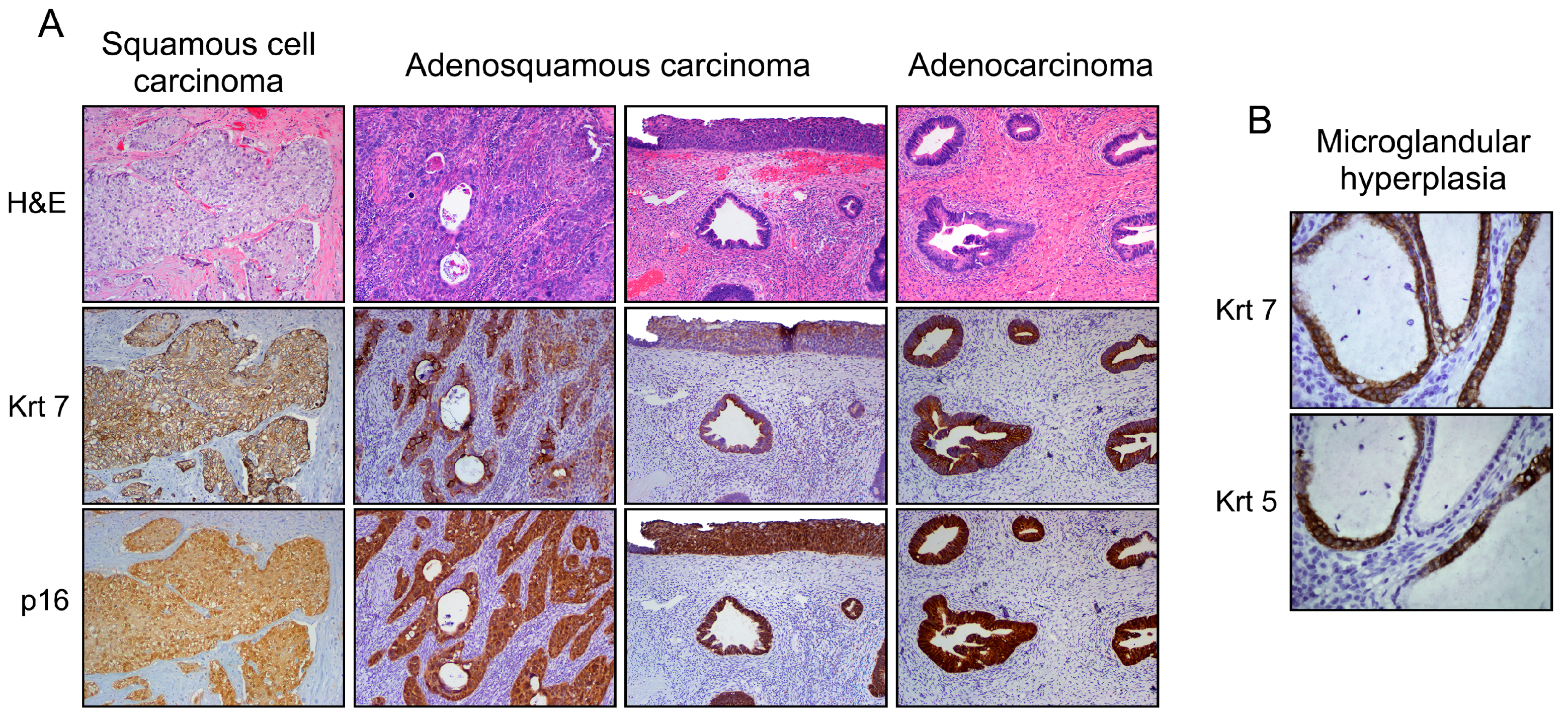{kind=link}
Abstract
:1. Introduction
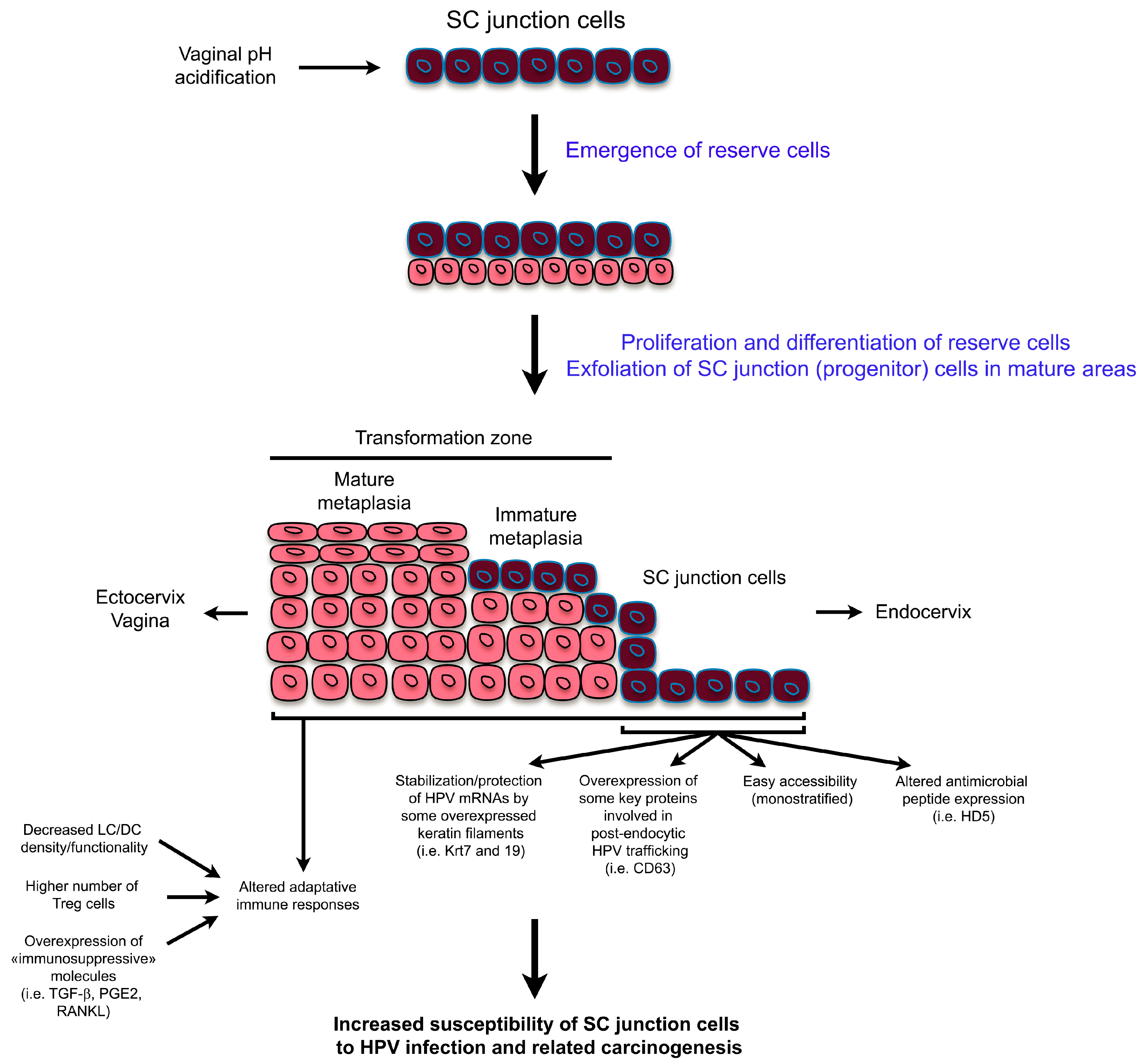
2. High Vulnerability of SC Junctions to HPV Infection and (Pre)Cancer Development: A Multifaceted Process
2.1. Altered Secretion of Antimicrobial Peptides
2.2. Overexpression of Key Proteins Implicated in Post-Endocytic HPV Trafficking
2.3. Possible Translational Regulation of HPV mRNAs by the Cytokeratin Filaments 7 and 19
2.4. Altered Adaptive Immune Responses in SC Junction Microenvironment
3. Dualistic Model of HPV-Related Carcinogenesis
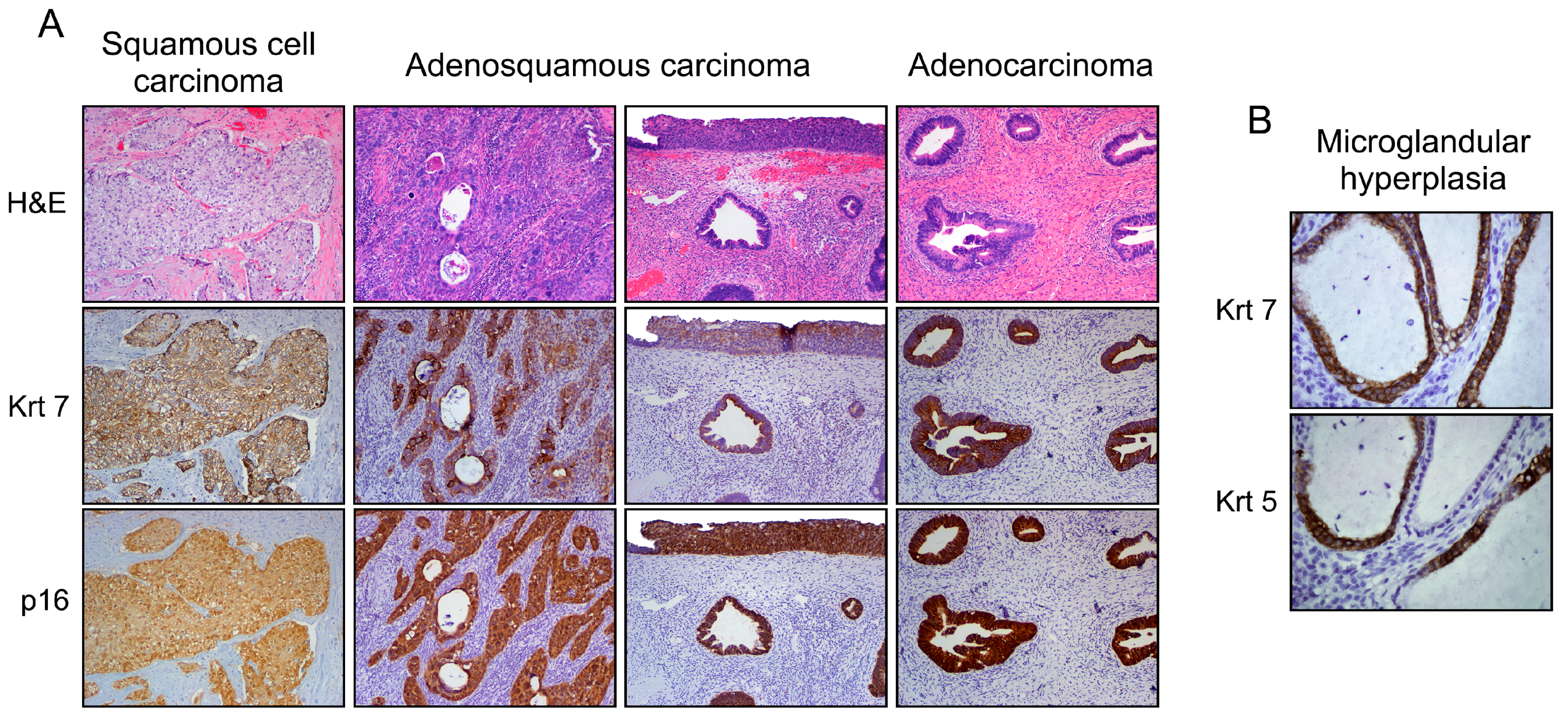
3.1. Explanation for Multiple Neoplastic Phenotypes
3.2. From the Bench to Cervical Cancer Prevention
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Woodman, C.B.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.N.; Buck, C.B.; Thompson, C.D.; Kines, R.; Bernardo, M.; Choyke, P.L.; Lowy, D.R.; Schiller, J.T. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat. Med. 2007, 13, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M. Original site of cervical carcinoma; topographical relationship of carcinoma of the cervix to the external os and to the squamocolumnar junction. Obstet. Gynecol. 1956, 7, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Richart, R.M. Cervical intraepithelial neoplasia. Pathol. Ann. 1973, 8, 301–328. [Google Scholar]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; Franceschi, S. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.E.; Arends, J.; Van der Linden, P.J.; De Boer, B.A.; Helmerhorst, T.J. Cytokeratin 17 and p63 are markers of the HPV target cell, the cervical stem cell. Anticancer Res. 2004, 24, 771–775. [Google Scholar] [PubMed]
- Martens, J.E.; Smedts, F.M.; Ploeger, D.; Helmerhorst, T.J.; Ramaekers, F.C.; Arends, J.W.; Hopman, A.H. Distribution pattern and marker profile show two subpopulations of reserve cells in the endocervical canal. Int. J. Gynecol. Pathol. 2009, 28, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Chin, N.; Platt, A.B.; Nuovo, G.J. Squamous intraepithelial lesions arising in benign endocervical polyps: A report of 9 cases with correlation to the Pap smears, HPV analysis, and immunoprofile. Int. J. Gynecol. Pathol. 2008, 27, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Yamamoto, Y.; Laury, A.; Wang, X.; Nucci, M.R.; McLaughlin-Drubin, M.E.; Munger, K.; Feldman, S.; McKeon, F.D.; Xian, W.; et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Vargas, S.O.; Yamamoto, Y.; Howitt, B.E.; Nucci, M.R.; Hornick, J.L.; McKeon, F.D.; Xian, W.; Crum, C.P. A novel blueprint for ‘top down’ differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. J. Pathol. 2013, 229, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ouyang, H.; Yamamoto, Y.; Kumar, P.A.; Wei, T.S.; Dagher, R.; Vincent, M.; Lu, X.; Bellizzi, A.M.; Ho, K.Y.; et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 2011, 145, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Parra-Herran, C.; Howitt, B.E.; Laury, A.R.; Nucci, M.R.; Feldman, S.; Jimenez, C.A.; McKeon, F.D.; Xian, W.; Crum, C.P. Cervical squamocolumnar junction-specific markers define distinct, clinically relevant subsets of low-grade squamous intraepithelial lesions. Am. J. Surg. Pathol. 2013, 37, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Paquette, C.; Mills, A.M.; Stoler, M.H. Predictive Value of Cytokeratin 7 Immunohistochemistry in Cervical Low-grade Squamous Intraepithelial Lesion as a Marker for Risk of Progression to a High-grade Lesion. Am. J. Surg. Pathol. 2016, 40, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.M.; Paquette, C.; Terzic, T.; Castle, P.E.; Stoler, M.H. CK7 Immunohistochemistry as a Predictor of CIN1 Progression: A Retrospective Study of Patients From the Quadrivalent HPV Vaccine Trials. Am. J. Surg. Pathol. 2017, 41, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.C.; Tomic, M.M.; Hanamornroongruang, S.; Meserve, E.E.; Herfs, M.; Crum, C.P. p16ink4 and cytokeratin 7 immunostaining in predicting HSIL outcome for low-grade squamous intraepithelial lesions: A case series, literature review and commentary. Mod. Pathol. 2016, 29, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wu, Z.; Yu, L.; Li, J.; Xu, W.; Chan, H.C.; Zhang, Y.; Hu, L. Overexpression of cystic fibrosis transmembrane conductance regulator (CFTR) is associated with human cervical cancer malignancy, progression and prognosis. Gynecol. Oncol. 2012, 125, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, H.; Cho, Y.K. Cytokeratin7 and cytokeratin19 expression in high grade cervical intraepithelial neoplasm and squamous cell carcinoma and their possible association in cervical carcinogenesis. Diagn. Pathol. 2017, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, J.; Howitt, B.E.; Roncarati, P.; Demoulin, S.; Suarez-Carmona, M.; Hubert, P.; McKeon, F.D.; Xian, W.; Li, A.; Delvenne, P.; et al. Carcinogenic HPV infection in the cervical squamo-columnar junction. J. Pathol. 2015, 236, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Yarbrough, V.L.; Winkle, S.; Herbst-Kralovetz, M.M. Antimicrobial peptides in the female reproductive tract: A critical component of the mucosal immune barrier with physiological and clinical implications. Hum. Reprod. Updat. 2015, 21, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Hubert, P.; Delvenne, P.; Herfs, M. Defensins: “Simple” antimicrobial peptides or broad-spectrum molecules? Cytokine Growth Factor Rev. 2015, 26, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Day, P.M.; Thompson, C.D.; Lubkowski, J.; Lu, W.; Lowy, D.R.; Schiller, J.T. Human α-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Herman, L.; Maillard, C.; Caberg, J.H.; Nikkels, A.; Pierard, G.; Foidart, J.M.; Noel, A.; Boniver, J.; Delvenne, P. Defensins induce the recruitment of dendritic cells in cervical human papillomavirus-associated (pre)neoplastic lesions formed in vitro and transplanted in vivo. FASEB J. 2007, 21, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhou, H.Y.; Li, H.; Yi, T.; Zhao, X. The therapeutic impact of HNP-1 in condyloma acuminatum. Int. J. Dermatol. 2015, 54, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Herman, L.; Roncarati, P.; Maillard, C.; Renoux, V.; Demoulin, S.; Erpicum, C.; Foidart, J.M.; Boniver, J.; Noel, A.; et al. Altered α-defensin 5 expression in cervical squamocolumnar junction: Implication in the formation of a viral/tumour-permissive microenvironment. J. Pathol. 2014, 234, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.E.; Smith, J.G. α-defensin HD5 inhibits furin cleavage of human papillomavirus 16 L2 to block infection. J. Virol. 2015, 89, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.K.; Nemerow, G.R.; Smith, J.G. Direct evidence from single-cell analysis that human α-defensins block adenovirus uncoating to neutralize infection. J. Virol. 2010, 84, 4041–4049. [Google Scholar] [CrossRef] [PubMed]
- Zins, S.R.; Nelson, C.D.; Maginnis, M.S.; Banerjee, R.; O’Hara, B.A.; Atwood, W.J. The human α-defensin HD5 neutralizes JC polyomavirus infection by reducing endoplasmic reticulum traffic and stabilizing the viral capsid. J. Virol. 2014, 88, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Nemerow, G.R. Mechanism of adenovirus neutralization by Human α-defensins. Cell Host Microbe 2008, 3, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.E.; Smith, J.G. α-Defensin HD5 Inhibits Human Papillomavirus 16 Infection via Capsid Stabilization and Redirection to the Lysosome. mBio 2017, 8, e02304–e02316. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Schelhaas, M. Concepts of papillomavirus entry into host cells. Curr. Opin. Virol. 2014, 4, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Grassel, L.; Fast, L.A.; Scheffer, K.D.; Boukhallouk, F.; Spoden, G.A.; Tenzer, S.; Boller, K.; Bago, R.; Rajesh, S.; Overduin, M.; et al. The CD63-Syntenin-1 Complex Controls Post-Endocytic Trafficking of Oncogenic Human Papillomaviruses. Sci. Rep. 2016, 6, 32337. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 2009, 384, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meyers, C.; Wang, H.K.; Chow, L.T.; Zheng, Z.M. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xue, Y.; Poidinger, M.; Lim, T.; Chew, S.H.; Pang, C.L.; Abastado, J.P.; Thierry, F. Mapping of HPV transcripts in four human cervical lesions using RNAseq suggests quantitative rearrangements during carcinogenic progression. Virology 2014, 462–463, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Nagasaka, K.; Kawana, K.; Hashimoto, K.; Kusumoto-Matsuo, R.; Plessy, C.; Thomas, M.; Nakamura, H.; Bonetti, A.; Oda, K.; et al. Characterization of novel transcripts of human papillomavirus type 16 using cap analysis gene expression technology. J. Virol. 2015, 89, 2448–2452. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Dalstein, V.; Waterboer, T.; Clavel, C.; Gissmann, L.; Pawlita, M. The HPV16 transcriptome in cervical lesions of different grades. Mol. Cell. Probes 2011, 25, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D. Translational regulation of human papillomavirus type 16 E7 mRNA by the peptide SEQIKA, shared by rabbit α1-globin and human cytokeratin 7. J. Virol. 2002, 76, 7040–7048. [Google Scholar] [CrossRef] [PubMed]
- Favia, G.; Kanduc, D.; Lo Muzio, L.; Lucchese, A.; Serpico, R. Possible association between HPV16 E7 protein level and cytokeratin 19. Int. J. Cancer 2004, 111, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Longuespee, R.; Quick, C.M.; Roncarati, P.; Suarez-Carmona, M.; Hubert, P.; Lebeau, A.; Bruyere, D.; Mazzucchelli, G.; Smargiasso, N.; et al. Proteomic signatures reveal a dualistic and clinically relevant classification of anal canal carcinoma. J. Pathol. 2017, 241, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Gorodeski, G.I.; Hopfer, U.; Liu, C.C.; Margles, E. Estrogen acidifies vaginal pH by up-regulation of proton secretion via the apical membrane of vaginal-ectocervical epithelial cells. Endocrinology 2005, 146, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Hubert, P.; Kholod, N.; Caberg, J.H.; Gilles, C.; Berx, G.; Savagner, P.; Boniver, J.; Delvenne, P. Transforming growth factor-β1-mediated Slug and Snail transcription factor up-regulation reduces the density of Langerhans cells in epithelial metaplasia by affecting E-cadherin expression. Am. J. Pathol. 2008, 172, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Caberg, J.H.; Hubert, P.M.; Begon, D.Y.; Herfs, M.F.; Roncarati, P.J.; Boniver, J.J.; Delvenne, P.O. Silencing of E7 oncogene restores functional E-cadherin expression in human papillomavirus 16-transformed keratinocytes. Carcinogenesis 2008, 29, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Caberg, J.H.; Gilles, C.; Bousarghin, L.; Franzen-Detrooz, E.; Boniver, J.; Delvenne, P. E-cadherin-dependent adhesion of dendritic and Langerhans cells to keratinocytes is defective in cervical human papillomavirus-associated (pre)neoplastic lesions. J. Pathol. 2005, 206, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Bloom, O.; Ono, S.; Cui, W.; Unternaehrer, J.; Jiang, S.; Whitney, J.A.; Connolly, J.; Banchereau, J.; Mellman, I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 2007, 27, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Mayumi, N.; Watanabe, E.; Norose, Y.; Watari, E.; Kawana, S.; Geijtenbeek, T.B.; Takahashi, H. E-cadherin interactions are required for Langerhans cell differentiation. Eur. J. Immunol. 2013, 43, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Herman, L.; Hubert, P.; Minner, F.; Arafa, M.; Roncarati, P.; Henrotin, Y.; Boniver, J.; Delvenne, P. High expression of PGE2 enzymatic pathways in cervical (pre)neoplastic lesions and functional consequences for antigen-presenting cells. Cancer Immunol. Immunother. CII 2009, 58, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, S.A.; Somja, J.; Duray, A.; Guenin, S.; Roncarati, P.; Delvenne, P.O.; Herfs, M.F.; Hubert, P.M. Cervical (pre)neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells via RANKL secretion. Oncoimmunology 2015, 4, e1008334. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, S.; Herfs, M.; Somja, J.; Roncarati, P.; Delvenne, P.; Hubert, P. HMGB1 secretion during cervical carcinogenesis promotes the acquisition of a tolerogenic functionality by plasmacytoid dendritic cells. Int. J. Cancer 2015, 137, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schroeder, J.M.; Bryan, J.T.; Stoler, M.H.; Fife, K.H. Detection of multiple human papillomavirus types in Condylomata acuminata lesions from otherwise healthy and immunosuppressed patients. J. Clin. Microbiol. 1999, 37, 3316–3322. [Google Scholar] [PubMed]
- Alves de Sousa, N.L.; Alves, R.R.; Martins, M.R.; Barros, N.K.; Ribeiro, A.A.; Zeferino, L.C.; Dufloth, R.M.; Rabelo-Santos, S.H. Cytopathic effects of human papillomavirus infection and the severity of cervical intraepithelial neoplasia: A frequency study. Diagn. Cytopathol. 2012, 40, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Vrdoljak-Mozetic, D.; Krasevic, M.; Versa Ostojic, D.; Stemberger-Papic, S.; Rubesa-Mihaljevic, R.; Bubonja-Sonje, M. HPV16 genotype, p16/Ki-67 dual staining and koilocytic morphology as potential predictors of the clinical outcome for cervical low-grade squamous intraepithelial lesions. Cytopathology 2015, 26, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, K.M.; Chapman, W.; Daya, D.; Julian, J.A.; Lytwyn, A.; McLachlin, C.M.; Elit, L. Reproducibility of the histological diagnosis of cervical dysplasia among pathologists from 4 continents. Int. J. Gynecol. Pathol. 2008, 27, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Dalla Palma, P.; Giorgi Rossi, P.; Collina, G.; Buccoliero, A.M.; Ghiringhello, B.; Gilioli, E.; Onnis, G.L.; Aldovini, D.; Galanti, G.; Casadei, G.; et al. The reproducibility of CIN diagnoses among different pathologists: Data from histology reviews from a multicenter randomized study. Am. J. Clin. Pathol. 2009, 132, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Crum, C.P. Laboratory management of cervical intraepithelial neoplasia: Proposing a new paradigm. Adv. Anat. Pathol. 2013, 20, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Sun, D.; Quade, B.J.; Flynn, C.; Sheets, E.E.; Yang, A.; McKeon, F.; Crum, C.P. Stratified mucin-producing intraepithelial lesions of the cervix: Adenosquamous or columnar cell neoplasia? Am. J. Surg. Pathol. 2000, 24, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P. Contemporary theories of cervical carcinogenesis: The virus, the host, and the stem cell. Mod. Pathol. 2000, 13, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Smotkin, D.; Berek, J.S.; Fu, Y.S.; Hacker, N.F.; Major, F.J.; Wettstein, F.O. Human papillomavirus deoxyribonucleic acid in adenocarcinoma and adenosquamous carcinoma of the uterine cervix. Obstet. Gynecol. 1986, 68, 241–244. [Google Scholar] [PubMed]
- Witkiewicz, A.K.; Hecht, J.L.; Cviko, A.; McKeon, F.D.; Ince, T.A.; Crum, C.P. Microglandular hyperplasia: A model for the de novo emergence and evolution of endocervical reserve cells. Hum. Pathol. 2005, 36, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Palefsky, J.M.; Giuliano, A.R.; Goldstone, S.; Moreira, E.D., Jr.; Aranda, C.; Jessen, H.; Hillman, R.; Ferris, D.; Coutlee, F.; Stoler, M.H.; et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N. Engl. J. Med. 2011, 365, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.; Paavonen, J.; Wheeler, C.M.; Jaisamrarn, U.; Garland, S.M.; Castellsague, X.; Skinner, S.R.; Apter, D.; Naud, P.; Salmeron, J.; et al. Overall efficacy of HPV-16/18 AS04-adjuvanted vaccine against grade 3 or greater cervical intraepithelial neoplasia: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 2012, 13, 89–99. [Google Scholar] [CrossRef]
- Munoz, N.; Kjaer, S.K.; Sigurdsson, K.; Iversen, O.E.; Hernandez-Avila, M.; Wheeler, C.M.; Perez, G.; Brown, D.R.; Koutsky, L.A.; Tay, E.H.; et al. Impact of human papillomavirus (HPV)-6/11/16/18 vaccine on all HPV-associated genital diseases in young women. J. Natl. Cancer Inst. 2010, 102, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Castellsague, X.; Garland, S.M. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 2012, 30 (Suppl. 5), F123–F138. [Google Scholar] [CrossRef] [PubMed]
- De Sanjose, S.; Diaz, M.; Castellsague, X.; Clifford, G.; Bruni, L.; Munoz, N.; Bosch, F.X. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: A meta-analysis. Lancet Infect. Dis. 2007, 7, 453–459. [Google Scholar] [CrossRef]
- Chesson, H.W.; Ekwueme, D.U.; Saraiya, M.; Watson, M.; Lowy, D.R.; Markowitz, L.E. Estimates of the annual direct medical costs of the prevention and treatment of disease associated with human papillomavirus in the United States. Vaccine 2012, 30, 6016–6019. [Google Scholar] [CrossRef] [PubMed]
- Massad, L.S.; Einstein, M.H.; Huh, W.K.; Katki, H.A.; Kinney, W.K.; Schiffman, M.; Solomon, D.; Wentzensen, N.; Lawson, H.W.; Conference, A.C.G. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. Obstet. Gynecol. 2013, 121, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Sagasta, A.; Castillo, P.; Saco, A.; Torne, A.; Esteve, R.; Marimon, L.; Ordi, J.; Del Pino, M. p16 staining has limited value in predicting the outcome of histological low-grade squamous intraepithelial lesions of the cervix. Mod. Pathol. 2016, 29, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.D.; Sellors, J.W.; Sun, H.K.; Zhang, X.; Bao, Y.P.; Jeronimo, J.; Chen, W.; Zhao, F.H.; Song, Y.; Cao, Z.; et al. p16INK4A immunohistochemical staining and predictive value for progression of cervical intraepithelial neoplasia grade 1: A prospective study in China. Int. J. Cancer 2014, 134, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Oki, A.; Furuta, R.; Maeda, H.; Yasugi, T.; Takatsuka, N.; Mitsuhashi, A.; Fujii, T.; Hirai, Y.; Iwasaka, T.; et al. Predicting the progression of cervical precursor lesions by human papillomavirus genotyping: A prospective cohort study. Int. J. Cancer 2011, 128, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, M.; Milicic-Juhas, V.; Milojkovic, M.; Topolovec, Z.; Curzik, D.; Mihaljevic, I. Assessment of HPV DNA test value in management women with cytological findings of ASC-US, CIN1 and CIN2. Coll. Antropol. 2010, 34, 81–86. [Google Scholar] [PubMed]
- Gravitt, P.E.; Kovacic, M.B.; Herrero, R.; Schiffman, M.; Bratti, C.; Hildesheim, A.; Morales, J.; Alfaro, M.; Sherman, M.E.; Wacholder, S.; et al. High load for most high risk human papillomavirus genotypes is associated with prevalent cervical cancer precursors but only HPV16 load predicts the development of incident disease. Int. J. Cancer 2007, 121, 2787–2793. [Google Scholar] [CrossRef] [PubMed]
- Dalstein, V.; Riethmuller, D.; Pretet, J.L.; Le Bail Carval, K.; Sautiere, J.L.; Carbillet, J.P.; Kantelip, B.; Schaal, J.P.; Mougin, C. Persistence and load of high-risk HPV are predictors for development of high-grade cervical lesions: A longitudinal French cohort study. Int. J. Cancer 2003, 106, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, A.T.; Berkhof, J.; Heideman, D.A.; Bulkmans, N.W.; van Tellingen, J.E.; Meijer, C.J.; Snijders, P.J. High-risk human papillomavirus DNA load in a population-based cervical screening cohort in relation to the detection of high-grade cervical intraepithelial neoplasia and cervical cancer. Int. J. Cancer 2009, 124, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Koutsky, L.A.; Castle, P.E.; Wheeler, C.M.; Galloway, D.A.; Mao, C.; Ho, J.; Kiviat, N.B. Human papillomavirus type 18 DNA load and 2-year cumulative diagnoses of cervical intraepithelial neoplasia grades 2–3. J. Natl. Cancer Inst. 2009, 101, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Holowaty, P.; Miller, A.B.; Rohan, T.; To, T. Natural history of dysplasia of the uterine cervix. J. Natl. Cancer Inst. 1999, 91, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, N.F.; Platt, R.W.; Duarte-Franco, E.; Costa, M.C.; Sobrinho, J.P.; Prado, J.C.; Ferenczy, A.; Rohan, T.E.; Villa, L.L.; Franco, E.L. Human papillomavirus infection and time to progression and regression of cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2003, 95, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Crum, C.P. Cervical cancer: Squamocolumnar junction ablation—Tying up loose ends? Nat. Rev. Clin. Oncol. 2015, 12, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Somja, J.; Howitt, B.E.; Suarez-Carmona, M.; Kustermans, G.; Hubert, P.; Doyen, J.; Goffin, F.; Kridelka, F.; Crum, C.P.; et al. Unique recurrence patterns of cervical intraepithelial neoplasia after excision of the squamocolumnar junction. Int. J. Cancer 2015, 136, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S. Past and future of prophylactic ablation of the cervical squamocolumnar junction. Ecancermedicalscience 2015, 9, 527. [Google Scholar] [PubMed]
- Younge, P.A. Cancer of the uterine cervix; a preventable disease. Obstet. Gynecol. 1957, 10, 469–481. [Google Scholar] [PubMed]
- Peyton, F.W.; Peyton, R.R.; Anderson, V.L.; Pavnica, P. The importance of cauterization to maintain a healthy cervix. Long-term study from a private gynecologic practice. Am. J. Obstet. Gynecol. 1978, 131, 374–380. [Google Scholar] [CrossRef]
- Kauraniemi, T.; Rasanen-Virtanen, U.; Hakama, M. Risk of cervical cancer among an electrocoagulated population. Am. J. Obstet. Gynecol. 1978, 131, 533–538. [Google Scholar] [CrossRef]
- Taylor, S.; Wang, C.; Wright, T.C.; Denny, L.; Tsai, W.Y.; Kuhn, L. Reduced acquisition and reactivation of human papillomavirus infections among older women treated with cryotherapy: Results from a randomized trial in South Africa. BMC Med. 2010, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Kocken, M.; Helmerhorst, T.J.; Berkhof, J.; Louwers, J.A.; Nobbenhuis, M.A.; Bais, A.G.; Hogewoning, C.J.; Zaal, A.; Verheijen, R.H.; Snijders, P.J.; et al. Risk of recurrent high-grade cervical intraepithelial neoplasia after successful treatment: A long-term multi-cohort study. Lancet Oncol. 2011, 12, 441–450. [Google Scholar] [CrossRef]
- Herfs, M.; Hubert, P.; Moutschen, M.; Delvenne, P. Mucosal junctions: Open doors to HPV and HIV infections? Trends Microbiol. 2011, 19, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Auvert, B.; Lissouba, P.; Cutler, E.; Zarca, K.; Puren, A.; Taljaard, D. Association of oncogenic and nononcogenic human papillomavirus with HIV incidence. J. Acquir. Immune Defic. Syndr. 2010, 53, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Lissouba, P.; Van de Perre, P.; Auvert, B. Association of genital human papillomavirus infection with HIV acquisition: A systematic review and meta-analysis. Sex. Transm. Infect. 2013, 89, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Averbach, S.H.; Gravitt, P.E.; Nowak, R.G.; Celentano, D.D.; Dunbar, M.S.; Morrison, C.S.; Grimes, B.; Padian, N.S. The association between cervical human papillomavirus infection and HIV acquisition among women in Zimbabwe. Aids 2010, 24, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Smith-McCune, K.K.; Shiboski, S.; Chirenje, M.Z.; Magure, T.; Tuveson, J.; Ma, Y.; Da Costa, M.; Moscicki, A.B.; Palefsky, J.M.; Makunike-Mutasa, R.; et al. Type-specific cervico-vaginal human papillomavirus infection increases risk of HIV acquisition independent of other sexually transmitted infections. PLoS ONE 2010, 5, e10094. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The biology of beta human papillomaviruses. Virus Res. 2017, 231, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Mueller-Decker, K.; Kloz, U.; Aengeneyndt, B.; Kopp-Schneider, A.; Grone, H.J.; Gheit, T.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011, 7, e1002125. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Pirog, E.C.; Kleter, B.; Olgac, S.; Bobkiewicz, P.; Lindeman, J.; Quint, W.G.; Richart, R.M.; Isacson, C. Prevalence of human papillomavirus DNA in different histological subtypes of cervical adenocarcinoma. Am. J. Pathol. 2000, 157, 1055–1062. [Google Scholar] [CrossRef]
- Kajitani, N.; Satsuka, A.; Kawate, A.; Sakai, H. Productive Lifecycle of Human Papillomaviruses that Depends Upon Squamous Epithelial Differentiation. Front. Microbiol. 2012, 3, 152. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herfs, M.; Soong, T.R.; Delvenne, P.; Crum, C.P. Deciphering the Multifactorial Susceptibility of Mucosal Junction Cells to HPV Infection and Related Carcinogenesis. Viruses 2017, 9, 85. https://doi.org/10.3390/v9040085
Herfs M, Soong TR, Delvenne P, Crum CP. Deciphering the Multifactorial Susceptibility of Mucosal Junction Cells to HPV Infection and Related Carcinogenesis. Viruses. 2017; 9(4):85. https://doi.org/10.3390/v9040085
Chicago/Turabian StyleHerfs, Michael, Thing R. Soong, Philippe Delvenne, and Christopher P. Crum. 2017. "Deciphering the Multifactorial Susceptibility of Mucosal Junction Cells to HPV Infection and Related Carcinogenesis" Viruses 9, no. 4: 85. https://doi.org/10.3390/v9040085
APA StyleHerfs, M., Soong, T. R., Delvenne, P., & Crum, C. P. (2017). Deciphering the Multifactorial Susceptibility of Mucosal Junction Cells to HPV Infection and Related Carcinogenesis. Viruses, 9(4), 85. https://doi.org/10.3390/v9040085