
Pressure for Pattern-Specific Intertypic Recombination between Sabin Polioviruses: Evolutionary Implications

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Set of the Viruses Investigated
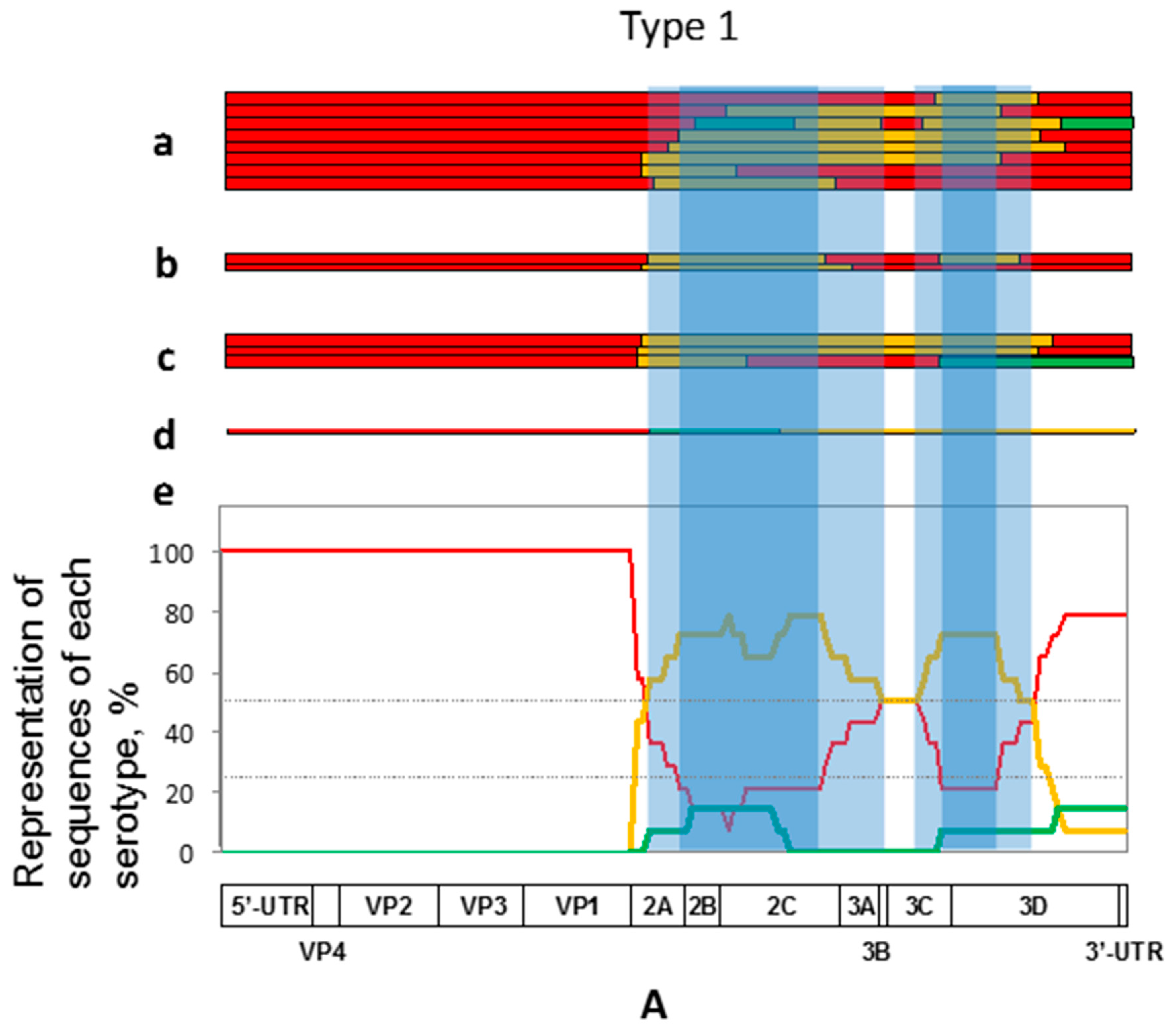
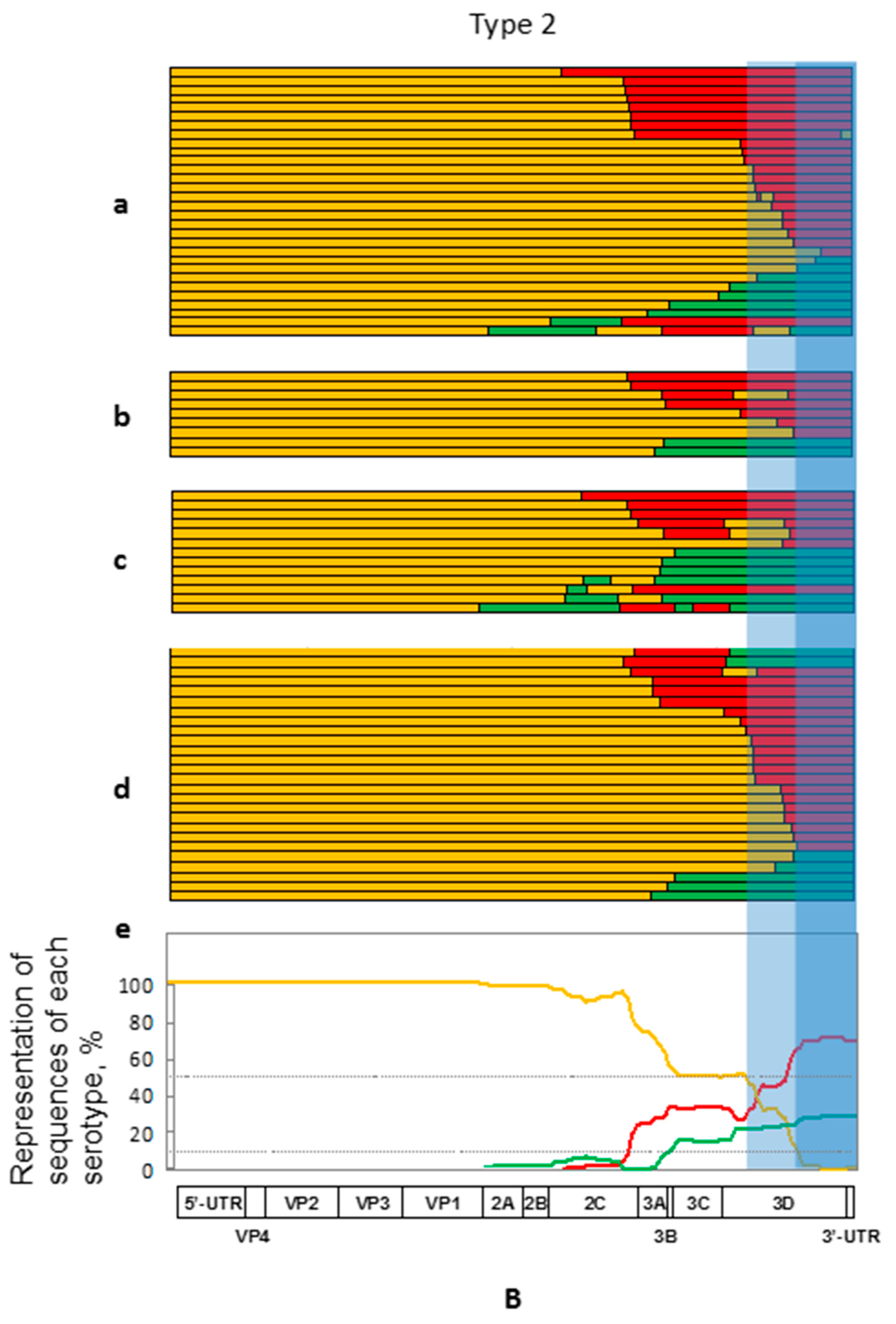
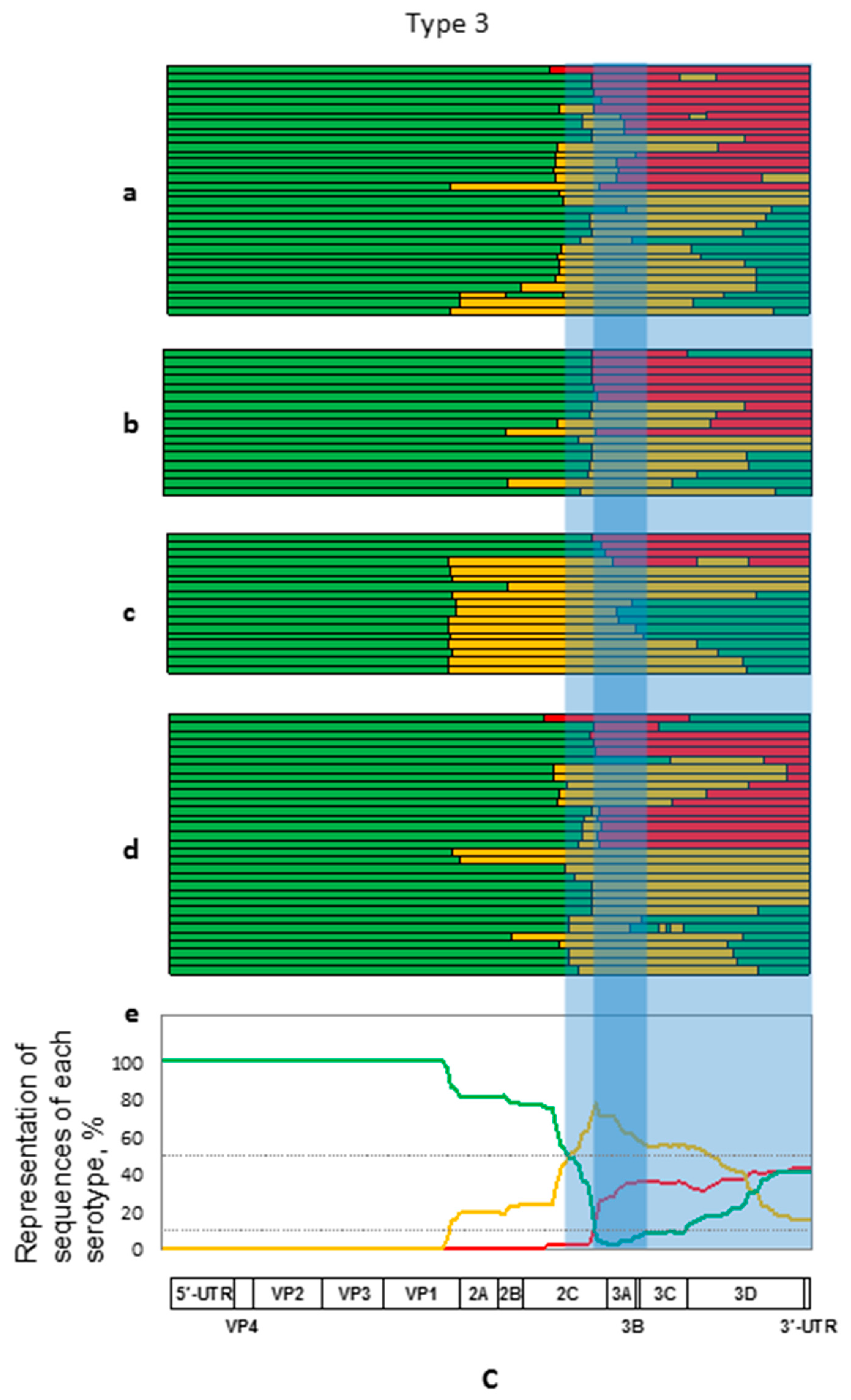
3.2. Serotype-Specific Patterns of Recombination
3.3. Preferential Type-Specific Location of the Lost Genomic Regions
3.4. Peculiarities of the Eliminated Genomic Regions
3.5. Characterization of the Crossover Sites
3.6. General Remarks: Possible Biological Relevance
4. Concluding Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perez-Losada, M.; Arenas, M.; Galan, J.C.; Palero, F.; Gonzalez-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Sabin, A.B.; Boulger, L.R. History of Sabin attenuated poliovirus oral live vaccine strains. J. Biol. Stand. 1973, 1, 115–118. [Google Scholar] [CrossRef]
- Minor, P.D.; Almond, J.W. Poliovirus vaccines: Molecular biology and immune response. In Molecular Biology of Picornaviruses; Semler, B.L., Wimmer, E., Eds.; ASM Press: Washington, DC, USA, 2002; pp. 381–390. [Google Scholar]
- World Health Assembly. Polio Eradication by the Year 2000; Resolution 41.28; World Health Organization: Geneva, Switzerland, 1988. [Google Scholar]
- Kew, O.M.; Sutter, R.W.; de Gourville, E.M.; Dowdle, W.R.; Pallansch, M.A. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 2005, 59, 587–635. [Google Scholar] [CrossRef] [PubMed]
- Agol, V.I. Vaccine-derived polioviruses. Biologicals 2006, 34, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Famulare, M.; Chang, S.; Iber, J.; Zhao, K.; Adeniji, J.A.; Bukbuk, D.; Baba, M.; Behrend, M.; Burns, C.C.; Oberste, M.S. Sabin vaccine reversion in the field: A comprehensive analysis of Sabin-like poliovirus isolates in Nigeria. J. Virol. 2016, 90, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Kew, O.M.; Nottay, B.K. Molecular epidemiology of polioviruses. Rev. Infect. Dis. 1984, 6, S499–S504. [Google Scholar] [CrossRef] [PubMed]
- Minor, P.D.; Ferguson, M.; Evans, D.M.A.; Almond, J.W.; Icenogle, J.P. Antigenic structure of polioviruses of serotypes 1, 2, and 3. J. Gen. Virol. 1986, 67, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Lipskaya, G.Y.; Muzychenko, A.R.; Kutitova, O.K.; Maslova, S.V.; Equestre, M.; Drozdov, S.G.; Bercoff, R.P.; Agol, V.I. Frequent isolation of intertypic poliovirus recombinants with serotype 2 specificity from vaccine-associated polio cases. J. Med. Virol. 1991, 35, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, N.S.; Guillot, S.; Romanenkova, N.; Combiescu, M.; Aubert-Combiescu, A.; Seghier, M.; Caro, V.; Crainic, R.; Delpeyroux, F. Genomic features of intertypic recombinant Sabin poliovirus strains excreted by primary vaccinees. J. Virol. 2001, 75, 5740–5751. [Google Scholar] [CrossRef] [PubMed]
- Savolainen-Kopra, C.; Blomqvist, S. Mechanisms of genetic variation in polioviruses. Rev. Med. Virol. 2010, 20, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.C.; Diop, O.M.; Sutter, R.W.; Kew, O.M. Vaccine-derived polioviruses. J. Infect. Dis. 2014, 210, S283–S293. [Google Scholar] [CrossRef] [PubMed]
- Combelas, N.; Holmblat, B.; Joffret, M.L.; Colbere-Garapin, F.; Delpeyroux, F. Recombination between poliovirus and Coxsackie A viruses of species C: A model of viral genetic plasticity and emergence. Viruses 2011, 3, 1460–1484. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Manual for the Virological Investigation of Poliomyelitis; WHO: Geneva, Switzerland, 1997. [Google Scholar]
- Simonyan, V.; Chumakov, K.; Dingerdissen, H.; Faison, W.; Goldweber, S.; Golikov, A.; Gulzar, N.; Karagiannis, K.; Vinh Nguyen Lam, P.; Maudru, T.; et al. High-performance integrated virtual environment (HIVE): A robust infrastructure for next-generation sequence data analysis. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.C. SimPlot for Windows (Version 1.6), Johns Hopkins University School of Medicine: Baltimore, MD, USA, 1998.
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Balanant, J.; Guillot, S.; Candréa, A.; Delpeyroux, F.; Crainic, R. The natural genomic variability of poliovirus analyzed by a restriction fragment polymorphism assay. Virology 1991, 184, 645–654. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Delpeyroux, F.; Tardy-Panit, M.; Balanant, J.; Combiescu, M.; Combiescu, A.A.; Guillot, S.; Crainic, R. High diversity of poliovirus strains isolated from the central nervous system from patients with vaccine-associated paralytic poliomyelitis. J. Virol. 1994, 68, 8089–8101. [Google Scholar] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. 1977. Biotechnology 1992, 24, 104–108. [Google Scholar] [PubMed]
- Furione, M.; Guillot, S.; Otelea, D.; Balanant, J.; Candrea, A.; Crainic, R. Polioviruses with natural recombinant genomes isolated from vaccine-associated poliomyelitis. Virology 1993, 196, 199–208. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Reporting and Classification of Vaccine-Derived Polioviruses: GPEI Guidelines; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Kew, O.; Morris-Glasgow, V.; Landaverde, M.; Burns, C.; Shaw, J.; Garib, Z.; Andre, J.; Blackman, E.; Freeman, C.J.; Jorba, J.; et al. Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 2002, 296, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Thorley, B.; Paladin, F.J.; Brussen, K.A.; Stambos, V.; Yuen, L.; Utama, A.; Tano, Y.; Arita, M.; Yoshida, H.; et al. Circulation of type 1 vaccine-derived poliovirus in the Philippines in 2001. J. Virol. 2004, 78, 13512–13521. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-F.; Naguib, T.; Yang, S.-J.; Nasr, E.; Jorba, J.; Ahmed, N.; Campagnoli, R.; van der Avoort, H.; Shimizu, H.; Yoneyama, T.; et al. Circulation of endemic type 2 vaccine-derived poliovirus in Egypt, 1983 to 1993. J. Virol. 2003, 77, 8366–8377. [Google Scholar] [CrossRef] [PubMed]
- Adu, F.D.; Iber, J.; Bukbuk, D.; Gumede, N.; Yang, S.-J.; Jorba, J.; Campagnoli, R.; Sule, W.F.; Yang, C.-F.; Burns, C.; et al. Isolation of recombinant type 2 vaccine-derived poliovirus (VDPV) from a Nigerian child. Virus Res. 2007, 127, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Joffret, M.L.; Jegouic, S.; Bessaud, M.; Balanant, J.; Tran, C.; Caro, V.; Holmblat, B.; Razafindratsimandresy, R.; Reynes, J.M.; Rakoto-Andrianarivelo, M.; et al. Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J. Infect. Dis. 2012, 205, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Tardy-Panit, M.; Blondel, B.; Martin, A.; Tekaia, F.; Horaud, F.; Delpeyroux, F. A mutation in the RNA polymerase of poliovirus type 1 contributes to attenuation in mice. J. Virol. 1993, 67, 4630–4638. [Google Scholar] [PubMed]
- Georgescu, M.M.; Delpeyroux, F.; Crainic, R. Tripartite genome organization of a natural type 2 vaccine/nonvaccine recombinant poliovirus. J. Gen. Virol. 1995, 76, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Rezapkin, G.V.; Fan, L.; Asher, D.M.; Fibi, M.R.; Dragunsky, E.M.; Chumakov, K.M. Mutations in Sabin 2 strain of poliovirus and stability of attenuated phenotype. Virology 1999, 258, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, G.V.; Cherkasova, E.A.; Lipskaya, G.Y.; Kew, O.M.; Agol, V.I. Evolution of circulating wild poliovirus and of vaccine-derived poliovirus in an immunodeficient patient: A unifying model. J. Virol. 2000, 74, 7381–7390. [Google Scholar] [CrossRef] [PubMed]
- Odoom, J.K.; Yunus, Z.; Dunn, G.; Minor, P.D.; Martin, J. Changes in population dynamics during long-term evolution of Sabin type 1 poliovirus in an immunodeficient patient. J.Virol. 2008, 82, 9179–9190. [Google Scholar] [CrossRef] [PubMed]
- Pilipenko, E.V.; Poperechny, K.V.; Maslova, S.V.; Melchers, W.J.; Slot, H.J.; Agol, V.I. Cis-element, oriR, involved in the initiation of (-) strand poliovirus RNA: A quasi-globular multi-domain RNA structure maintained by tertiary (‘kissing’) interactions. EMBO J. 1996, 15, 5428–5436. [Google Scholar] [PubMed]
- Agol, V.I. Picornaviruses as a Model for studying the nature of RNA recombination. In Picornaviruses: Molecular Biology, Evolution, and Pathogenesis; Domingo, E., Ehrenfeld, E., Roos, R.P., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 239–252. [Google Scholar]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Romanova, L.I.; Blinov, V.M.; Tolskaya, E.A.; Viktorova, E.G.; Kolesnikova, M.S.; Guseva, E.A.; Agol, V.I. The primary structure of crossover regions of intertypic poliovirus recombinants: A model of recombination between RNA genomes. Virology 1986, 155, 202–213. [Google Scholar] [CrossRef]
- Kuge, S.; Saito, I.; Nomoto, A. Primary structure of poliovirus defective-interfering particle genomes and possible generation mechanisms of the particles. J. Mol. Biol. 1986, 192, 473–487. [Google Scholar] [CrossRef]
- Gmyl, A.P.; Belousov, E.V.; Maslova, S.V.; Khitrina, E.V.; Chetverin, A.B.; Agol, V.I. Nonreplicative RNA recombination in poliovirus. J. Virol. 1999, 73, 8958–8965. [Google Scholar] [PubMed]
- Gmyl, A.P.; Korshenko, S.A.; Belousov, E.V.; Khitrina, E.V.; Agol, V.I. Nonreplicative homologous RNA recombination: Promiscuous joining of RNA pieces? RNA 2003, 9, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Polio Global Eradication Initiative. This Week. Available online: http://polioeradication.org/polio-today/polio-now/this-week/ (accessed on 27 September 2017).
- Chumakov, K.; Ehrenfeld, E.; Wimmer, E.; Agol, V.I. Vaccination against polio should not be stopped. Nat. Rev. 2007, 5, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Duintjer Tebbens, R.J.; Thompson, K.M. Poliovirus vaccination during the endgame: Insights from integrated modeling. Expert Rev. Vaccines 2017, 16, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Yaari, R.; Kaliner, E.; Grotto, I.; Katriel, G.; Moran-Gilad, J.; Sofer, D.; Mendelson, E.; Miller, E.; Huppert, A.; POG Group; et al. Modeling the spread of polio in an IPV-vaccinated population: Lessons learned from the 2013 silent outbreak in southern Israel. BMC Med. 2016, 14, 95. [Google Scholar]
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Jones, J.O.; Andino, R. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Knowlson, S.; Burlison, J.; Giles, E.; Fox, H.; Macadam, A.J.; Minor, P.D. New Strains Intended for the production of inactivatedpPolio vaccine at low-containment after eradication. PLoS Pathog. 2015, 11, e1005316. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, H.; Sein, C.; Hamidi, A.; Bakker, W.A.; Sutter, R.W. Development of inactivated poliovirus vaccine from Sabin strains: A progress report. Biologicals 2016, 44, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Fox, H.; Knowlson, S.; Minor, P.D.; Macadam, A.J. Genetically thermo-stabilised, immunogenic poliovirus empty capsids; a strategy for non-replicating vaccines. PLoS Pathog. 2017, 13, e1006117. [Google Scholar] [CrossRef] [PubMed]
- Savolainen-Kopra, C.; Samoilovich, E.; Kahelin, H.; Hiekka, A.K.; Hovi, T.; Roivainen, M. Comparison of poliovirus recombinants: Accumulation of point mutations provides further advantages. J. Gen. Virol. 2009, 90, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Moustafa, I.M.; Arnold, J.J. Fidelity of nucleotide incorporation by the RNA-dependent RNA Polymerase from poliovirus. Enzymes 2016, 39, 293–323. [Google Scholar] [PubMed]
- Borderia, A.V.; Rozen-Gagnon, K.; Vignuzzi, M. Fidelity variants and RNA quasispecies. Curr. Top. Microbiol. Immunol. 2016, 392, 303–322. [Google Scholar] [PubMed]
- Agol, V.I. Molecular mechanisms of poliovirus variation and evolution. Curr. Top. Microbiol. Immunol. 2006, 299, 211–259. [Google Scholar] [PubMed]
- Poirier, E.Z.; Vignuzzi, M. Virus population dynamics during infection. Curr. Opin. Virol. 2017, 23, 82–87. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Our Study | Published Data | Total | |||
|---|---|---|---|---|---|
| Methods of Analysis | Full Genome Sequencing | Partial Genome Sequencing | Full Genome Sequencing | Partial Genome Sequencing | |
| Number of isolates | 70 | 28 | 33 | 58 | 189 |
| Number (%) of type 1, type 2, and type 3 isolates | 8 (11), | 2 (7), | 3 (9), | 1 (2), | 14 (8), |
| 30 (43), | 9 (32), | 13 (39), | 26 (45), | 78 (41), | |
| 32 (46) | 17 (61) | 17 (52) | 31 (53) | 97 (51) | |
| Number (%) of VDPV | 5 (7) | ? | 15 (45) | ? | ? |
| Number (%) of Sabin-like strains | 65 (93) | ? | 18 (55) | ? | ? |
| Number (%) of AFP cases | 40 (57) | 20 (71) | 24 (73) | 20 (34) | 104 (55) |
| Number (%) of isolates from patients with other diagnoses | 6 (9) | 4 (14) | 1 (3) | - | 11 (6) |
| Number (%) of isolates from healthy persons | 12 (17) | 1 (4) | 7 (21) | 16 (28) | 36 (19) |
| Number (%) of isolates from sewage | 9 (13) | 3 (11) | 1 (3) | 11 (19) | 24 (13) |
| Number (%) of strains of unknown origin | 3 (4) | - | - | 11 (19) | 14 (7) |
| Serotype | Location of Mutation | Attenuating Mutations Lost, % | |
|---|---|---|---|
| In the Retained Part of the Genome | In the Removed Part of the Genome | ||
| 1 | 480 | 67 | - |
| 525 | 11 | - | |
| 935 | 22 | - | |
| 2438 | 33 | - | |
| 2741 | 22 | - | |
| 2795 | 67 | - | |
| 2879 | 0 | - | |
| 6203 | 9 | 82 | |
| 7071 | 8 | 25 | |
| 2 | 398 | 49 | - |
| 481 | 100 | - | |
| 2908 | 5 | - | |
| 2909 | 85 | - | |
| 3 | 472 | 98 | - |
| 2034 | 20 | - | |
| 2493 a | 100 | - | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korotkova, E.; Laassri, M.; Zagorodnyaya, T.; Petrovskaya, S.; Rodionova, E.; Cherkasova, E.; Gmyl, A.; Ivanova, O.E.; Eremeeva, T.P.; Lipskaya, G.Y.; et al. Pressure for Pattern-Specific Intertypic Recombination between Sabin Polioviruses: Evolutionary Implications. Viruses 2017, 9, 353. https://doi.org/10.3390/v9110353
Korotkova E, Laassri M, Zagorodnyaya T, Petrovskaya S, Rodionova E, Cherkasova E, Gmyl A, Ivanova OE, Eremeeva TP, Lipskaya GY, et al. Pressure for Pattern-Specific Intertypic Recombination between Sabin Polioviruses: Evolutionary Implications. Viruses. 2017; 9(11):353. https://doi.org/10.3390/v9110353
Chicago/Turabian StyleKorotkova, Ekaterina, Majid Laassri, Tatiana Zagorodnyaya, Svetlana Petrovskaya, Elvira Rodionova, Elena Cherkasova, Anatoly Gmyl, Olga E. Ivanova, Tatyana P. Eremeeva, Galina Y. Lipskaya, and et al. 2017. "Pressure for Pattern-Specific Intertypic Recombination between Sabin Polioviruses: Evolutionary Implications" Viruses 9, no. 11: 353. https://doi.org/10.3390/v9110353
APA StyleKorotkova, E., Laassri, M., Zagorodnyaya, T., Petrovskaya, S., Rodionova, E., Cherkasova, E., Gmyl, A., Ivanova, O. E., Eremeeva, T. P., Lipskaya, G. Y., Agol, V. I., & Chumakov, K. (2017). Pressure for Pattern-Specific Intertypic Recombination between Sabin Polioviruses: Evolutionary Implications. Viruses, 9(11), 353. https://doi.org/10.3390/v9110353