
HBV DNA Integration: Molecular Mechanisms and Clinical Implications



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Hepatitis B Virus Infection
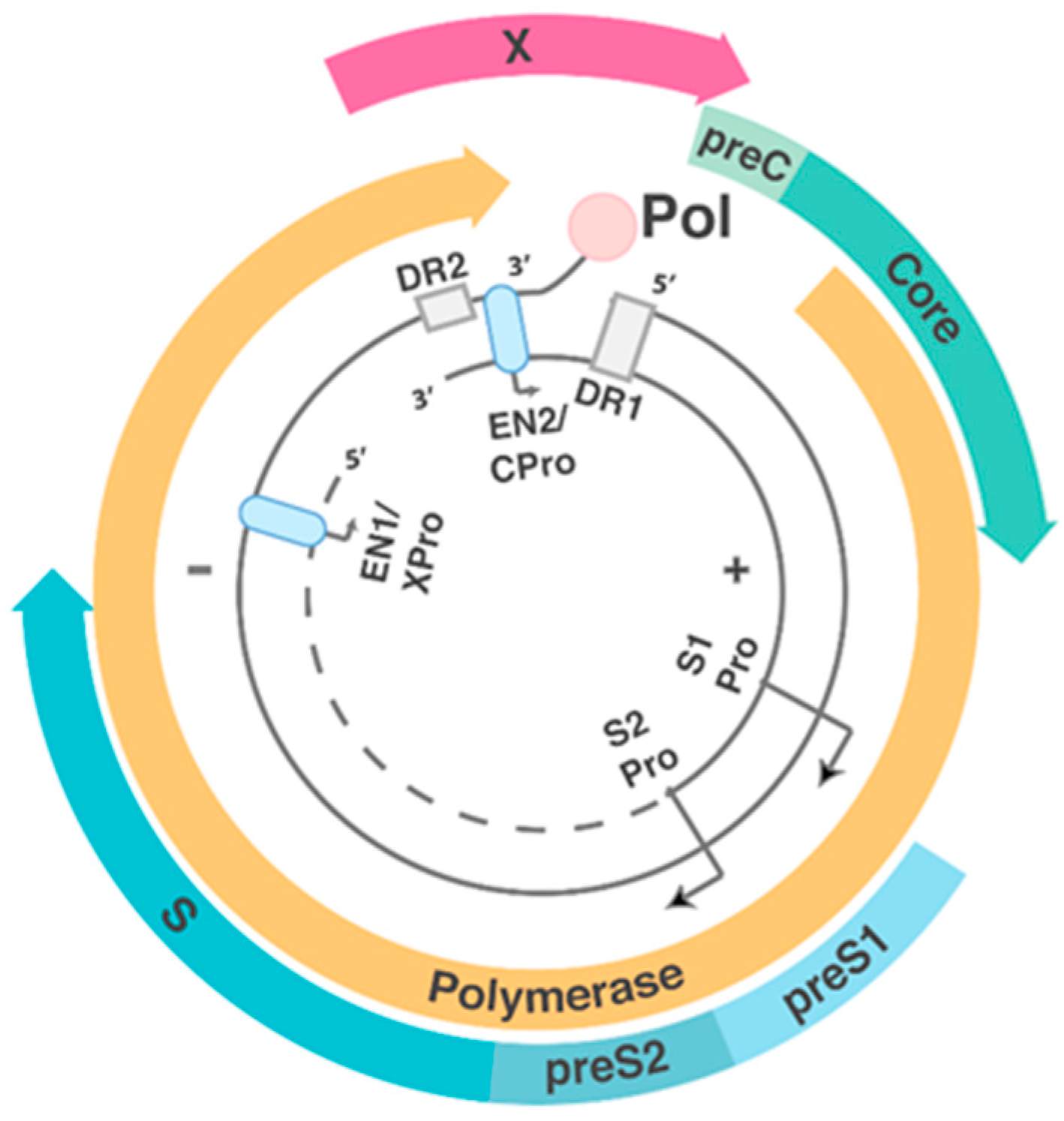
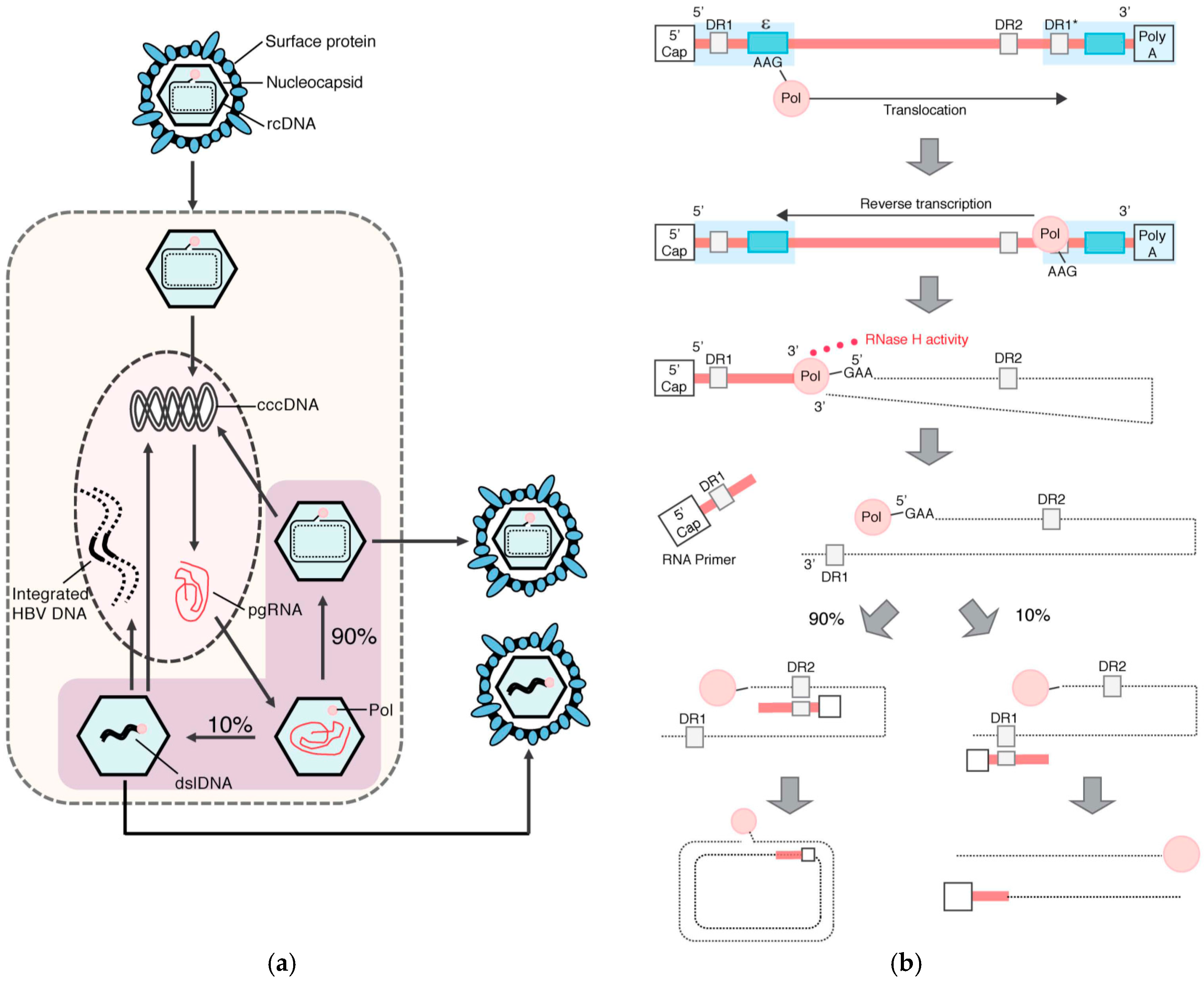
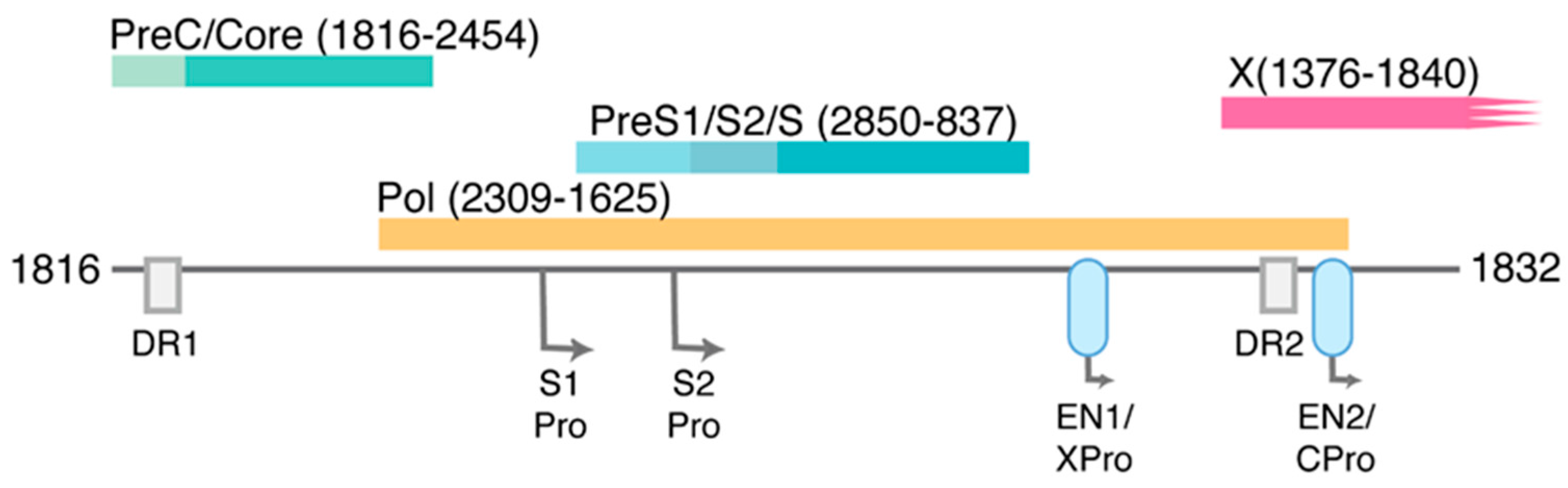
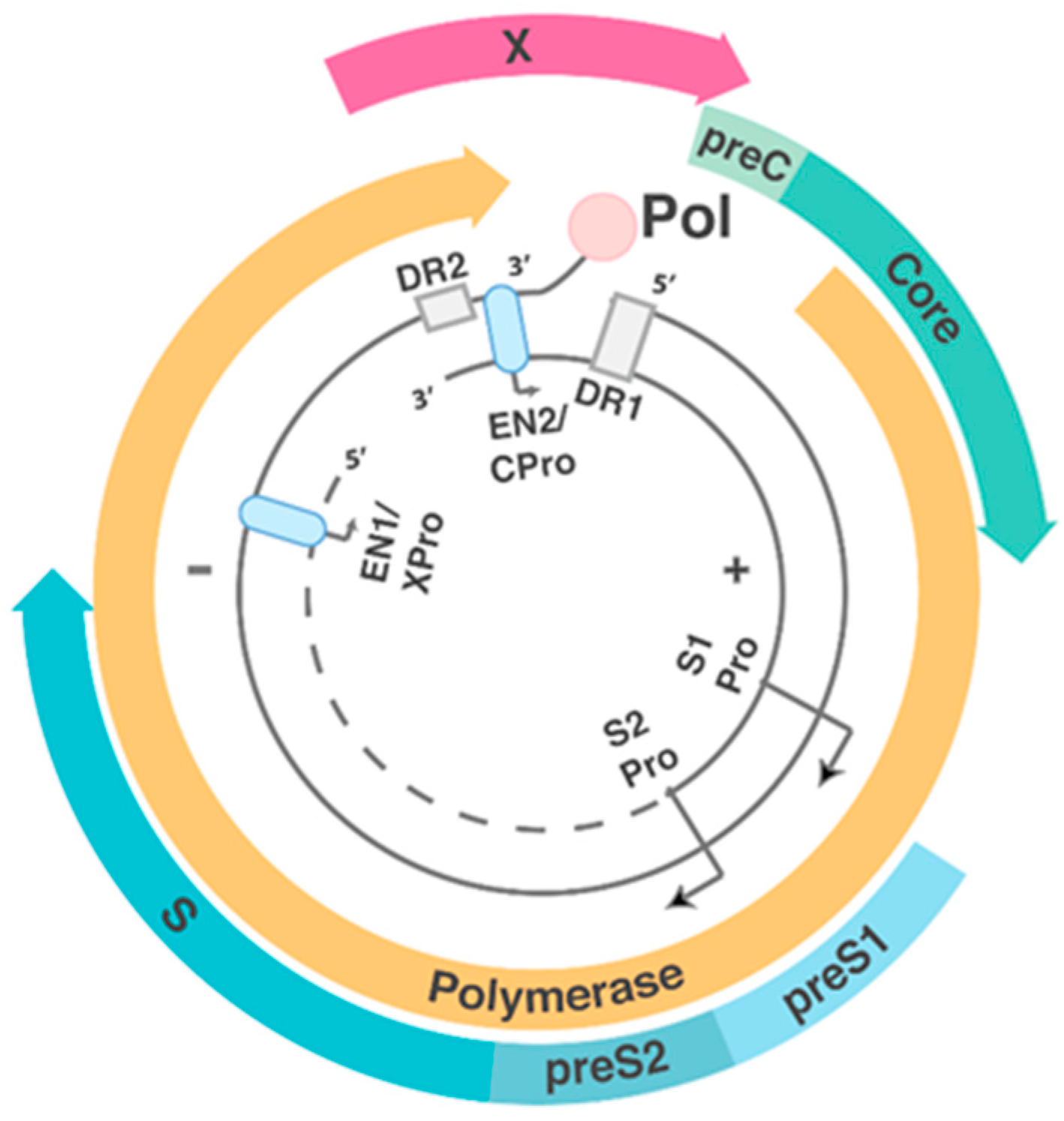
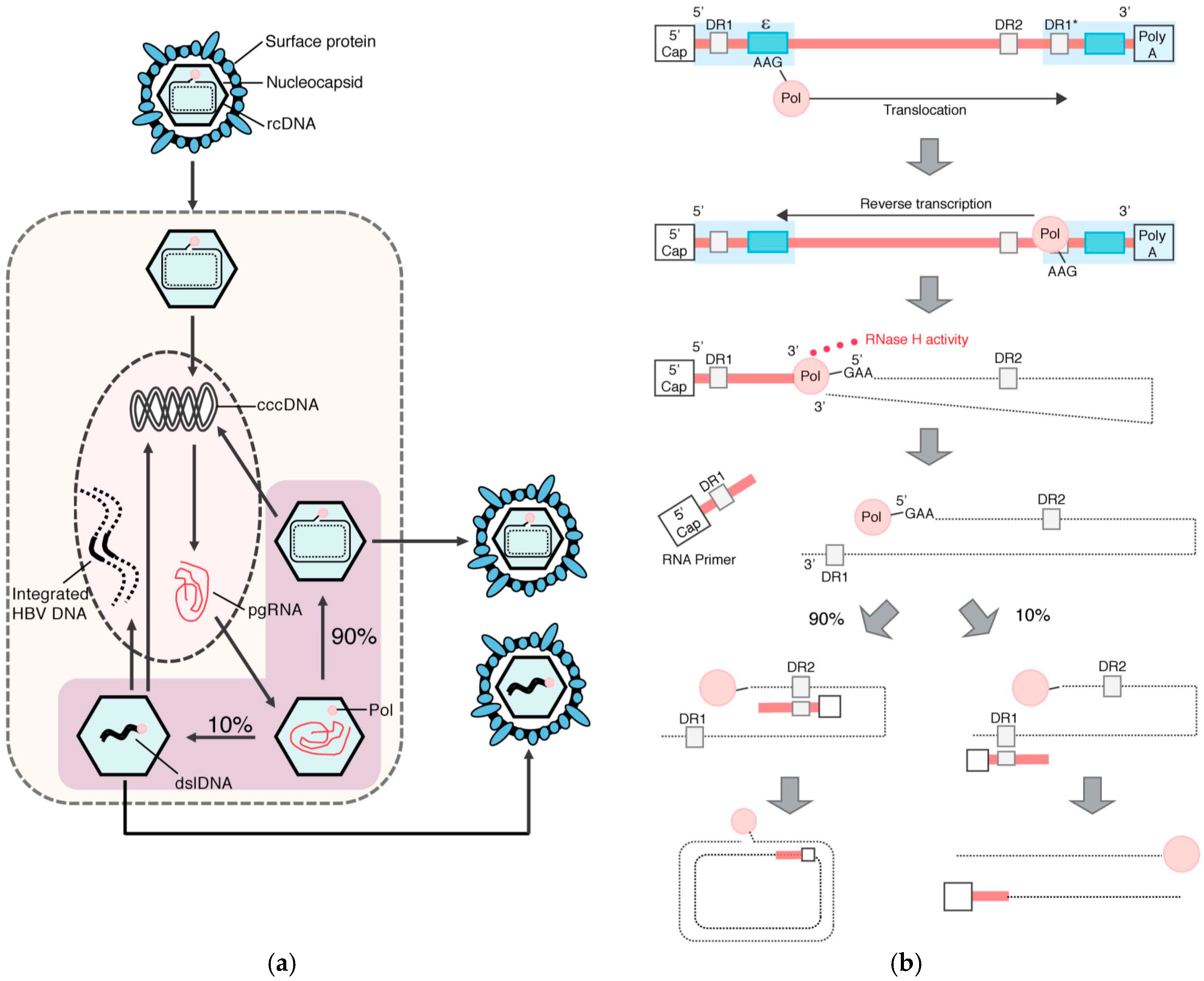
1.2. HBV Structure and Replication Cycle
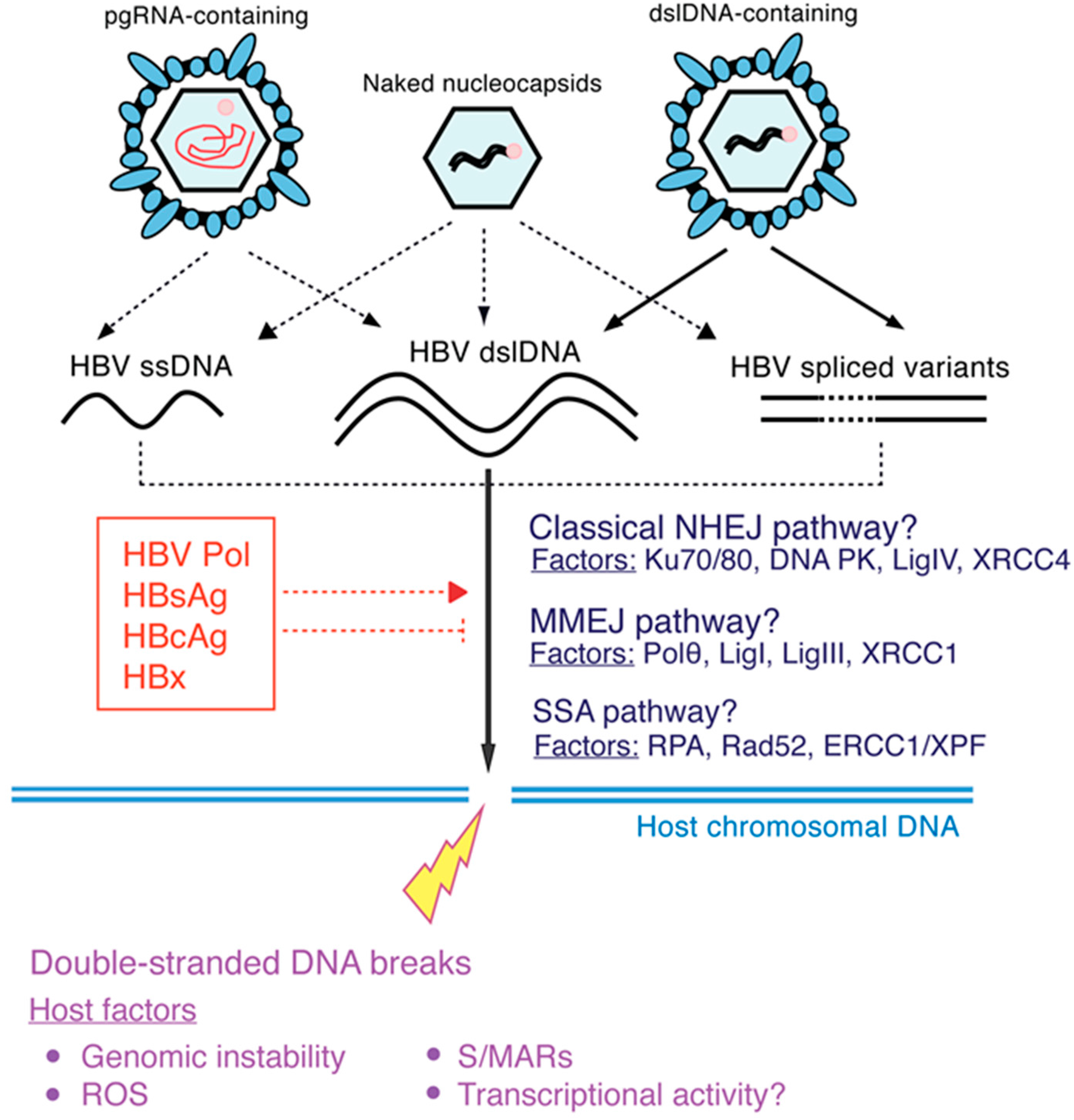
2. HBV DNA Integration
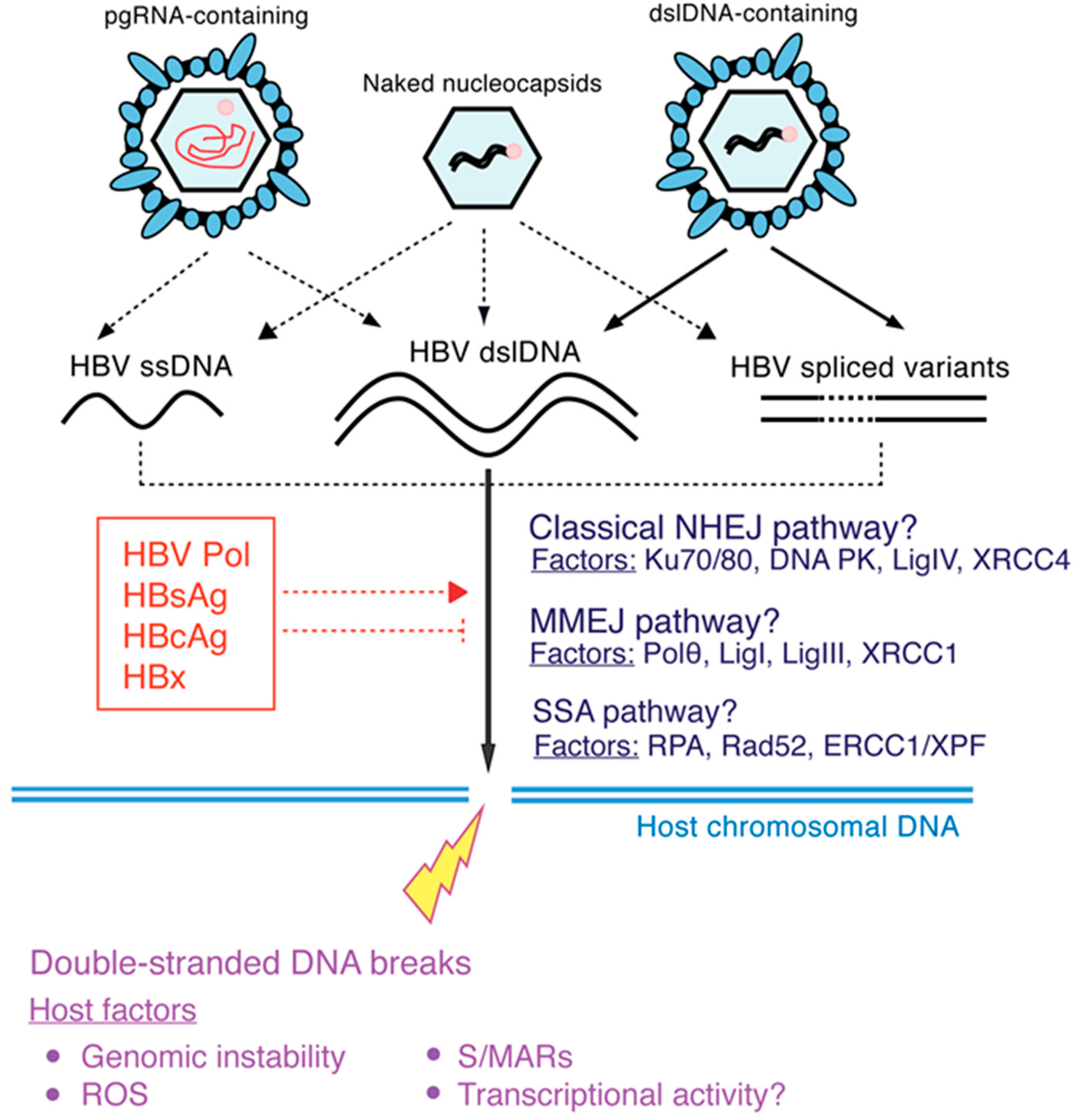
2.1. Molecular Aspects of HBV DNA Integration
2.2. The Role of HBV Integration in Viral Infection
2.3. The Role of HBV Integration in HCC Initiation and Progression
2.3.1. Insertional Mutagenesis
2.3.2. Chromosomal Instability
2.3.3. Expression of HBV Proteins with Oncogenic Potential from Integrated HBV DNA
3. Unanswered Questions
3.1. What Are the Dynamics of HBV DNA Integration throughout HBV Infection?
3.2. How Does Integration Occur? What Cellular and Viral Factors Affect Integration?
3.3. To What Extent Does Integration Contribute to HBV Replication and Persistence?
3.4. Do Integrated Forms Persisting after Functional Cure of CHB Contribute to HCC Risk?
3.5. Can Integrated HBV DNA Contribute to the Replication of Hepatitis Delta Virus (HDV)?
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the global burden of disease study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis b and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host-virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. Easl clinical practice guidelines: Management of chronic hepatitis B virus infection. J. Hepatol. 2012, 57, 167–185. [Google Scholar]
- Terrault, N.A.; Bzowej, N.H.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Murad, M.H. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016, 63, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Chien, M.H.; Huang, H.P.; Chang, H.C.; Wu, C.C.; Chen, P.J.; Chang, M.H.; Chen, D.S. Distinct hepatitis B virus dynamics in the immunotolerant and early immunoclearance phases. J. Virol. 2010, 84, 3454–3463. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.T.; Sandalova, E.; Jo, J.; Gill, U.; Ushiro-Lumb, I.; Tan, A.T.; Naik, S.; Foster, G.R.; Bertoletti, A. Preserved T-cell function in children and young adults with immune-tolerant chronic hepatitis B. Gastroenterology 2012, 143, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Sandalova, E.; Low, D.; Gehring, A.J.; Fieni, S.; Amadei, B.; Urbani, S.; Chong, Y.S.; Guccione, E.; Bertoletti, A. Trained immunity in newborn infants of HBV-infected mothers. Nat. Commun. 2015, 6, 6588. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Kennedy, P.T. The immune tolerant phase of chronic HBV infection: New perspectives on an old concept. Cell. Mol. Immunol. 2015, 12, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Liaw, Y.F.; Chu, C.M. Immune tolerance phase of chronic hepatitis B. Gastroenterology 2017, 152, 1245–1246. [Google Scholar] [CrossRef] [PubMed]
- Milich, D.R. The concept of immune tolerance in chronic hepatitis B virus infection is alive and well. Gastroenterology 2016, 151, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Guidotti, L.G. Hepatitis B virus immunopathogenesis. In Hepatitis B Virus in Human Diseases; Liaw, Y.-F., Zoulim, F., Eds.; Humana Press: New York, NY, USA, 2016; pp. 79–93. [Google Scholar]
- Invernizzi, F.; Vigano, M.; Grossi, G.; Lampertico, P. The prognosis and management of inactive HBV carriers. Liver Int. 2016, 36 (Suppl. 1), 100–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, H.I.; Lee, M.H.; Jen, C.L.; Batrla-Utermann, R.; Lu, S.N.; Wang, L.Y.; You, S.L.; Chen, C.J. Serum levels of hepatitis B surface antigen and DNA can predict inactive carriers with low risk of disease progression. Hepatology 2016, 64, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Liaw, Y.F. Hepatitis b surface antigen seroclearance during chronic HBV infection. Antivir. Ther. 2010, 15, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Raffetti, E.; Fattovich, G.; Donato, F. Incidence of hepatocellular carcinoma in untreated subjects with chronic hepatitis B: A systematic review and meta-analysis. Liver Int. 2016, 36, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Urban, S.; Knoop, E.V.; Cag, N.; Krass, P.; Grun, S.; Bulavaite, A.; Sasnauskas, K.; Gerlich, W.H. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 2005, 129, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Le Duff, Y.; Blanchet, M.; Sureau, C. The pre-s1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J. Virol. 2009, 83, 12443–12451. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Beterams, G.; Nassal, M. Significant interference with hepatitis B virus replication by a core-nuclease fusion protein. J. Biol. Chem. 2001, 276, 8875–8883. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, T.; Huang, X.; Kumar, G.R.; Chen, X.; Zeng, Z.; Zhang, R.; Chen, R.; Li, T.; Zhang, T.; et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J. Hepatol. 2016, 65, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shin, M.K.; Lee, H.J.; Yoon, G.; Ryu, W.S. Three novel cis-acting elements required for efficient plus-strand DNA synthesis of the hepatitis B virus genome. J. Virol. 2004, 78, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.L.; Yang, J.R.; Lin, S.Z.; Ma, H.; Guo, F.; Yang, R.F.; Zhang, H.H.; Han, J.C.; Wei, L.; Pan, X.B. Serum viral duplex-linear DNA proportion increases with the progression of liver disease in patients infected with HBV. Gut 2016, 65, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Lewellyn, E.B.; Loeb, D.D. Base pairing between cis-acting sequences contributes to template switching during plus-strand DNA synthesis in human hepatitis B virus. J. Virol. 2007, 81, 6207–6215. [Google Scholar] [CrossRef] [PubMed]
- Haines, K.M.; Loeb, D.D. The sequence of the RNA primer and the DNA template influence the initiation of plus-strand DNA synthesis in hepatitis B virus. J. Mol. Biol. 2007, 370, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Infection of ducklings with virus particles containing linear double-stranded duck hepatitis B virus DNA: Illegitimate replication and reversion. J. Virol. 1998, 72, 8710–8717. [Google Scholar] [PubMed]
- Yang, W.; Summers, J. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J. Virol. 1995, 69, 4029–4036. [Google Scholar] [PubMed]
- Yang, W.; Mason, W.S.; Summers, J. Covalently closed circular viral DNA formed from two types of linear DNA in woodchuck hepatitis virus-infected liver. J. Virol. 1996, 70, 4567–4575. [Google Scholar] [PubMed]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [PubMed]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Mason, W.S. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc. Natl. Acad. Sci. USA 2004, 101, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal expansion of normal-appearing human hepatocytes during chronic hepatitis B virus infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Edman, J.C.; Gray, P.; Valenzuela, P.; Rall, L.B.; Rutter, W.J. Integration of hepatitis B virus sequences and their expression in a human hepatoma cell. Nature 1980, 286, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Twist, E.M.; Clark, H.F.; Aden, D.P.; Knowles, B.B.; Plotkin, S.A. Integration pattern of hepatitis B virus DNA sequences in human hepatoma cell lines. J. Virol. 1981, 37, 239–243. [Google Scholar] [PubMed]
- Lee, J.H.; Ku, J.L.; Park, Y.J.; Lee, K.U.; Kim, W.H.; Park, J.G. Establishment and characterization of four human hepatocellular carcinoma cell lines containing hepatitis B virus DNA. World J. Gastroenterol. 1999, 5, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Shamay, M.; Agami, R.; Shaul, Y. HBV integrants of hepatocellular carcinoma cell lines contain an active enhancer. Oncogene 2001, 20, 6811–6819. [Google Scholar] [CrossRef] [PubMed]
- Schluter, V.; Meyer, M.; Hofschneider, P.H.; Koshy, R.; Caselmann, W.H. Integrated hepatitis B virus X and 3’ truncated preS/S sequences derived from human hepatomas encode functionally active transactivators. Oncogene 1994, 9, 3335–3344. [Google Scholar] [PubMed]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A truncated mutant (residues 58–140) of the hepatitis B virus X protein retains transactivation function. Proc. Natl. Acad. Sci. USA 1996, 93, 5647–5652. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.B.; Liu, M.S.; Chang, P.C.; Wu, S.M.; Su, M.W.; Pan, C.C.; Han, S.H. Analysis of six distinct integrated hepatitis B virus sequences cloned from the cellular DNA of a human hepatocellular carcinoma. Virology 1986, 154, 405–408. [Google Scholar] [CrossRef]
- Koch, S.; von Loringhoven, A.F.; Kahmann, R.; Hofschneider, P.H.; Koshy, R. The genetic organization of integrated hepatitis B virus DNA in the human hepatoma cell line PLC/PRF/5. Nucleic Acids Res. 1984, 12, 6871–6886. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Kobayashi, M.; Mizusawa, H.; Yoshida, E.; Yaginuma, K.; Taira, M. Rearrangement of the surface antigen gene of hepatitis B virus integrated in the human hepatoma cell lines. Nucleic Acids Res. 1983, 11, 5391–5402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Z.; Butel, J.S.; Li, P.J.; Finegold, M.J.; Melnick, J.L. Integrated state of subgenomic fragments of hepatitis B virus DNA in hepatocellular carcinoma from mainland china. J. Natl. Cancer Inst. 1987, 79, 223–231. [Google Scholar] [PubMed]
- Takada, S.; Gotoh, Y.; Hayashi, S.; Yoshida, M.; Koike, K. Structural rearrangement of integrated hepatitis B virus DNA as well as cellular flanking DNA is present in chronically infected hepatic tissues. J. Virol. 1990, 64, 822–828. [Google Scholar] [PubMed]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.S.; Jensen, A.D.; Chang, C.J.; Rogler, C.E. Double-stranded linear duck hepatitis B virus (DHBV) stably integrates at a higher frequency than wild-type DHBV in LMH chicken hepatoma cells. J. Virol. 1999, 73, 1492–1502. [Google Scholar] [PubMed]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens. N. Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Hino, O.; Kitagawa, T.; Koike, K.; Kobayashi, M.; Hara, M.; Mori, W.; Nakashima, T.; Hattori, N.; Sugano, H. Detection of hepatitis B virus DNA in hepatocellular carcinomas in Japan. Hepatology 1984, 4, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J.; Greenfield, C.; Chu, C.M.; Karayiannis, P.; Dunk, A.; Lok, A.S.; Lai, C.L.; Yeoh, E.K.; Monjardino, J.P.; Wankya, B.M.; et al. Integration of HBV-DNA may not be a prerequisite for the maintenance of the state of malignant transformation. An analysis of 110 liver biopsies. J. Hepatol. 1986, 2, 218–229. [Google Scholar] [CrossRef]
- Chen, J.Y.; Harrison, T.J.; Lee, C.S.; Chen, D.S.; Zuckerman, A.J. Detection of hepatitis B virus DNA in hepatocellular carcinoma: Analysis by hybridization with subgenomic DNA fragments. Hepatology 1988, 8, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Esumi, M.; Tanaka, Y.; Tozuka, S.; Shikata, T. Clonal state of human hepatocellular carcinoma and non-tumorous hepatocytes. Cancer Chemother. Pharmacol. 1989, 23 (Suppl. 1), S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Brechot, C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: Old and new paradigms. Gastroenterology 2004, 127, S56–S61. [Google Scholar] [CrossRef] [PubMed]
- Paterlini-Brechot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Brechot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Koshy, R.; Maupas, P.; Muller, R.; Hofschneider, P.H. Detection of hepatitis B virus-specific DNA in the genomes of human hepatocellular carcinoma and liver cirrhosis tissues. J. Gen. Virol. 1981, 57, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Scotto, J.; Hadchouel, M.; Hery, C.; Alvarez, F.; Yvart, J.; Tiollais, P.; Bernard, O.; Brechot, C. Hepatitis B virus DNA in children’s liver diseases: Detection by blot hybridisation in liver and serum. Gut 1983, 24, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, K.; Kobayashi, H.; Kobayashi, M.; Morishima, T.; Matsuyama, K.; Koike, K. Multiple integration site of hepatitis B virus DNA in hepatocellular carcinoma and chronic active hepatitis tissues from children. J. Virol. 1987, 61, 1808–1813. [Google Scholar] [PubMed]
- Kimbi, G.C.; Kramvis, A.; Kew, M.C. Integration of hepatitis B virus DNA into chromosomal DNA during acute hepatitis B. World J. Gastroenterol. 2005, 11, 6416–6421. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Jilbert, A.R. Conceptual models for the initiation of hepatitis B virus-associated hepatocellular carcinoma. Liver Int. 2015, 35, 1786–1800. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Fourel, G.; Ponzetto, A.; Silvestro, M.; Tiollais, P.; Buendia, M.A. Hepadnavirus integration: Mechanisms of activation of the N-myc2 retrotransposon in woodchuck liver tumors. J. Virol. 1992, 66, 5265–5276. [Google Scholar] [PubMed]
- Wei, Y.; Ponzetto, A.; Tiollais, P.; Buendia, M.A. Multiple rearrangements and activated expression of c-myc induced by woodchuck hepatitis virus integration in a primary liver tumour. Res. Virol. 1992, 143, 89–96. [Google Scholar] [CrossRef]
- Bruni, R.; D’Ugo, E.; Giuseppetti, R.; Argentini, C.; Rapicetta, M. Activation of the N-myc2 oncogene by woodchuck hepatitis virus integration in the linked downstreamb3nLocus in woodchuck hepatocellular carcinoma. Virology 1999, 257, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Popper, H.; Roth, L.; Purcell, R.H.; Tennant, B.C.; Gerin, J.L. Hepatocarcinogenicity of the woodchuck hepatitis virus. Proc. Natl. Acad. Sci. USA 1987, 84, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Yang, Z.; Kang, J.; Jiang, S.; Zhang, T.; Chen, T.; Li, M.; Lv, Q.; Chen, X.; et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J. Hepatol. 2014, 60, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Fujimoto, A.; Furuta, M.; Tanaka, H.; Chiba, K.; Boroevich, K.A.; Abe, T.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS ONE 2014, 9, e114263. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Lou, X.; Hua, D.; Yu, W.; Li, L.; Wang, J.; Gao, F.; Zhao, N.; Ren, G.; Li, L.; et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet 2012, 8, e1003065. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yang, Z.; Li, W.; Li, X.; Wang, Y.; Zhang, J.; Xu, C.; Chen, P.J.; Hou, J.; McCrae, M.A.; et al. Re-evaluation of the carcinogenic significance of hepatitis B virus integration in hepatocarcinogenesis. PLoS ONE 2012, 7, e40363. [Google Scholar] [CrossRef] [PubMed]
- Budzinska, M.A.; Tu, T.; d’Avigdor, W.M.; McCaughan, G.W.; Luciani, F.; Shackel, N.A. Accumulation of deleterious passenger mutations is associated with the progression of hepatocellular carcinoma. PLoS ONE 2016, 11, e0162586. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Tokino, T. Integration of hepatitis B virus DNA and its implications for hepatocarcinogenesis. Mol. Biol. Med. 1990, 7, 243–260. [Google Scholar] [PubMed]
- Dong, H.; Zhang, L.; Qian, Z.; Zhu, X.; Zhu, G.; Chen, Y.; Xie, X.; Ye, Q.; Zang, J.; Ren, Z.; et al. Identification of HBV-MLL4 integration and its molecular basis in chinese hepatocellular carcinoma. PLoS ONE 2015, 10, e0123175. [Google Scholar] [CrossRef] [PubMed]
- Saigo, K.; Yoshida, K.; Ikeda, R.; Sakamoto, Y.; Murakami, Y.; Urashima, T.; Asano, T.; Kenmochi, T.; Inoue, I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum. Mutat. 2008, 29, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Shera, K.A.; Shera, C.A.; McDougall, J.K. Small tumor virus genomes are integrated near nuclear matrix attachment regions in transformed cells. J. Virol. 2001, 75, 12339–12346. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Seo, D.; Andersen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Imbeaud, S.; Datta, S.; Zucman-Rossi, J. Authors’ response: Virus-host interactions in HBV-related hepatocellular carcinoma: More to be revealed? Gut 2015, 64, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; D’Ugo, E.; Argentini, C.; Giuseppetti, R.; Rapicetta, M. Scaffold attachment region located in a locus targeted by hepadnavirus integration in hepatocellular carcinomas. Cancer Detect. Prev. 2003, 27, 175–181. [Google Scholar] [CrossRef]
- Levy, L.; Renard, C.A.; Wei, Y.; Buendia, M.A. Genetic alterations and oncogenic pathways in hepatocellular carcinoma. Ann. N. Y. Acad. Sci. 2002, 963, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Bonilla Guerrero, R.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Huang, W.; Lai, M.D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- Fan, Y.F.; Lu, C.C.; Chen, W.C.; Yao, W.J.; Wang, H.C.; Chang, T.T.; Lei, H.Y.; Shiau, A.L.; Su, I.J. Prevalence and significance of hepatitis B virus (HBV) pre-s mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001, 33, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Huang, W.; Lai, M.D.; Su, I.J. Aberrant cyclin a expression and centrosome overduplication induced by hepatitis B virus pre-S2 mutants and its implication in hepatocarcinogenesis. Carcinogenesis 2012, 33, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Su, I.J.; Wang, H.C.; Wu, H.C.; Huang, W.Y. Ground glass hepatocytes contain pre-s mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, C.; Bhavani, K.; Chisari, F.V. Multiple oncogenes and tumor suppressor genes are structurally and functionally intact during hepatocarcinogenesis in hepatitis B virus transgenic mice. Cancer Res. 1992, 52, 2823–2829. [Google Scholar] [PubMed]
- Chisari, F.V.; Filippi, P.; Buras, J.; McLachlan, A.; Popper, H.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc. Natl. Acad. Sci. USA 1987, 84, 6909–6913. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.Y.; Chai, S.; Tong, M.; Guan, X.Y.; Lin, C.H.; Ching, Y.P.; Xie, D.; Cheng, A.S.; Ma, S. C-terminal truncated hepatitis B virus X protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 2016, 7, 24005–24017. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Bonura, C.; Giannini, C.; Mouly, H.; Soussan, P.; Kew, M.; Paterlini-Brechot, P.; Brechot, C.; Kremsdorf, D. Biological impact of natural cooh-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res. 2001, 61, 7803–7810. [Google Scholar] [PubMed]
- Ma, N.F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.C.; Fung, J.; Bai, X.; et al. Cooh-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, W.; Liu, Q.; Zhang, X.; Lv, N.; Ye, L.; Zhang, X. A mutant of hepatitis B virus X protein (hbxdelta127) promotes cell growth through a positive feedback loop involving 5-lipoxygenase and fatty acid synthase. Neoplasia 2010, 12, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Sze, K.M.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus X protein is associated with metastasis and enhances invasiveness by c-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.; Ou, J.H.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBX) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Villarreal, D.; Shim, E.Y.; Lee, S.E. Risky business: Microhomology-mediated end joining. Mutat. Res. 2016, 788, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Abraham, T.M.; Lewellyn, E.B.; Haines, K.M.; Loeb, D.D. Characterization of the contribution of spliced RNAs of hepatitis B virus to DNA synthesis in transfected cultures of Huh7 and HepG2 cells. Virology 2008, 379, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Zhang, C.; Han, Q.; Zhang, J.; Tian, Z. Therapeutic recovery of hepatitis B virus (HBV)-induced hepatocyte-intrinsic immune defect reverses systemic adaptive immune tolerance. Hepatology 2013, 58, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Michler, T.; Grosse, S.; Mockenhaupt, S.; Roder, N.; Stuckler, F.; Knapp, B.; Ko, C.; Heikenwalder, M.; Protzer, U.; Grimm, D. Blocking sense-strand activity improves potency, safety and specificity of anti-hepatitis B virus short hairpin RNA. EMBO Mol. Med. 2016, 8, 1082–1098. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sun, S.; Li, M.; Cheng, X.; Li, H.; Kang, F.; Kang, J.; Dornbrack, K.; Nassal, M.; Sun, D. Suppression of hepatitis B virus antigen production and replication by wild-type HBV dependently replicating HBV shRNA vectors in vitro and in vivo. Antivir. Res. 2016, 134, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Poussin, K.; Dienes, H.; Sirma, H.; Urban, S.; Beaugrand, M.; Franco, D.; Schirmacher, P.; Brechot, C.; Paterlini Brechot, P. Expression of mutated hepatitis B virus X genes in human hepatocellular carcinomas. Int J Cancer 1999, 80, 497–505. [Google Scholar] [CrossRef]
- Su, Q.; Schroder, C.H.; Hofmann, W.J.; Otto, G.; Pichlmayr, R.; Bannasch, P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology 1998, 27, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Coppola, N.; Onorato, L.; Iodice, V.; Starace, M.; Minichini, C.; Farella, N.; Liorre, G.; Filippini, P.; Sagnelli, E.; Stefano, G. Occult HBV infection in HCC and cirrhotic tissue of HBsAg-negative patients: A virological and clinical study. Oncotarget 2016, 7, 62706–62714. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Squadrito, G.; Cerenzia, G.; Cacciola, I.; Raffa, G.; Craxi, A.; Farinati, F.; Missale, G.; Smedile, A.; Tiribelli, C.; et al. Hepatitis B virus maintains its pro-oncogenic properties in the case of occult HBV infection. Gastroenterology 2004, 126, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, G.; Caccamo, G.; Filomia, R.; Pollicino, T. Occult HBV infection. Semin. Immunopathol. 2013, 35, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis delta virus: Insights into a peculiar pathogen and novel treatment options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Bortolotti, F.; Donato, F. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J. Hepatol. 2008, 48, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Giustina, G.; Christensen, E.; Pantalena, M.; Zagni, I.; Realdi, G.; Schalm, S.W. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European concerted action on viral hepatitis (eurohep). Gut 2000, 46, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Freitas, N.; Cunha, C.; Menne, S.; Gudima, S.O. Envelope proteins derived from naturally integrated hepatitis B virus DNA support assembly and release of infectious hepatitis delta virus particles. J. Virol. 2014, 88, 5742–5754. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. https://doi.org/10.3390/v9040075
Tu T, Budzinska MA, Shackel NA, Urban S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses. 2017; 9(4):75. https://doi.org/10.3390/v9040075
Chicago/Turabian StyleTu, Thomas, Magdalena A. Budzinska, Nicholas A. Shackel, and Stephan Urban. 2017. "HBV DNA Integration: Molecular Mechanisms and Clinical Implications" Viruses 9, no. 4: 75. https://doi.org/10.3390/v9040075
APA StyleTu, T., Budzinska, M. A., Shackel, N. A., & Urban, S. (2017). HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses, 9(4), 75. https://doi.org/10.3390/v9040075