How to Name and Classify Your Phage: An Informal Guide


{kind=link}
Abstract
:1. Introduction
2. A Short, Informal Guide to Naming and Classifying Your Phage
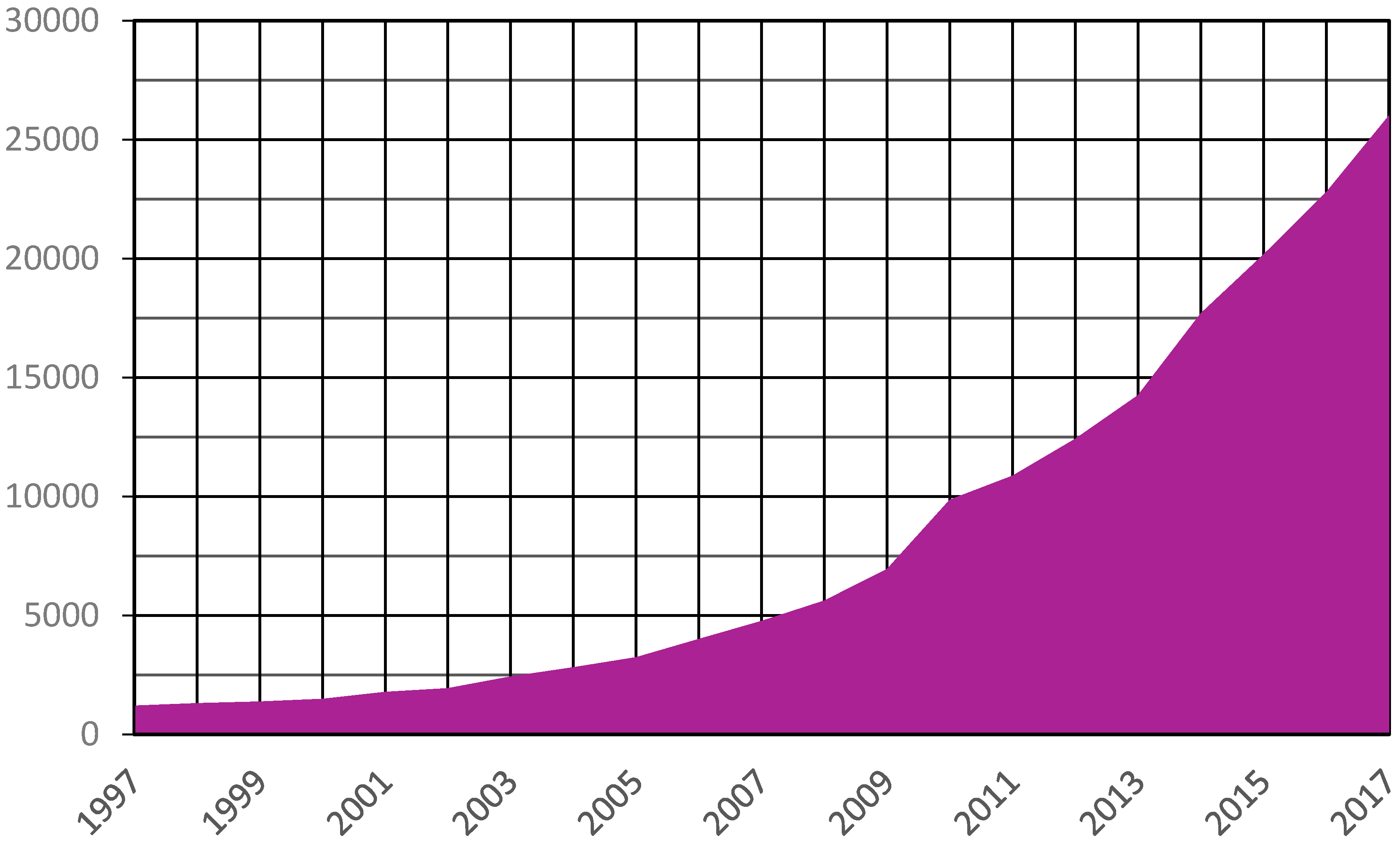
2.1. Congratulations, You Have Isolated a Bacteriophage!
- Always use the complete host genus name, followed by a space, followed by the word “phage”, followed by a space, followed by a unique identifier (e.g., Escherichia phage T4).
- Use only the isolation host genus in the name, rather than higher order taxa names—such as Enterobacteria, Pseudomonad, or the generic Bacteriophage—or lower order taxa names like Staphylococcus aureus DSM 1234. This approach to species naming was officially adopted by the BAVS in 2015 and should also be used in isolate names [18].
- Do not combine the host genus and the word “phage” into a single word, for example, Mycobacteriophage, Mycophage, etc.
- Do not use an existing unique identifier in the name.
- Do not use Greek letters in the unique identifier.
- Do not start the unique identifier with a numeral and do not use only use only a single letter. Identifiers should include enough complexity to easily distinguish your bacteriophage from others.
- Do not use hyphens, slashes, or any type of special character (e.g., %, $, @, etc.). You may use underscores to separate parts of the designation (e.g., vs_p123_233), but these underscores cannot be carried over into official taxon names (see Section 2.4).
- Do not use controversial names/phrases, profanity, names of prominent people, and trademarked names/phrases as unique identifiers.
- Please do contact the friendly folks on the BAVS if you have any questions.
2.2. What Is the Relationship between Bacteriophage Isolate Names and Taxa Names?
2.3. Now It Is Time to Publish Your Phage Sequence
- Do include lineage information for all submitted sequences. Even if your bacteriophage is novel and does not belong to a described species, provide the most accurate lineage information possible that places the sequence including genus and/or family using the criteria discussed in this manuscript.
- Do include accurate genomic composition information when no other lineage information is available or can be inferred. In most cases, it should be possible to place a new isolate within the higher-order dsDNA, ssDNA, dsRNA, or ssRNA lineage groupings.
- Do identify prophages using the “proviral” location descriptor.
- If you have questions about sequence submission to INSDC databases, please see The GenBank Submissions Handbook [27].
- If you still have questions, contact GenBank or another INSDC database.
2.4. Classifying Bacteriophage
2.4.1. Does My Phage Represent a New Species?
2.4.2. Is My Phage a Member of an Existing Genus?
2.4.3. What about Subfamilies and Families?
2.4.4. My Phage/Virus Does Not Fit in Anywhere, What Now?
2.5. Proposed Software to Use
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses (ICTV). Available online: http://www.ictvonline.org/ (accessed on 23 February 2017).
- Bacterial and Archaeal Viruses Subcommittee. Available online: https://talk.ictvonline.org/information/w/members/441/bacterial-and-archaeal-viruses-subcommittee (accessed on 23 February 2017).
- Bradley, D.E. Ultrastructure of bacteriophage and bacteriocins. Bacteriol. Rev. 1967, 31, 230–314. [Google Scholar] [PubMed]
- Ackermann, H.-W. Classification of bacteriophages. In The Bacteriophages, 2nd ed.; Calendar, R., Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 8–17. [Google Scholar]
- Francki, R.; Fauquet, C.; Knudson, D.; Brown, F. Classification and Nomenclature of Viruses: Fifth Report of the International Committee on the Taxonomy of Viruses; Springer: Wien, NY, USA, 1991. [Google Scholar]
- Fauquet, M.C.; Mayo, A.M. The 7th ICTV Report. Arch. Virol. 2001, 146, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Fauquet, C.; Mayo, M.; Maniloff, J.; Desselberger, U.; Ball, L. Virus Taxonomy: Eighth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: San Diego, CA, USA, 2005. [Google Scholar]
- Nelson, D. Phage taxonomy: We agree to disagree. J. Bacteriol. 2004, 186, 7029–7031. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A genome-based taxonomy for phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 2008, 25, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Pride, D.T.; Wassenaar, T.M.; Ghose, C.; Blaser, M.J. Evidence of host-virus co-evolution in tetranucleotide usage patterns of bacteriophages and eukaryotic viruses. BMC Genom. 2006, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Asare, P.T.; Jeong, T.; Ryu, S.; Klumpp, J.; Loessner, M.J.; Merrill, B.D.; Kim, K. Putative type 1 thymidylate synthase and dihydrofolate reductase as signature genes of a novel bastille-like group of phages in the subfamily Spounavirinae. BMC Genom. 2015, 16, 582. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2014, 468–470, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Edwards, R.; Nash, J.H.E.; Mahadevan, P.; Seto, D.; Ackermann, H.-W.; Lavigne, R.; Kropinski, A.M. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology 2015, 477, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.-W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 4, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Krupovic, M.; Knezevic, P.; Ackermann, H.-W.; Barylski, J.; Brister, J.R.; Clokie, M.R.C.; Duffy, S.; Dutilh, B.E.; Edwards, R.A.; et al. Taxonomy of prokaryotic viruses: 2016 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Virus Taxonomy: 2015 Release. Available online: http://talk.ictvonline.org/taxonomy/ (accessed on 23 February 2017).
- Karsch-Mizrachi, I.; Nakamura, Y.; Cochrane, G. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2012, 40, D33–D37. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W.; Abedon, S.T. Bacteriophage Names 2000. Available online: http://www.phage.org/names/2000/ (accessed on 23 February 2017).
- Abedon, S.T. Phage Name Check. Available online: http://www.phage.org/phage_name_check.html (accessed on 28 March 2017).
- NCBI Nucleotide Database. Available online: http://www.ncbi.nlm.nih.gov/nuccore (accessed on 24 February 2017).
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: the creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef] [PubMed]
- ICTV Code. Available online: http://talk.ictvonline.org/information/w/ictv-information/383/ictv-code (accessed on 23 February 2017).
- National Center for Biotechnology Information (US). The GenBank Submissions Handbook. Available online: https://www.ncbi.nlm.nih.gov/books/NBK51157/ (accessed on 23 February 2017).
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef] [PubMed]
- ICTV Taxonomy Proposals Templates. Available online: https://talk.ictvonline.org/files/taxonomy-proposal-templates/ (accessed on 23 February 2017).
- Petrov, V.M.; Ratnayaka, S.; Nolan, J.M.; Miller, E.S.; Karam, J.D. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 2010, 7, 292. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Ravet, V.; Pereira, O.; Sullivan, M.B. Genomic characteristics and environmental distributions of the uncultivated Far-T4 phages. Front. Microbiol. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Zafar, N.; Mazumder, R.; Seto, D. CoreGenes: A computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinform. 2002, 3, 12. [Google Scholar] [CrossRef]
- Mahadevan, P.; King, J.F.; Seto, D. CGUG: In silico proteome and genome parsing tool for the determination of “core” and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC Res. Notes 2009, 2, 168. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2016, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adriaenssens, E.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. https://doi.org/10.3390/v9040070
Adriaenssens E, Brister JR. How to Name and Classify Your Phage: An Informal Guide. Viruses. 2017; 9(4):70. https://doi.org/10.3390/v9040070
Chicago/Turabian StyleAdriaenssens, Evelien, and J. Rodney Brister. 2017. "How to Name and Classify Your Phage: An Informal Guide" Viruses 9, no. 4: 70. https://doi.org/10.3390/v9040070
APA StyleAdriaenssens, E., & Brister, J. R. (2017). How to Name and Classify Your Phage: An Informal Guide. Viruses, 9(4), 70. https://doi.org/10.3390/v9040070