Host Cell Restriction Factors that Limit Influenza A Infection

{kind=link}
{kind=link}
Abstract
1. Influenza Virus
2. IAV Infection of Host Cells
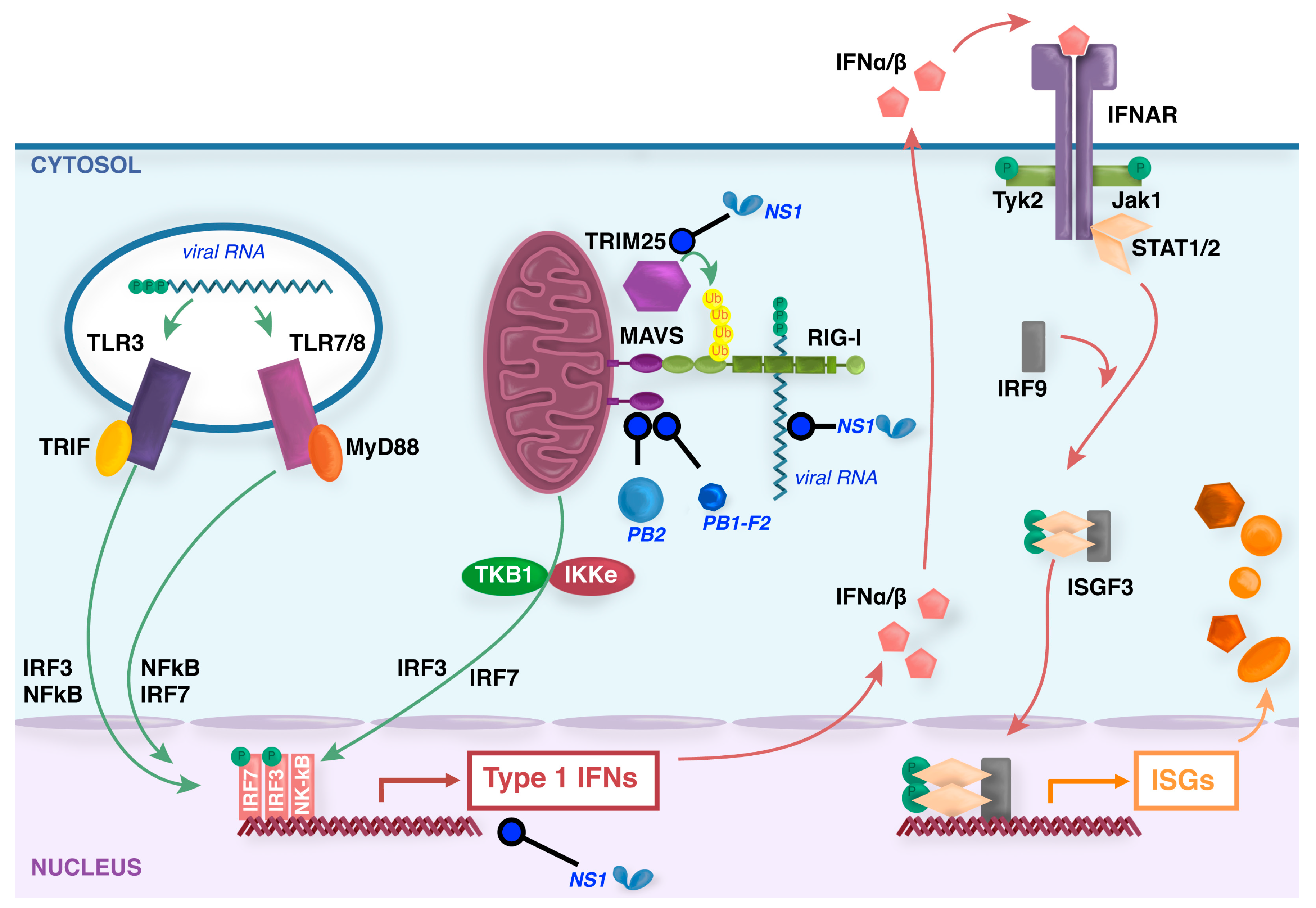
3. Induction of Type I Interferons and Innate Immunity Following IAV Infection
4. Induction of Cellular Restriction Factors: ISGs and Other Antiviral Factors
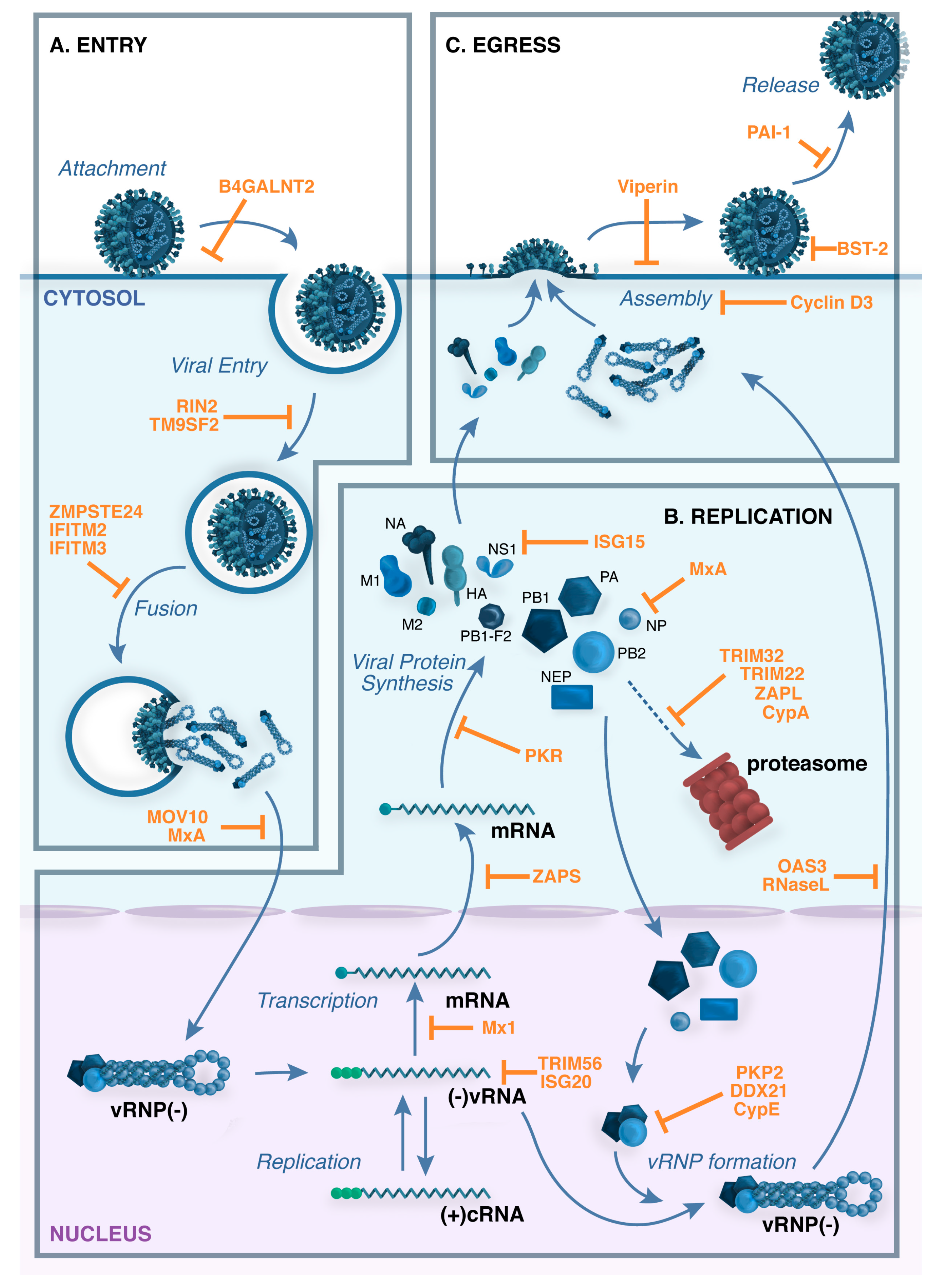
5. Restriction Factors That Target IAV Entry
6. Restriction Factors That Interfere with Genomic Transcription and Replication
6.1. Mx Proteins and Other GTPases
6.2. Protein Kinase R
6.3. OAS-Family Proteins
6.4. Other Restriction Factors That Interact Directly with Viral RNA
6.5. Restriction Factors That Target Viral Proteins
7. Blocking Virus Assembly and Egress
8. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The mother of all pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Schotsaert, M.; Garcia-Sastre, A. Inactivated influenza virus vaccines: The future of TIV and QIV. Curr. Opin. Virol. 2017, 23, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Houser, K.; Subbarao, K. Influenza vaccines: Challenges and solutions. Cell Host Microbe 2015, 17, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.A.; Medina, M.J.; Xu, X.; Perez-Oronoz, G.; Wallis, T.R.; Davis, X.M.; Povinelli, L.; Cox, N.J.; Klimov, A.I. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: A cause for concern. Lancet 2005, 366, 1175–1181. [Google Scholar] [CrossRef]
- Heneghan, C.J.; Onakpoya, I.; Jones, M.A.; Doshi, P.; Del Mar, C.B.; Hama, R.; Thompson, M.J.; Spencer, E.A.; Mahtani, K.R.; Nunan, D.; et al. Neuraminidase inhibitors for influenza: A systematic review and meta-analysis of regulatory and mortality data. Health Technol. Assess. 2016, 20, 1–242. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Brooks, A.G.; Reading, P.C.; Londrigan, S.L. The fate of influenza A virus after infection of human macrophages and dendritic cells. J. Gen. Virol. 2012, 93, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Londrigan, S.L.; Short, K.R.; Ma, J.; Gillespie, L.; Rockman, S.P.; Brooks, A.G.; Reading, P.C. Infection of Mouse Macrophages by Seasonal Influenza Viruses Can Be Restricted at the Level of Virus Entry and at a Late Stage in the Virus Life Cycle. J. Virol. 2015, 89, 12319–12329. [Google Scholar] [CrossRef] [PubMed]
- De Conto, F.; Covan, S.; Arcangeletti, M.C.; Orlandini, G.; Gatti, R.; Dettori, G.; Chezzi, C. Differential infectious entry of human influenza A/NWS/33 virus (H1N1) in mammalian kidney cells. Virus Res. 2011, 155, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Boulo, S.; Akarsu, H.; Ruigrok, R.W.; Baudin, F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007, 124, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Husser, L.; Alves, M.P.; Ruggli, N.; Summerfield, A. Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus Res. 2011, 159, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011, 162, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef] [PubMed]
- Graef, K.M.; Vreede, F.T.; Lau, Y.F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 2010, 84, 8433–8445. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Shiozaki, T.; Kawai, T.; Akira, S.; Kawaoka, Y.; Takada, A.; Kida, H.; Miyazaki, T. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon β promoter stimulator 1. J. Biol. Chem. 2010, 285, 32064–32074. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, P.; Gaumond, M.H.; Salois, P.; Sellin, K.; Bessette, M.C.; Godin, E.; de Oliveira, P.T.; Atkins, G.J.; Nanci, A.; Thomas, G. Bril: A novel bone-specific modulator of mineralization. J. Bone Miner. Res. 2008, 23, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Huang, I.C.; Kam, C.; Farzan, M. IFITM3 limits the severity of acute influenza in mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [PubMed]
- Chesarino, N.M.; Compton, A.A.; McMichael, T.M.; Kenney, A.D.; Zhang, L.; Soewarna, V.; Davis, M.; Schwartz, O.; Yount, J.S. IFITM3 requires an amphipathic helix for antiviral activity. EMBO Rep. 2017, 18, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Li, S.; Dorf, M.E. ZMPSTE24 defends against influenza and other pathogenic viruses. J. Exp. Med. 2017, 214, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Heaton, B.E.; Kennedy, E.M.; Dumm, R.E.; Harding, A.T.; Sacco, M.T.; Sachs, D.; Heaton, N.S. A CRISPR Activation Screen Identifies a Pan-avian Influenza Virus Inhibitory Host Factor. Cell Rep. 2017, 20, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Hulpiau, P.; Saelens, X. Mx proteins: Antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 2013, 77, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Howard, J. The interferon-inducible GTPases. Ann. Rev. Cell Dev. Biol. 2006, 22, 559–589. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Deeg, C.M.; Hassan, E.; Mutz, P.; Rheinemann, L.; Gotz, V.; Magar, L.; Schilling, M.; Kallfass, C.; Nurnberger, C.; Soubies, S.; et al. In vivo evasion of MxA by avian influenza viruses requires human signature in the viral nucleoprotein. J. Exp. Med. 2017, 214, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Tumpey, T.M.; Szretter, K.J.; van Hoeven, N.; Katz, J.M.; Kochs, G.; Haller, O.; Garcia-Sastre, A.; Staeheli, P. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J. Virol. 2007, 81, 10818–10821. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, S.R.; Carroll, T.D.; Dutra, J.C.; Ma, Z.M.; Miller, C.J. Myxovirus resistance gene A (MxA) expression suppresses influenza A virus replication in alpha interferon-treated primate cells. J. Virol. 2013, 87, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Killip, M.J.; Staeheli, P.; Randall, R.E.; Jackson, D. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J. Virol. 2013, 87, 13053–13058. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, J.; Haller, O.; Staeheli, P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 1992, 66, 2564–2569. [Google Scholar] [PubMed]
- Zimmermann, P.; Manz, B.; Haller, O.; Schwemmle, M.; Kochs, G. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J. Virol. 2011, 85, 8133–8140. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Pavlovic, J.; Staeheli, P.; Krystal, M. Overexpression of the influenza virus polymerase can titrate out inhibition by the murine Mx1 protein. J. Virol. 1992, 66, 4154–4160. [Google Scholar] [PubMed]
- Stranden, A.M.; Staeheli, P.; Pavlovic, J. Function of the mouse Mx1 protein is inhibited by overexpression of the PB2 protein of influenza virus. Virology 1993, 197, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Place, D.E.; Kuriakose, T.; Kanneganti, T.D. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J. Leukoc. Biol. 2017, 101, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Shi, Z.; Yan, W.; Wei, J.; Shao, D.; Deng, X.; Wang, S.; Li, B.; Tong, G.; Ma, Z. Nonstructural protein 1 of influenza A virus interacts with human guanylate-binding protein 1 to antagonize antiviral activity. PLoS ONE 2013, 8, e55920. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, A.; Wixler, L.; Boergeling, Y.; Wixler, V.; Ludwig, S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 2012, 26, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Cao, Z.; Wang, L.; Wan, Y.; Peng, N.; Wang, Q.; Chen, X.; Zhou, Y.; Zhu, Y. Inducible GBP5 Mediates the Antiviral Response via Interferon-Related Pathways during Influenza A Virus Infection. J. Innate Immun. 2017, 9, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Wolff, T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses 2009, 1, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Abraham, N.; Stojdl, D.F.; Duncan, P.I.; Methot, N.; Ishii, T.; Dube, M.; Vanderhyden, B.C.; Atkins, H.L.; Gray, D.A.; McBurney, M.W.; et al. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 1999, 274, 5953–5962. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 2007, 81, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Schneider, J.; Wolff, T. Double-stranded RNA binding of influenza B virus nonstructural NS1 protein inhibits protein kinase R but is not essential to antagonize production of alpha/beta interferon. J. Virol. 2006, 80, 11667–11677. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.; Saito, S.; Fukuda, R. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J. Virol. 1999, 73, 2425–2433. [Google Scholar] [PubMed]
- Lee, T.G.; Tang, N.; Thompson, S.; Miller, J.; Katze, M.G. The 58,000-dalton cellular inhibitor of the interferon-induced double-stranded RNA-activated protein kinase (PKR) is a member of the tetratricopeptide repeat family of proteins. Mol. Cell. Biol. 1994, 14, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, Y.; Zhang, Z.; Ouyang, J.; Wang, Y.; Yan, R.; Huang, S.; Gao, G.F.; Guo, G.; Chen, J.L. Robust expression of vault RNAs induced by influenza A virus plays a critical role in suppression of PKR-mediated innate immunity. Nucleic Acids Res. 2015, 43, 10321–10337. [Google Scholar] [CrossRef] [PubMed]
- Gusho, E.; Baskar, D.; Banerjee, S. New advances in our understanding of the “unique” RNase L in host pathogen interaction and immune signaling. Cytokine 2016. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Lietzen, N.; Ohman, T.; Rintahaka, J.; Julkunen, I.; Aittokallio, T.; Matikainen, S.; Nyman, T.A. Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog. 2011, 7, e1001340. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.A.; Gorna, M.W.; Baumann, C.L.; Burkard, T.R.; Burckstummer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.K.; Williams, G.D.; Szretter, K.J.; White, J.P.; Proenca-Modena, J.L.; Liu, G.; Olejnik, J.; Brien, J.D.; Ebihara, H.; Muhlberger, E.; et al. Human and Murine IFIT1 Proteins Do Not Restrict Infection of Negative-Sense RNA Viruses of the Orthomyxoviridae, Bunyaviridae, and Filoviridae Families. J. Virol. 2015, 89, 9465–9476. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.V.; George, C.X.; Welch, M.J.; Liou, L.Y.; Hahm, B.; Lewicki, H.; de la Torre, J.C.; Samuel, C.E.; Oldstone, M.B. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 331–336. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Aublin-Gex, A.; Ruggieri, A.; Meyniel-Schicklin, L.; Pradezynski, F.; Davoust, N.; Chantier, T.; Tafforeau, L.; Mangeot, P.E.; Ciancia, C.; et al. The interactomes of influenza virus NS1 and NS2 proteins identify new host factors and provide insights for ADAR1 playing a supportive role in virus replication. PLoS Pathog. 2013, 9, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, F.; Tan, L.; Bai, C.; Chen, B.; Liu, J.; Liang, J.; Liu, C.; Zhang, S.; Lu, G.; et al. Host Protein Moloney Leukemia Virus 10 (MOV10) Acts as a Restriction Factor of Influenza A Virus by Inhibiting the Nuclear Import of the Viral Nucleoprotein. J. Virol. 2016, 90, 3966–3980. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, C.H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, B.; Li, W.; Patil, G.; Liu, L.; Dorf, M.E.; Li, S. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat. Commun. 2017, 8, 13876. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Degols, G.; Gongora, C.; Blondel, D.; Williams, B.R.; Silverman, R.H.; Mechti, N. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 2003, 278, 16151–16158. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Li, J.; Yang, L.; Sun, L.; Liu, W.; He, H. Influenza A Virus-induced expression of ISG20 inhibits viral replication by interacting with nucleoprotein. Virus Genes 2016, 52, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, Z.; Liu, W. Insights into the roles of cyclophilin A during influenza virus infection. Viruses 2013, 5, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, L.; Yu, M.; Wang, Z.; Xu, C.; Xue, Q.; Zhang, K.; Ye, X.; Kitamura, Y.; Liu, W. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell. Microbiol. 2009, 11, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, Z.; Xu, C.; Sun, L.; Chen, J.; Zhang, L.; Liu, W. Cyclophilin A restricts influenza A virus replication through degradation of the M1 protein. PLoS ONE 2012, 7, e31063. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, C.; Wong, G.; Dong, W.; Zheng, W.; Li, Y.; Sun, L.; Zhang, L.; Gao, G.F.; Bi, Y.; et al. Cyclophilin A protects mice against infection by influenza A virus. Sci. Rep. 2016, 6, 28978. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, X.; Zhao, Z.; Xu, C.; Zhang, K.; Chen, C.; Sun, L.; Gao, G.F.; Ye, X.; Liu, W. Cyclophilin E functions as a negative regulator to influenza virus replication by impairing the formation of the viral ribonucleoprotein complex. PLoS ONE 2011, 6, e22625. [Google Scholar] [CrossRef] [PubMed]
- Kerns, J.A.; Emerman, M.; Malik, H.S. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Zhou, L.; Chen, G.; Krug, R.M. Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proc. Natl. Acad. Sci. USA 2015, 112, 14048–14053. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wang, X.; Gao, G. The Short Form of the Zinc Finger Antiviral Protein Inhibits Influenza A Virus Protein Expression and Is Antagonized by the Virus-Encoded NS1. J. Virol. 2017, 91, e01909-16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Collins, M.N.; Hsiang, T.Y.; Krug, R.M. Interferon-induced ISG15 pathway: An ongoing virus-host battle. Trends Microbiol. 2013, 21, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Hsiang, T.Y.; Kuo, R.L.; Krug, R.M. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhong, G.; Zhu, L.; Liu, X.; Shan, Y.; Feng, H.; Bu, Z.; Chen, H.; Wang, C. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J. Immunol. 2010, 184, 5777–5790. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, H.; Zhao, C.; Krug, R.M. Species specificity of the NS1 protein of influenza B virus: NS1 binds only human and non-human primate ubiquitin-like ISG15 proteins. J. Biol. Chem. 2010, 285, 7852–7856. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.M.; Chen, C.J.; Chang, S.C.; Liu, T.J.; Chen, Y.H.; Huang, S.Y.; Shih, S.R. Inhibition of Avian Influenza A Virus Replication in Human Cells by Host Restriction Factor TUFM Is Correlated with Autophagy. mBio 2017, 8, e00481-17. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mok, C.K.; Chan, M.C.; Zhang, Y.; Nal, B.; Kien, F.; Bruzzone, R.; Sanyal, S. Cell Cycle-independent Role of Cyclin D3 in Host Restriction of Influenza Virus Infection. J. Biol. Chem. 2017, 292, 5070–5088. [Google Scholar] [CrossRef] [PubMed]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. The role of BST-2/Tetherin in host protection and disease manifestation. Immun. Inflamm. Dis. 2016, 4, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Leser, G.P.; Lamb, R.A. Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 2011, 417, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Yondola, M.A.; Fernandes, F.; Belicha-Villanueva, A.; Uccelini, M.; Gao, Q.; Carter, C.; Palese, P. Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J. Virol. 2011, 85, 2480–2491. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Grado, V.H.; Hai, R.; Fernandes, F.; Belicha-Villanueva, A.; Carter, C.; Yondola, M.A. Modulation of an ectodomain motif in the influenza A virus neuraminidase alters tetherin sensitivity and results in virus attenuation in vivo. J. Mol. Biol. 2014, 426, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Cavagliotti, L.; Lehmann, M.; Gers-Huber, G.; Kaur, I.; Thomas, Y.; Kaiser, L.; Piguet, V. Influenza virus partially counteracts restriction imposed by tetherin/BST-2. J. Biol. Chem. 2012, 287, 22015–22029. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, M.; Hoffmann, H.H.; Scull, M.A.; Gilmore, R.H.; Bell, K.L.; Ciancanelli, M.; Wilson, S.J.; Crotta, S.; Yu, Y.; Flatley, B.; et al. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell 2015, 160, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Gnirss, K.; Zmora, P.; Blazejewska, P.; Winkler, M.; Lins, A.; Nehlmeier, I.; Gartner, S.; Moldenhauer, A.S.; Hofmann-Winkler, H.; Wolff, T.; et al. Tetherin Sensitivity of Influenza A Viruses Is Strain Specific: Role of Hemagglutinin and Neuraminidase. J. Virol. 2015, 89, 9178–9188. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.; Oh, J.; Giao, N.Q.; Oh, S.; Park, S.H. Enhanced production of enveloped viruses in BST-2-deficient cell lines. Biotechnol. Bioeng. 2017, 114, 2289–2297. [Google Scholar] [CrossRef] [PubMed]
- Bruce, E.A.; Abbink, T.E.; Wise, H.M.; Rollason, R.; Galao, R.P.; Banting, G.; Neil, S.J.; Digard, P. Release of filamentous and spherical influenza A virus is not restricted by tetherin. J. Gen. Virol. 2012, 93, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Londrigan, S.L.; Tate, M.D.; Job, E.R.; Moffat, J.M.; Wakim, L.M.; Gonelli, C.A.; Purcell, D.F.; Brooks, A.G.; Villadangos, J.A.; Reading, P.C.; et al. Endogenous Murine BST-2/Tetherin Is Not a Major Restriction Factor of Influenza A Virus Infection. PLoS ONE 2015, 10, e0142925. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Bertram, S.; Gnirss, K.; Nehlmeier, I.; Gawanbacht, A.; Kirchhoff, F.; Ehrhardt, C.; Ludwig, S.; Kiene, M.; Moldenhauer, A.S.; et al. Influenza A virus does not encode a tetherin antagonist with Vpu-like activity and induces IFN-dependent tetherin expression in infected cells. PLoS ONE 2012, 7, e43337. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yin, L.; Mei, S.; Li, J.; Xu, F.; Sun, H.; Liu, X.; Cen, S.; Liang, C.; Li, A.; et al. BST-2 restricts IAV release and is countered by the viral M2 protein. Biochem. J. 2017, 474, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Wang, Y.; Gilfillan, S.; Lenschow, D.J.; Colonna, M. Cutting edge: Paradoxical roles of BST2/tetherin in promoting type I IFN response and viral infection. J. Immunol. 2012, 188, 2488–2492. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The role of viperin in the innate antiviral response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Olfat, F.; Phoon, M.C.; Hsu, J.P.; Howe, J.L.; Seet, J.E.; Chin, K.C.; Chow, V.T. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol. 2012, 93, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.K.; Schmolke, M.; Hofmann, H.; Ehrhardt, C.; Flory, E.; Munk, C.; Ludwig, S. High level expression of the anti-retroviral protein APOBEC3G is induced by influenza A virus but does not confer antiviral activity. Retrovirology 2009, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zeng, H.; Kumar, A.; Belser, J.A.; Maines, T.R.; Tumpey, T.M. Constitutively Expressed IFITM3 Protein in Human Endothelial Cells Poses an Early Infection Block to Human Influenza Viruses. J. Virol. 2016, 90, 11157–11167. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, K.; Smith, M.S.; Malouli, D.; Mansouri, M.; Nelson, J.A.; Fruh, K. BST2/Tetherin enhances entry of human cytomegalovirus. PLoS Pathog. 2011, 7, e1002332. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Yaneva, R.; Hinson, E.R.; Cresswell, P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 2011, 332, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanz, R.; Mata, M.; Bermejo-Martin, J.; Alvarez, A.; Cortijo, J.; Melero, J.A.; Martinez, I. ISG15 Is Upregulated in Respiratory Syncytial Virus Infection and Reduces Virus Growth through Protein ISGylation. J. Virol. 2016, 90, 3428–3438. [Google Scholar] [CrossRef] [PubMed]
- Jumat, M.R.; Huong, T.N.; Ravi, L.I.; Stanford, R.; Tan, B.H.; Sugrue, R.J. Viperin protein expression inhibits the late stage of respiratory syncytial virus morphogenesis. Antivir. Res. 2015, 114, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, L.; Zan, Y.; Du, N.; Yang, Y.; Tien, P. Human respiratory syncytial virus infection is inhibited by IFN-induced transmembrane proteins. J. Gen. Virol. 2015, 96, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Ribaudo, M.; Guo, J.T.; Barik, S. Identification of Interferon-Stimulated Gene Proteins That Inhibit Human Parainfluenza Virus Type 3. J. Virol. 2016, 90, 11145–11156. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Meyniel-Schicklin, L.; Vonderscher, J.; Andre, P.; Lotteau, V. Virus-host interactomics: New insights and opportunities for antiviral drug discovery. Genome Med. 2014, 6, 115. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalón-Letelier, F.; Brooks, A.G.; Saunders, P.M.; Londrigan, S.L.; Reading, P.C. Host Cell Restriction Factors that Limit Influenza A Infection. Viruses 2017, 9, 376. https://doi.org/10.3390/v9120376
Villalón-Letelier F, Brooks AG, Saunders PM, Londrigan SL, Reading PC. Host Cell Restriction Factors that Limit Influenza A Infection. Viruses. 2017; 9(12):376. https://doi.org/10.3390/v9120376
Chicago/Turabian StyleVillalón-Letelier, Fernando, Andrew G. Brooks, Philippa M. Saunders, Sarah L. Londrigan, and Patrick C. Reading. 2017. "Host Cell Restriction Factors that Limit Influenza A Infection" Viruses 9, no. 12: 376. https://doi.org/10.3390/v9120376
APA StyleVillalón-Letelier, F., Brooks, A. G., Saunders, P. M., Londrigan, S. L., & Reading, P. C. (2017). Host Cell Restriction Factors that Limit Influenza A Infection. Viruses, 9(12), 376. https://doi.org/10.3390/v9120376