
Targeting TNF and TNF Receptor Pathway in HIV-1 Infection: from Immune Activation to Viral Reservoirs



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
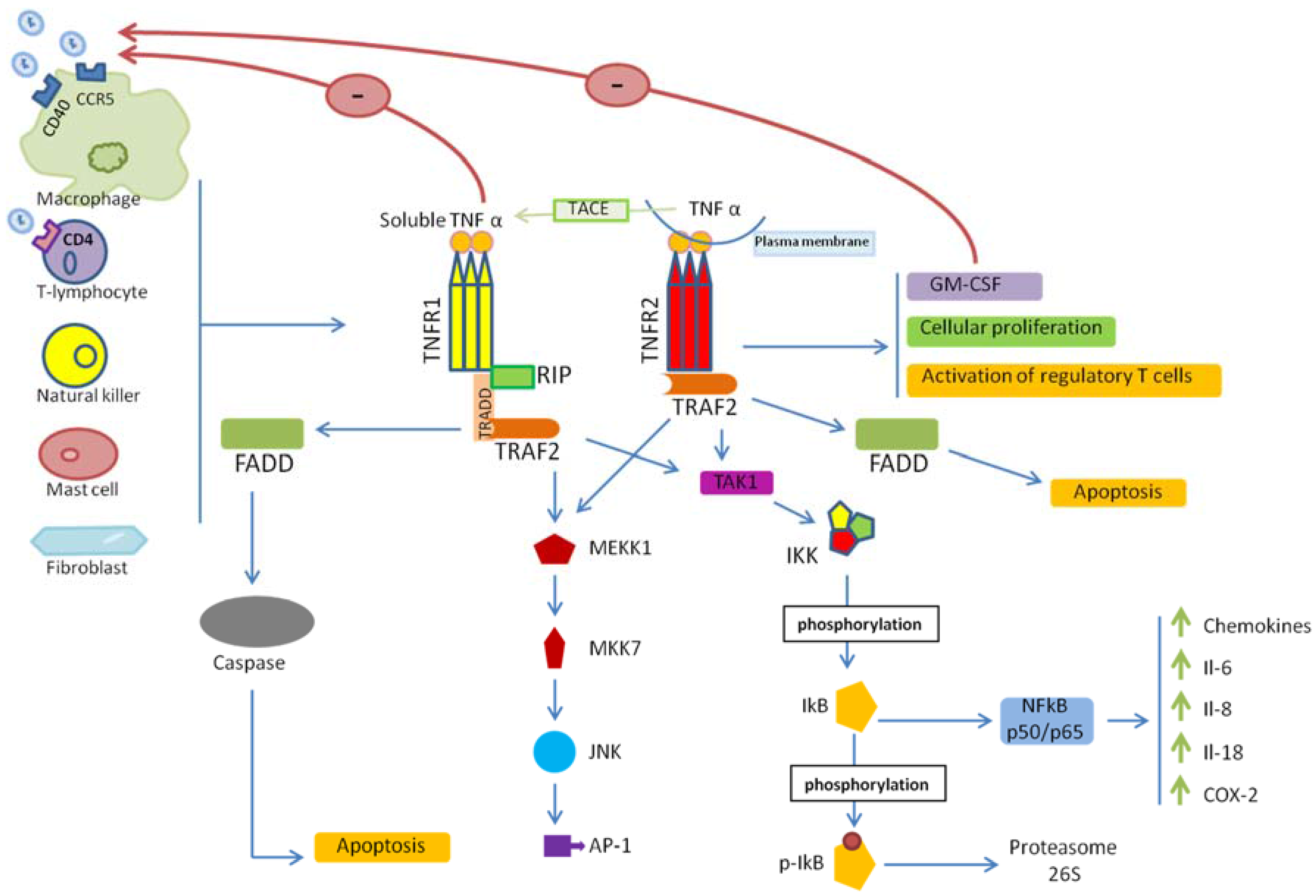
2. TNF/TNFR Signaling
3. TNF/TNFRs and HIV-1 Entry
4. TNF/TNFR Signaling in HIV-1 Infection
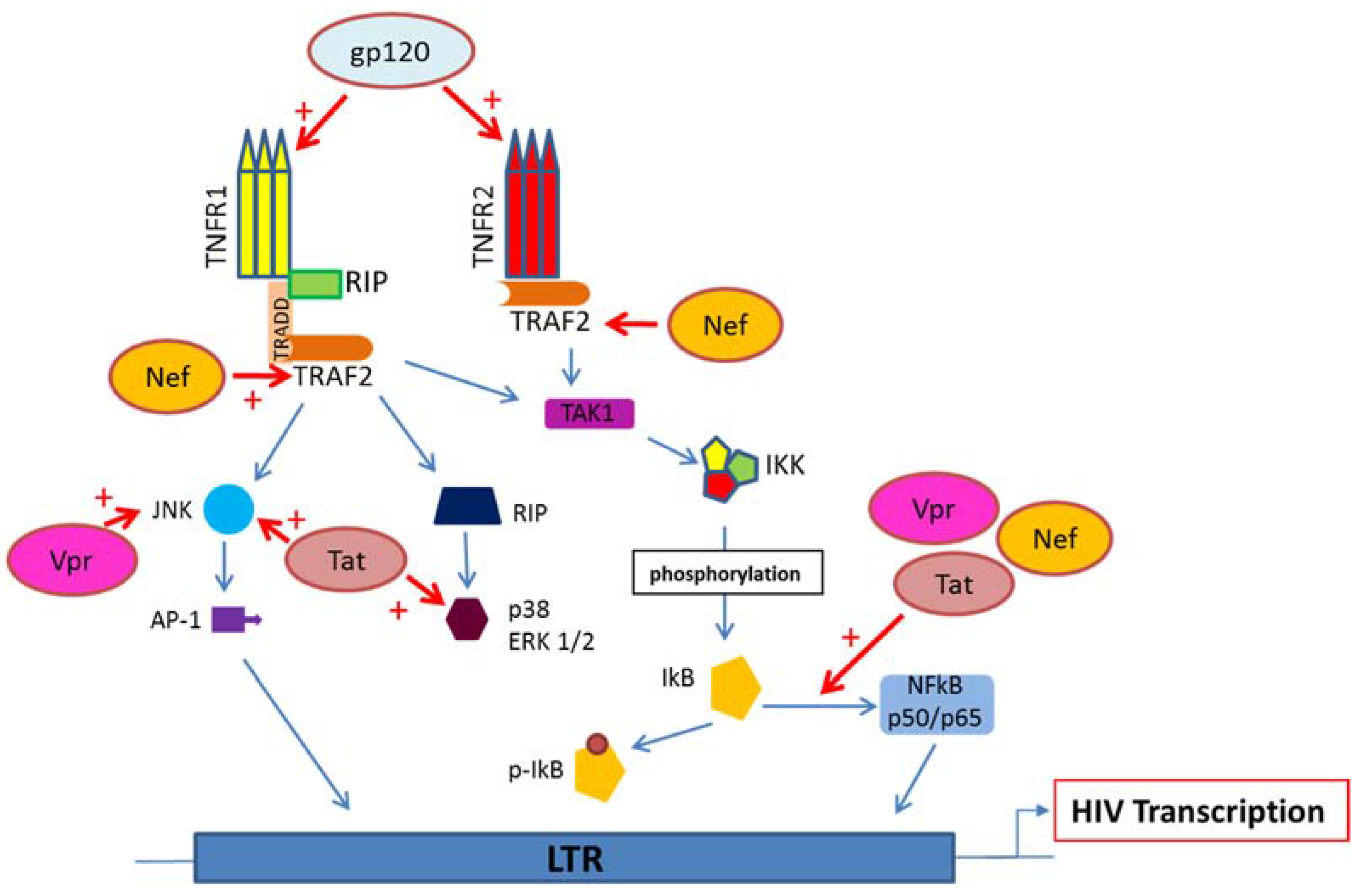
5. HIV Proteins Interaction with TNFR and Downstream Signaling Pathways
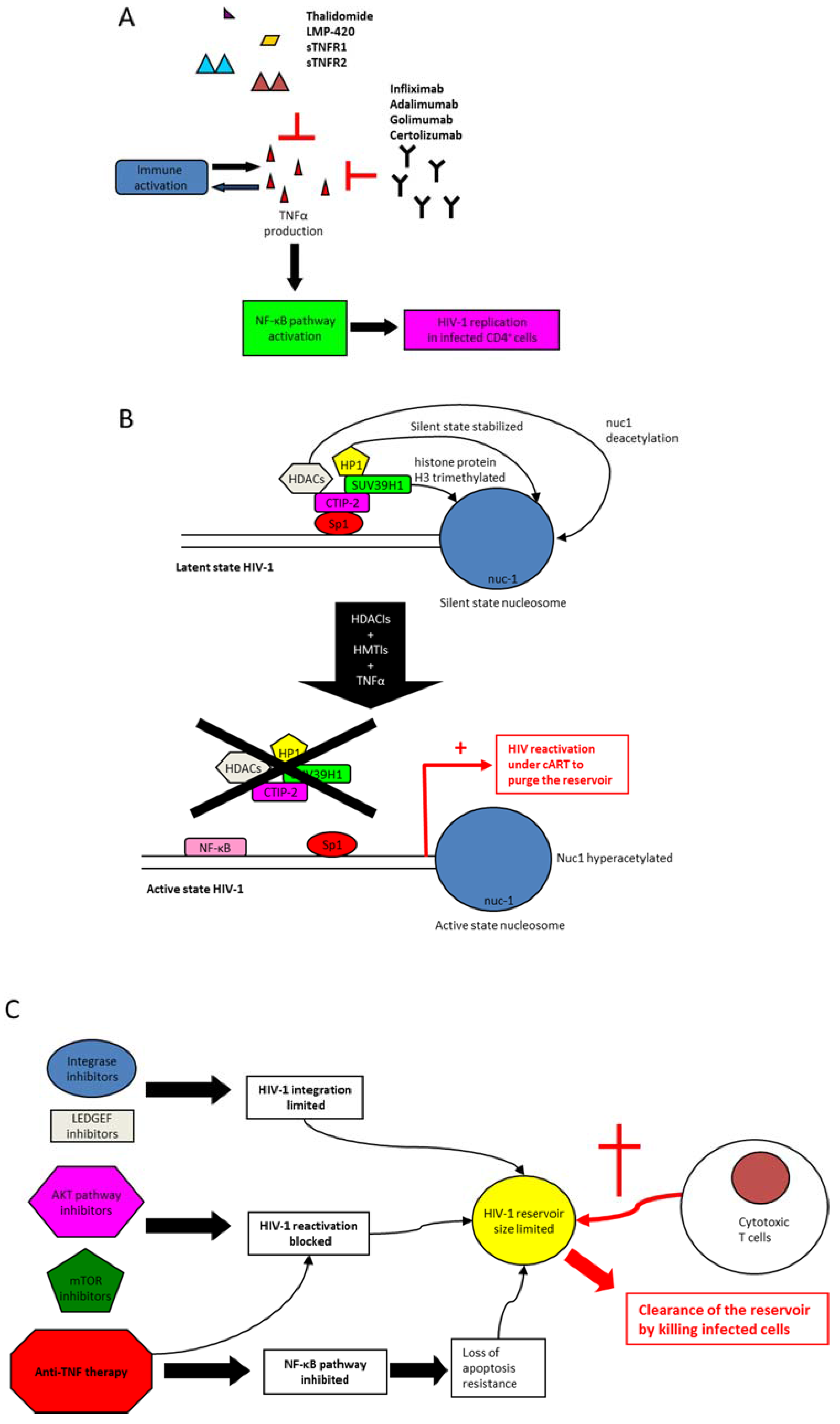
6. Anti-TNF Medications
7. Anti-TNF Therapy and Control of Immune Activation in HIV-1 Infection
8. Modulation of TNF Activity to Clear HIV-1 Reservoirs
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Cerami, A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 1986, 320, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Moelants, E.A.V.; Mortier, A.; Van Damme, J.; Proost, P. Regulation of TNF-α with a focus on rheumatoid arthritis. Immunol. Cell Biol. 2013, 91, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D.V. Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nature 1984, 312, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Kelker, H.C.; Oppenheim, J.D.; Stone-Wolff, D.; Henriksen-DeStefano, D.; Aggarwal, B.B.; Stevenson, H.C.; Vilcek, J. Characterization of human tumor necrosis factor produced by peripheral blood monocytes and its separation from lymphotoxin. Int. J. Cancer 1985, 36, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; O’Brien, W.A. Tumor necrosis factor (TNF)-αand TNF receptors in viral pathogenesis. Proc. Soc. Exp. Biol. Med. 2000, 223, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; van Huffel, C. Unraveling function in the TNF ligand and receptor families. Science 1994, 264, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Wajant, H.; Zimmermann, G.; Scheurich, P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1998, 95, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Hsu, H.; Shu, H.B.; Pan, M.G.; Goeddel, D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84, 299–308. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature 1997, 388, 548–554. [Google Scholar] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IκB kinases essential for NF-κB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Devin, A.; Cook, A.; Lin, Y.; Rodriguez, Y.; Kelliher, M.; Liu, Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity 2000, 12, 419–429. [Google Scholar] [CrossRef]
- Natoli, G.; Costanzo, A.; Moretti, F.; Fulco, M.; Balsano, C.; Levrero, M. Tumor necrosis factor (TNF) receptor 1 signaling downstream of TNF receptor-associated factor 2. NUCLEAR FACTOR ΚB (NFΚB)-INDUCING KINASE REQUIREMENT FOR ACTIVATION OF ACTIVATING PROTEIN 1 AND NFΚB BUT NOT OF C-JUN N-TERMINAL KINASE/STRESS-ACTIVATED PROTEIN KINASE. J. Biol. Chem. 1997, 272, 26079–26082. [Google Scholar] [PubMed]
- Grell, M.; Becke, F.M.; Wajant, H.; Männel, D.N.; Scheurich, P. TNF receptor type 2 mediates thymocyte proliferation independently of TNF receptor type 1. Eur. J. Immunol. 1998, 28, 257–263. [Google Scholar] [CrossRef]
- Gehr, G.; Gentz, R.; Brockhaus, M.; Loetscher, H.; Lesslauer, W. Both tumor necrosis factor receptor types mediate proliferative signals in human mononuclear cell activation. J. Immunol. 1992, 149, 911–917. [Google Scholar] [PubMed]
- Zheng, L.; Fisher, G.; Miller, R.E.; Peschon, J.; Lynch, D.H.; Lenardo, M.J. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995, 377, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Montaner, L.J.; Gordon, S. Tumor necrosis factor alpha inhibits entry of human immunodeficiency virus type 1 into primary human macrophages: A selective role for the 75-kilodalton receptor. J. Virol. 1996, 70, 7388–7397. [Google Scholar] [PubMed]
- Karsten, V.; Gordon, S.; Kirn, A.; Herbein, G. HIV-1 envelope glycoprotein gp120 down-regulates CD4 expression in primary human macrophages through induction of endogenous tumour necrosis factor-α. Immunology 1996, 88, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Doyle, A.G.; Montaner, L.J.; Gordon, S. Lipopolysaccharide (LPS) down-regulates CD4 expression in primary human macrophages through induction of endogenous tumour necrosis factor (TNF) and IL-1 β. Clin. Exp. Immunol. 1995, 102, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Di Marzio, P.; Tse, J.; Landau, N.R. Chemokine receptor regulation and HIV type 1 tropism in monocyte-macrophages. AIDS Res. Hum. Retrovir. 1998, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Gordon, S. 55- and 75-kilodalton tumor necrosis factor receptors mediate distinct actions in regard to human immunodeficiency virus type 1 replication in primary human macrophages. J. Virol. 1997, 71, 4150–4156. [Google Scholar] [PubMed]
- Coffey, M.J.; Woffendin, C.; Phare, S.M.; Strieter, R.M.; Markovitz, D.M. RANTES inhibits HIV-1 replication in human peripheral blood monocytes and alveolar macrophages. Am. J. Physiol. 1997, 272, L1025–L1029. [Google Scholar] [PubMed]
- Simmons, G.; Clapham, P.R.; Picard, L.; Offord, R.E.; Rosenkilde, M.M.; Schwartz, T.W.; Buser, R.; Wells, T.N.; Proudfoot, A.E. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 1997, 276, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.R.; Markovitz, D.M.; Woodford, N.L.; Rochford, R.; Strieter, R.M.; Coffey, M.J. TNF-α inhibits HIV-1 replication in peripheral blood monocytes and alveolar macrophages by inducing the production of RANTES and decreasing C-C chemokine receptor 5 (CCR5) expression. J. Immunol. 1999, 163, 3653–3661. [Google Scholar] [PubMed]
- McManus, C.M.; Brosnan, C.F.; Berman, J.W. Cytokine induction of MIP-1 α and MIP-1 β in human fetal microglia. J. Immunol. 1998, 160, 1449–1455. [Google Scholar] [PubMed]
- Kitai, R.; Zhao, M.L.; Zhang, N.; Hua, L.L.; Lee, S.C. Role of MIP-1beta and RANTES in HIV-1 infection of microglia: Inhibition of infection and induction by IFNβ. J. Neuroimmunol. 2000, 110, 230–239. [Google Scholar] [CrossRef]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Kuprash, D.V.; Liu, Z.-G.; Nedospasov, S.A. Intracellular signals and events activated by cytokines of the tumor necrosis factor superfamily: From simple paradigms to complex mechanisms. Int. Rev. Cytol. 2006, 252, 129–161. [Google Scholar] [PubMed]
- Nakano, H. Signaling crosstalk between NF-κB and JNK. Trends Immunol. 2004, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Maury, W.J.; Folks, T.M.; Fauci, A.S.; Rabson, A.B. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-κB sites in the long terminal repeat. Proc. Natl. Acad. Sci. USA 1989, 86, 5974–5978. [Google Scholar] [CrossRef] [PubMed]
- Griffin, G.E.; Leung, K.; Folks, T.M.; Kunkel, S.; Nabel, G.J. Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B. Nature 1989, 339, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Matsuyama, T.; Mori, S.; Hamamoto, Y.; Kobayashi, N.; Yamamoto, N.; Josephs, S.F.; Wong-Staal, F.; Shimotohno, K. Augmentation of human immunodeficiency virus type 1 gene expression by tumor necrosis factor α. AIDS Res. Hum. Retrovir. 1989, 5, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Osborn, L.; Kunkel, S.; Nabel, G.J. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 1989, 86, 2336–2340. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Tanaka, Y.; Yamashita, A.; Koyanagi, Y.; Nakamura, M.; Yamamoto, N. OX40 stimulation by gp34/OX40 ligand enhances productive human immunodeficiency virus type 1 infection. J. Virol. 2001, 75, 6748–6757. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Galli, M.; Bosis, S.; Gervasoni, C.; Moroni, M.; Norbiato, G. Immunoendocrinologic abnormalities in human immunodeficiency virus infection. Ann. N. Y. Acad. Sci. 2000, 917, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Khan, K.A. Is HIV infection a TNF receptor signalling-driven disease? Trends Immunol. 2008, 29, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Arokium, H.; Kamata, M.; Chen, I. Virion-associated Vpr of human immunodeficiency virus type 1 triggers activation of apoptotic events and enhances fas-induced apoptosis in human T cells. J. Virol. 2009, 83, 11283–11297. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Kim, B.; Jacquot, G.; Benichou, S.; Planelles, V. HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathog. 2006, 2, e127. [Google Scholar] [CrossRef] [PubMed]
- Kogan, M.; Deshmane, S.; Sawaya, B.E.; Gracely, E.J.; Khalili, K.; Rappaport, J. Inhibition of NF-κB activity by HIV-1 Vpr is dependent on Vpr binding protein. J. Cell. Physiol. 2013, 228, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Rainaldi, G.; Matarrese, P.; Varano, B.; Rivabene, R.; Columba, S.; Sato, A.; Belardelli, F.; Malorni, W.; Gessani, S. The HIV-1 VPR protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: Possible implications for the pathogenesis of AIDS. J. Exp. Med. 1998, 187, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, S.; Konishi, M.; Mori, M.; Shimura, M.; Nishitani, C.; Kuroki, Y.; Koyanagi, Y.; Kano, S.; Itabe, H.; Ishizaka, Y. HIV-1 Vpr induces TLR4/MyD88-mediated IL-6 production and reactivates viral production from latency. J. Leukoc. Biol. 2010, 87, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Decrion, A.-Z.; Sabbah, E.; Quivy, V.; Sire, J.; Van Lint, C.; Roques, B.P.; Aggarwal, B.B.; Herbein, G. Synthetic Vpr protein activates activator protein-1, c-Jun N-terminal kinase, and NF-κB and stimulates HIV-1 transcription in promonocytic cells and primary macrophages. J. Biol. Chem. 2005, 280, 42557–42567. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.; Alfieri, C.; Hrimech, M.; Cohen, E.A.; Tanner, J.E. Activation of transcription factors NF-κB and NF-IL-6 by human immunodeficiency virus type 1 protein R (VPR) induces interleukin-8 expression. J. Virol. 2000, 74, 4658–4665. [Google Scholar] [CrossRef] [PubMed]
- Roesch, F.; Richard, L.; Rua, R.; Porrot, F.; Casartelli, N.; Schwartz, O. VPR Enhances Tumor Necrosis Factor Production by HIV-1-Infected T Cells. J. Virol. 2015, 89, 12118–12130. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, B.E.; Khalili, K.; Gordon, J.; Taube, R.; Amini, S. Cooperative interaction between HIV-1 regulatory proteins Tat and VPR modulates transcription of the viral genome. J. Biol. Chem. 2000, 275, 35209–35214. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Choo, A.Y.; Zong, W.-X.; Madesh, M.; Hwang, D.S.; Premkumar, A.; Thieu, K.P.; Emmanuel, J.; Kumar, S.; Thompson, C.B.; et al. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat. Cell Biol. 2006, 8, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G. TNF and HIV-1 Nef: An Intimate Interplay. EBioMedicine 2016, 13, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV NEF is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Manna, S.K.; Quivy, V.; Decrion, A.-Z.; Van Lint, C.; Herbein, G.; Aggarwal, B.B. Exogenous Nef protein activates NF-κB, AP-1, and c-Jun N-terminal kinase and stimulates HIV transcription in promonocytic cells. Role in AIDS pathogenesis. J. Biol. Chem. 2003, 278, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Olivetta, E.; Percario, Z.; Fiorucci, G.; Mattia, G.; Schiavoni, I.; Dennis, C.; Jäger, J.; Harris, M.; Romeo, G.; Affabris, E.; et al. HIV-1 Nef induces the release of inflammatory factors from human monocyte/macrophages: Involvement of Nef endocytotic signals and NF-κB activation. J. Immunol. 2003, 170, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Mangino, G.; Percario, Z.A.; Fiorucci, G.; Vaccari, G.; Manrique, S.; Romeo, G.; Federico, M.; Geyer, M.; Affabris, E. In vitro treatment of human monocytes/macrophages with myristoylated recombinant Nef of human immunodeficiency virus type 1 leads to the activation of mitogen-activated protein kinases, IκB kinases, and interferon regulatory factor 3 and to the release of beta interferon. J. Virol. 2007, 81, 2777–2791. [Google Scholar] [PubMed]
- Olivetta, E.; Tirelli, V.; Chiozzini, C.; Scazzocchio, B.; Romano, I.; Arenaccio, C.; Sanchez, M. HIV-1 Nef impairs key functional activities in human macrophages through CD36 downregulation. PLoS ONE 2014, 9, e93699. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Varin, A.; Larbi, A.; Fortin, C.; Mahlknecht, U.; Fulop, T.; Aggarwal, B. NEF and TNFα are Coplayers that Favor HIV-1 Replication in Monocytic Cells and Primary Macrophages. Curr. HIV Res. 2008, 6, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Abbas, W.; Varin, A.; Kumar, A.; Di Martino, V.; Dichamp, I.; Herbein, G. HIV-1 Nef interacts with HCV Core, recruits TRAF2, TRAF5 and TRAF6, and stimulates HIV-1 replication in macrophages. J. Innate Immun. 2013, 5, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Ostalecki, C.; Wittki, S.; Lee, J.-H.; Geist, M.M.; Tibroni, N.; Harrer, T.; Schuler, G.; Fackler, O.T.; Baur, A.S. HIV NEF- and Notch1-dependent Endocytosis of ADAM17 Induces Vesicular TNF Secretion in Chronic HIV Infection. EBioMedicine 2016, 13, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Schorey, J.S.; Harding, C.V. Extracellular vesicles and infectious diseases: New complexity to an old story. J. Clin. Investig. 2016, 126, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Chiozzini, C.; Columba-Cabezas, S.; Manfredi, F.; Federico, M. Cell activation and HIV-1 replication in unstimulated CD4+ T lymphocytes ingesting exosomes from cells expressing defective HIV-1. Retrovirology 2014, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Chiozzini, C.; Columba-Cabezas, S.; Manfredi, F.; Affabris, E.; Baur, A.; Federico, M. Exosomes from human immunodeficiency virus type 1 (HIV-1)-infected cells license quiescent CD4+ T lymphocytes to replicate HIV-1 through a Nef- and ADAM17-dependent mechanism. J. Virol. 2014, 88, 11529–11539. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected cells stimulate production of pro-inflammatory cytokines through trans-activating response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Manna, S.K.; Dhawan, S.; Aggarwal, B.B. HIV-Tat protein activates c-Jun N-terminal kinase and activator protein-1. J. Immunol. 1998, 161, 776–781. [Google Scholar] [PubMed]
- Planès, R.; Ben Haij, N.; Leghmari, K.; Serrero, M.; BenMohamed, L.; Bahraoui, E. HIV-1 Tat protein activates both the MyD88 and TRIF pathways to induce tumor necrosis factor α and interleukin-10 in human monocytes. J. Virol. 2016, 90, 5886–5898. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Herbein, G. T-Cell Signaling in HIV-1 Infection. Open Virol. J. 2013, 7, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, P.; Poli, G.; Fox, C.H.; Kehrl, J.H.; Fauci, A.S. Recombinant gp120 specifically enhances tumor necrosis factor-α production and Ig secretion in B lymphocytes from HIV-infected individuals but not from seronegative donors. J. Immunol. 1991, 147, 2922–2927. [Google Scholar] [PubMed]
- Merrill, J.E.; Koyanagi, Y.; Chen, I.S. Interleukin-1 and tumor necrosis factor α can be induced from mononuclear phagocytes by human immunodeficiency virus type 1 binding to the CD4 receptor. J. Virol. 1989, 63, 4404–4408. [Google Scholar] [PubMed]
- Khanna, K.V.; Yu, X.F.; Ford, D.H.; Ratner, L.; Hildreth, J.K.; Markham, R.B. Differences among HIV-1 variants in their ability to elicit secretion of TNF-α. J. Immunol. 2000, 164, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Tomkowicz, B.; Freedman, B.D.; Collman, R.G. HIV-1 gp120-induced TNF-{α} production by primary human macrophages is mediated by phosphatidylinositol-3 (PI-3) kinase and mitogen-activated protein (MAP) kinase pathways. J. Leukoc. Biol. 2005, 78, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Ouyang, H.; Zheng, X.; Liu, S.; Mata, M.; Fink, D.J.; Hao, S. Glial TNFα in the spinal cord regulates neuropathic pain induced by HIV gp120 application in rats. Mol. Pain 2011, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Furler, R.L.; Uittenbogaart, C.H. Signaling through the P38 and ERK pathways: A common link between HIV replication and the immune response. Immunol. Res. 2010, 48, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Rychert, J.; Strick, D.; Bazner, S.; Robinson, J.; Rosenberg, E. Detection of HIV gp120 in plasma during early HIV infection is associated with increased proinflammatory and immunoregulatory cytokines. AIDS Res. Hum. Retrovir. 2010, 26, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Mahlknecht, U.; Batliwalla, F.; Gregersen, P.; Pappas, T.; Butler, J.; O’Brien, W.A.; Verdin, E. Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature 1998, 395, 189–194. [Google Scholar] [PubMed]
- Alfano, M.; Alfano, M.; Banki, Z.; Borde, C.; Bristow, C.L.; Caruz, A.; Chang, T.L.; Cortes, J.; Eugen-Olsen, J.; Fibla, J.; et al. Soluble Factors Mediating Innate Immune Responses to HIV Infection; Bentham Science Publishers: Chennai, India, 2010. [Google Scholar]
- Zangerle, R.; Gallati, H.; Sarcletti, M.; Wachter, H.; Fuchs, D. Tumor necrosis factor α and soluble tumor necrosis factor receptors in individuals with human immunodeficiency virus infection. Immunol. Lett. 1994, 41, 229–234. [Google Scholar] [CrossRef]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Herbein, G. TNFα antagonist therapy in patients with joint disease and chronic viral infection. Jt. Bone Spine 2007, 74, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.M.; Trinh, H.; Le, J.; Siegel, S.; Shealy, D.; McDonough, M.; Scallon, B.; Moore, M.A.; Vilcek, J.; Daddona, P. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol. Immunol. 1993, 30, 1443–1453. [Google Scholar] [CrossRef]
- Goel, N.; Stephens, S. Certolizumab pegol. MAbs 2010, 2, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Aukrust, P.; Liabakk, N.B.; Müller, F.; Lien, E.; Espevik, T.; Frøland, S.S. Serum levels of tumor necrosis factor-alpha (TNF α) and soluble TNF receptors in human immunodeficiency virus type 1 infection--correlations to clinical, immunologic, and virologic parameters. J. Infect. Dis. 1994, 169, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Whalen, C.; Horsburgh, C.R.; Hom, D.; Lahart, C.; Simberkoff, M.; Ellner, J. Accelerated course of human immunodeficiency virus infection after tuberculosis. Am. J. Respir. Crit. Care Med. 1995, 151, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Sereti, I.; Krebs, S.J.; Phanuphak, N.; Fletcher, J.L.; Slike, B.; Pinyakorn, S.; O’Connell, R.J.; Rupert, A.; Chomont, N.; Valcour, V.; et al. Persistent, Albeit Reduced, Chronic Inflammation in Persons Starting Antiretroviral Therapy in Acute HIV Infection. Clin. Infect. Dis. 2017, 64, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.P.; Singh, A.; Hullsiek, K.H.; Gibson, D.; Henry, W.K.; Lichtenstein, K.; Önen, N.F.; Kojic, E.; Patel, P.; Brooks, J.T.; et al. Study to understand the natural history of HIV/AIDS in the era of effective therapy (SUN study) investigators monocyte-activation phenotypes are associated with biomarkers of inflammation and coagulation in chronic HIV infection. J. Infect. Dis. 2014, 210, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Mancilla, J.R.; Brown, T.T.; Erlandson, K.M.; Palella, F.J.; Gardner, E.M.; Macatangay, B.J. C.; Breen, E.C.; Jacobson, L.P.; Anderson, P.L.; Wada, N.I. Suboptimal adherence to combination antiretroviral therapy is associated with higher levels of inflammation despite HIV suppression. Clin. Infect. Dis. 2016, 63, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.I.; Jacobson, L.P.; Margolick, J.B.; Breen, E.C.; Macatangay, B.; Penugonda, S.; Martínez-Maza, O.; Bream, J.H. The effect of HAART-induced HIV suppression on circulating markers of inflammation and immune activation. AIDS 2015, 29, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Dezube, B.J.; Lederman, M.M.; Chapman, B.; Georges, D.L.; Dogon, A.L.; Mudido, P.; Reis-Lishing, J.; Cheng, S.L.; Silberman, S.L.; Crumpacker, C.S. The effect of tenidap on cytokines, acute-phase proteins, and virus load in human immunodeficiency virus (HIV)-infected patients: Correlation between plasma HIV-1 RNA and proinflammatory cytokine levels. J. Infect. Dis. 1997, 176, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Valdez, H.; Lederman, M.M. Cytokines and cytokine therapies in HIV infection. AIDS Clin. Rev. 1997, 187–228. [Google Scholar]
- Beyer, M.; Abdullah, Z.; Chemnitz, J.M.; Maisel, D.; Sander, J.; Lehmann, C.; Thabet, Y.; Shinde, P.V.; Schmidleithner, L.; Köhne, M.; et al. Tumor-necrosis factor impairs CD4(+) T cell-mediated immunological control in chronic viral infection. Nat. Immunol. 2016, 17, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Aderka, D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996, 7, 231–240. [Google Scholar] [CrossRef]
- Brockhaus, M. Soluble TNF receptor: What is the significance? Intensive Care Med. 1997, 23, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Decrion, A.Z.; Dichamp, I.; Varin, A.; Herbein, G. HIV and inflammation. Curr. HIV Res. 2005, 3, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Marriott, J.B.; Cookson, S.; Carlin, E.; Youle, M.; Hawkins, D.A.; Nelson, M.; Pearson, M.; Vaughan, A.N.; Gazzard, B.; Dalgleish, A.G. A double-blind placebo-controlled phase II trial of thalidomide in asymptomatic HIV-positive patients: Clinical tolerance and effect on activation markers and cytokines. AIDS Res. Hum. Retrovir. 1997, 13, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Nsubuga, P.; Whalen, C.; Mugerwa, R.D.; Okwera, A.; Oette, D.; Jackson, J.B.; Johnson, J.L.; Ellner, J.J. Pentoxifylline therapy in human immunodeficiency virus-seropositive persons with tuberculosis: A randomized, controlled trial. J. Infect. Dis. 1996, 174, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, S.; Day, N.K.; Kamchaisatian, W.; Beigier-Pompadre, M.; Stenger, S.; Tangsinmankong, N.; Sleasman, J.W.; Pizzo, S.V.; Cianciolo, G.J. LMP-420, a small-molecule inhibitor of TNF-α, reduces replication of HIV-1 and Mycobacterium tuberculosis in human cells. AIDS Res. Ther. 2006, 3, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ting, P.T.; Koo, J.Y. Use of etanercept in human immunodeficiency virus (HIV) and acquired immunodeficiency syndrome (AIDS) patients. Int. J. Dermatol. 2006, 45, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.H.; Zein, N.; Vassilopoulos, D. Safety of antitumour necrosis factor (anti-TNF) therapy in patients with chronic viral infections: Hepatitis C, hepatitis B, and HIV infection. Ann. Rheum. Dis. 2004, 63 (Suppl. 2), ii18–ii24. [Google Scholar] [CrossRef] [PubMed]
- Sha, B.E.; Valdez, H.; Gelman, R.S.; Landay, A.L.; Agosti, J.; Mitsuyasu, R.; Pollard, R.B.; Mildvan, D.; Namkung, A.; Ogata-Arakaki, D.M.; et al. Effect of etanercept (Enbrel) on interleukin 6, tumor necrosis factor α, and markers of immune activation in HIV-infected subjects receiving interleukin 2. AIDS Res. Hum. Retrovir. 2002, 18, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Kyambadde, P.; Johnson, J.L.; Horter, L.; Kittle, R.; Pohle, M.; Ducar, C.; Millard, M.; Mayanja-Kizza, H.; Whalen, C.; et al. A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV-1-associated tuberculosis. AIDS 2004, 18, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.E.; Spooner, K.M.; Kelly, G.; McCloskey, R.V.; Woody, J.N.; Falloon, J.; Baseler, M.; Piscitelli, S.C.; Davey, R.T.; Polis, M.A.; et al. Inhibition of immunoreactive tumor necrosis factor-α by a chimeric antibody in patients infected with human immunodeficiency virus type 1. J. Infect. Dis. 1996, 174, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Gaylis, N. Infliximab in the treatment of an HIV positive patient with Reiter’s syndrome. J. Rheumatol. 2003, 30, 407–411. [Google Scholar] [PubMed]
- Gallitano, S.M.; McDermott, L.; Brar, K.; Lowenstein, E. Use of tumor necrosis factor (TNF) inhibitors in patients with HIV/AIDS. J. Am. Acad. Dermatol. 2016, 74, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, E.J.; Williams, F.M.; Ishimori, M.L.; Weisman, M.H.; Reveille, J.D. The use of anti-tumour necrosis factor therapy in HIV-positive individuals with rheumatic disease. Ann. Rheum. Dis. 2008, 67, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Wangsiricharoen, S.; Ligon, C.; Gedmintas, L.; Dehrab, A.; Tungsiripat, M.; Bingham, C.; Lozada, C.; Calabrese, L. The rates of serious infections in HIV-infected patients who received tumor necrosis factor (TNF)-α inhibitor therapy for concomitant autoimmune diseases. Arthritis Care Res. 2016, 69, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Fauci, A.S. Latent reservoirs of HIV: Obstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA 1999, 96, 10958–10961. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Herbein, G. Molecular understanding of HIV-1 latency. Adv. Virol. 2012, 2012, 574967. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the effort to cure HIV infection: Going beyond N = 1. J. Clin. Investig. 2016, 126, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.; Lu, H.K.; Evans, V.; Harisson, D.; Zhou, J.; Jaworowski, A.; Sallmann, G.; Cheong, K.Y.; Mota, T.M.; Tennakoon, S.; et al. HIV integration and the establishment of latency in CCL19-treated resting CD4(+) T cells require activation of NF-κB. Retrovirology 2016, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Khan, K.A.; Kumar, A.; Tripathy, M.K.; Dichamp, I.; Keita, M.; Mahlknecht, U.; Rohr, O.; Herbein, G. Blockade of BFA-mediated apoptosis in macrophages by the HIV-1 Nef protein. Cell Death Dis. 2014, 5, e1080. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Tariq, M.; Iqbal, M.; Kumar, A.; Herbein, G. Eradication of HIV-1 from the macrophage reservoir: An uncertain goal? Viruses 2015, 7, 1578–1598. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Engel, D.; Mizell, S.B.; Ehler, L.A.; Fauci, A.S. Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J. Exp. Med. 1998, 188, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.-S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.L.; Greene, W.C. NF-κB/Rel: Agonist and antagonist roles in HIV-1 latency. Curr. Opin. HIV AIDS 2011, 6, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Lim, K.-I.; Calafi, A.; Rossi, J.J.; Schaffer, D.V.; Arkin, A.P. Combinatorial latency reactivation for HIV-1 subtypes and variants. J. Virol. 2010, 84, 5958–5974. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef] [PubMed]
- Quivy, V.; Adam, E.; Collette, Y.; Demonte, D.; Chariot, A.; Vanhulle, C.; Berkhout, B.; Castellano, R.; de Launoit, Y.; Burny, A.; et al. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-κB and inhibitors of deacetylases: Potential perspectives for the development of therapeutic strategies. J. Virol. 2002, 76, 11091–11103. [Google Scholar] [CrossRef] [PubMed]
- Vandergeeten, C.; Quivy, V.; Moutschen, M.; Van Lint, C.; Piette, J.; Legrand-Poels, S. HIV-1 protease inhibitors do not interfere with provirus transcription and host cell apoptosis induced by combined treatment TNF-α+ TSA. Biochem. Pharmacol. 2007, 73, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Demonté, D.; Quivy, V.; Colette, Y.; Van Lint, C. Administration of HDAC inhibitors to reactivate HIV-1 expression in latent cellular reservoirs: Implications for the development of therapeutic strategies. Biochem. Pharmacol. 2004, 68, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- El Kharroubi, A.; Piras, G.; Zensen, R.; Martin, M.A. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol. Cell. Biol. 1998, 18, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Adam, E.; Quivy, V.; Bex, F.; Chariot, A.; Collette, Y.; Vanhulle, C.; Schoonbroodt, S.; Goffin, V.; Nguyên, T.L.-A.; Gloire, G.; et al. Potentiation of tumor necrosis factor-induced NF-κB activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of IκBα. Mol. Cell. Biol. 2003, 23, 6200–6209. [Google Scholar] [CrossRef] [PubMed]
- Bouchat, S.; Gatot, J.-S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: Activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.-C.; Manucci, J.L.; Bhat, S.; Zhang, H.; Margolick, J.B.; Quinn, T.C.; et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- International AIDS Society Scientific Working Group on HIV Cure; Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.-W.; Churchill, M.; Di Mascio, M.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Moir, S.; Fauci, A.S. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 2015, 16, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Colin, L.; Khan, K.A.; Bouchat, S.; Varin, A.; Larbi, A.; Gatot, J.-S.; Kabeya, K.; Vanhulle, C.; et al. Tuning of AKT-pathway by Nef and its blockade by protease inhibitors results in limited recovery in latently HIV infected T-cell line. Sci. Rep. 2016, 6, 24090. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Bouchat, S.; Gatot, J.-S.; Pasquereau, S.; Kabeya, K.; Clumeck, N.; De Wit, S.; Van Lint, C.; Herbein, G. Limited HIV-1 reactivation in resting CD4(+) T cells from aviremic patients under protease inhibitors. Sci. Rep. 2016, 6, 38313. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-mediated inhibition of integrase-LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, P.; Wensing, A.M.J.; Fun, A.; Nijhuis, M.; Brusselaers, N.; Vandekerckhove, L. Clinical use of HIV integrase inhibitors: A systematic review and meta-analysis. PLoS ONE 2013, 8, e52562. [Google Scholar] [CrossRef] [PubMed]
- Withers, D.R.; Jaensson, E.; Gaspal, F.; McConnell, F.M.; Eksteen, B.; Anderson, G.; Agace, W.W.; Lane, P.J.L. The survival of memory CD4+ T cells within the gut lamina propria requires OX40 and CD30 signals. J. Immunol. 2009, 183, 5079–5084. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, U.; Deng, C.; Lu, M.C.; Greenough, T.C.; Sullivan, J.L.; O’Brien, W.A.; Herbein, G. Resistance to apoptosis in HIV-infected CD4+ T lymphocytes is mediated by macrophages: Role for Nef and immune activation in viral persistence. J. Immunol. 2000, 165, 6437–6446. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquereau, S.; Kumar, A.; Herbein, G. Targeting TNF and TNF Receptor Pathway in HIV-1 Infection: from Immune Activation to Viral Reservoirs. Viruses 2017, 9, 64. https://doi.org/10.3390/v9040064
Pasquereau S, Kumar A, Herbein G. Targeting TNF and TNF Receptor Pathway in HIV-1 Infection: from Immune Activation to Viral Reservoirs. Viruses. 2017; 9(4):64. https://doi.org/10.3390/v9040064
Chicago/Turabian StylePasquereau, Sébastien, Amit Kumar, and Georges Herbein. 2017. "Targeting TNF and TNF Receptor Pathway in HIV-1 Infection: from Immune Activation to Viral Reservoirs" Viruses 9, no. 4: 64. https://doi.org/10.3390/v9040064
APA StylePasquereau, S., Kumar, A., & Herbein, G. (2017). Targeting TNF and TNF Receptor Pathway in HIV-1 Infection: from Immune Activation to Viral Reservoirs. Viruses, 9(4), 64. https://doi.org/10.3390/v9040064