The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis

{kind=link}
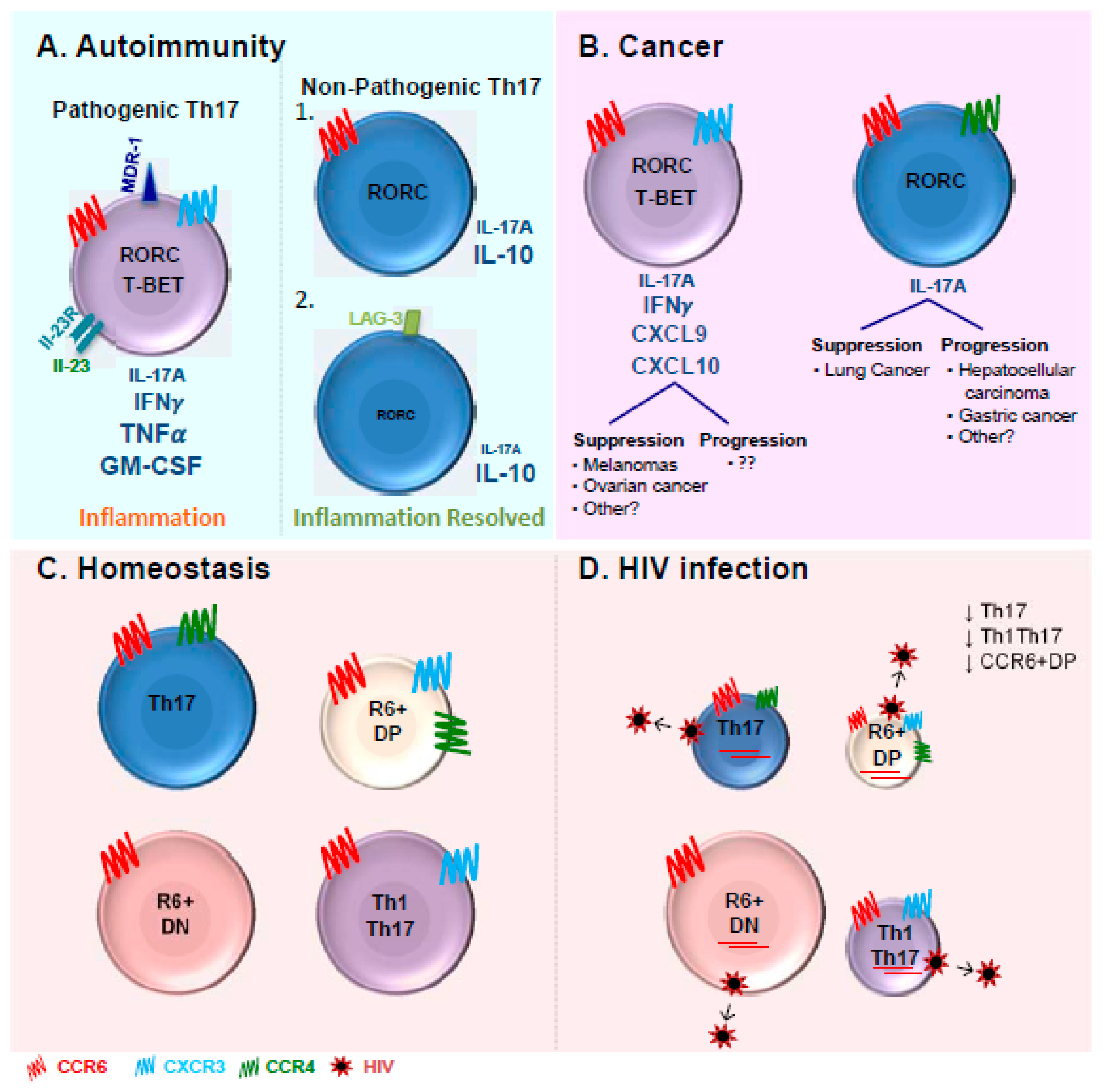
Abstract
1. History of Th17 Cell Discovery
2. Role of Th17 Cells in Promoting Immunity at Barrier Surfaces
2.1. IL-17A
2.2. IL-17F
2.3. IL-22
2.4. IL-26
3. Transcriptional Regulation of Th17 Differentiation
3.1. Positive Regulators
3.1.1. Retinoic Acid-Related Orphan Receptor (ROR) Gamma t (RORγt)
3.1.2. Signal Transduction and Activation of Transcription 3 (STAT3)
3.1.3. Basic Leucine Zipper ATF-Like Transcriptional Factor (BATF)
3.1.4. Interferon Regulatory Factor 4 (IRF4)
3.1.5. Aryl Hydrocarbon Receptor (AhR)
3.1.6. Other Transcription Factors Involved in Th17 Polarization
3.2. Negative Regulators
4. Cytokines Involved in Th17 Lineage Polarization
5. Th17 Lineage Plasticity
6. Surface Markers Defining Human Th17 Subsets
7. Th17 Regulation Mechanisms
8. Natural Th17 Cells
9. Th17 Pathogenicity
10. The Discovery of Long-Lived Th17 Cells in Cancer
11. Role of Th17 Cells in HIV-1/SIV Pathogenesis
12. Pathogenic Versus Non-Pathogenic Th17 Cells during HIV/SIV Infections
13. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dong, C. TH17 cells in development: An updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 2008, 8, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.W.; Netea, M.G. A salty taste to autoimmunity. N. Engl. J. Med. 2013, 368, 2520–2521. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Hatton, R.D.; Weaver, C.T. The Th17 family: Flexibility follows function. Immunol. Rev. 2013, 252, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- Basso, A.S.; Cheroutre, H.; Mucida, D. More stories on Th17 cells. Cell Res. 2009, 19, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Schnurr, M.; Toy, T.; Shin, A.; Wagner, M.; Cebon, J.; Maraskovsky, E. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: A novel role for the cAMP pathway. Blood 2005, 105, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Sheibanie, A.F.; Tadmori, I.; Jing, H.; Vassiliou, E.; Ganea, D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. 2004, 18, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and biology of IL-23 and IL-27: Related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ghilardi, N.; Xie, M.H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Happel, K.I.; Dubin, P.J.; Zheng, M.; Ghilardi, N.; Lockhart, C.; Quinton, L.J.; Odden, A.R.; Shellito, J.E.; Bagby, G.J.; Nelson, S.; et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 2005, 202, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Rudensky, A.Y. Foxp3 in control of the regulatory T cell lineage. Nat. Immunol. 2007, 8, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.T.; Ye, J.J.; Alonso, M.N.; Landrigan, A.; Cheung, R.K.; Engleman, E.; Utz, P.J. Regulation of human Th9 differentiation by type I interferons and IL-21. Immunol. Cell Biol. 2010, 88, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Chung, Y.; Hwang, D.; Yang, X.O.; Kang, H.S.; Ma, L.; Wang, Y.H.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Flavell, R.A. Life, death, and miracles: Th17 cells in the intestine. Eur. J. Immunol. 2012, 42, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytometry Part A 2014, 85, 36–42. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, D.B.; Palmer, M.T.; Lee, Y.K.; Weaver, C.T. Emergence of the Th17 pathway and its role in host defense. Adv. Immunol. 2008, 99, 115–163. [Google Scholar] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; O’Connor, W., Jr.; Rongvaux, A.; van Rooijen, N.; Haberman, A.M.; et al. Control of TH17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; de Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Lecuyer, E.; Rakotobe, S.; Lengline-Garnier, H.; Lebreton, C.; Picard, M.; Juste, C.; Fritzen, R.; Eberl, G.; McCoy, K.D.; Macpherson, A.J.; et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014, 40, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Nishimura, J.; Shima, T.; Umesaki, Y.; Yamamoto, M.; Onoue, M.; Yagita, H.; Ishii, N.; Evans, R.; Honda, K.; et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008, 455, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, Y.; Saito, M.; Nagasawa, M.; Takada, H.; Hara, T.; Tsuchiya, S.; Agematsu, K.; Yamada, M.; Kawamura, N.; Ariga, T.; et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J. Exp. Med. 2009, 206, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, J.; Liu, C.; Novotny, M.; Li, X.; Hamilton, T. IL-17 enhances chemokine gene expression through mRNA stabilization. J. Immunol. 2007, 179, 4135–4141. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Chen, K.; Kolls, J.K. Th17 cell based vaccines in mucosal immunity. Curr. Opin. Immunol. 2013, 25, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.P.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F.; et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Long, A.J.; Bennett, F.; Whitters, M.J.; Karim, R.; Collins, M.; Goldman, S.J.; Dunussi-Joannopoulos, K.; Williams, C.M.; Wright, J.F.; et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 2007, 179, 7791–7799. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W., Jr.; Zenewicz, L.A.; Flavell, R.A. The dual nature of T(H)17 cells: Shifting the focus to function. Nat. Immunol. 2010, 11, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Karow, M.; Flavell, R.A. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007, 27, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Bernard, F.X.; Garcia, M.; Gurney, A.L.; Lecron, J.C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [PubMed]
- Meller, S.; Di Domizio, J.; Voo, K.S.; Friedrich, H.C.; Chamilos, G.; Ganguly, D.; Conrad, C.; Gregorio, J.; Le Roy, D.; Roger, T.; et al. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat. Immunol. 2015, 16, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Cua, D.J. IL-26 AMPs up the T(H)17 arsenal. Nat. Immunol. 2015, 16, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Bell, G.K.; Pearl, J.E.; Fountain, J.J.; Rangel-Moreno, J.; Cilley, G.E.; Shen, F.; Eaton, S.M.; Gaffen, S.L.; Swain, S.L.; et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 2007, 8, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Larussa, T.; Leone, I.; Suraci, E.; Imeneo, M.; Luzza, F. Helicobacter pylori and T helper cells: Mechanisms of immune escape and tolerance. J. Immunol. Res. 2015, 2015, 981328. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Manni, M.L.; Biswas, P.S.; Alcorn, J.F. Clinical consequences of targeting IL-17 and TH17 in autoimmune and allergic disorders. Curr. Allergy Asthma Rep. 2013, 13, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F. Heterogeneity of human CD4(+) T cells against microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Rev. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Nunez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Douek, D.C. Microbial translocation across the GI tract. Annu. Rev. Immunol. 2012, 30, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Brenchley, J.M. Th17 cell dynamics in HIV infection. Curr. Opin. HIV AIDS 2010, 5, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Ancuta, P.; Monteiro, P.; RSekaly, R.P. Th17 lineage commitment and HIV-1 pathogenesis. Curr. Opin. HIV AIDS 2010, in press. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Srinivasan, N.; Pott, J.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.G.; Sefik, E.; Geva-Zatorsky, N.; Kua, L.; Naskar, D.; Teng, F.; Pasman, L.; Ortiz-Lopez, A.; Jupp, R.; Wu, H.J.; et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8141–E8150. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Torchinsky, M.B.; Gobert, M.; Xiong, H.; Xu, M.; Linehan, J.L.; Alonzo, F.; Ng, C.; Chen, A.; Lin, X.; et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 2014, 510, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Unutmaz, D. RORC2: The master of human Th17 cell programming. Eur. J. Immunol. 2009, 39, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Al-Muhsen, S.; Halwani, R.; Ma, C.S.; et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 2015, 349, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Schraml, B.U.; Hildner, K.; Ise, W.; Lee, W.L.; Smith, W.A.; Solomon, B.; Sahota, G.; Sim, J.; Mukasa, R.; Cemerski, S.; et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 2009, 460, 405–409. [Google Scholar] [PubMed]
- Brustle, A.; Heink, S.; Huber, M.; Rosenplanter, C.; Stadelmann, C.; Yu, P.; Arpaia, E.; Mak, T.W.; Kamradt, T.; Lohoff, M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 2007, 8, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links T(H)17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR α and ROR γ. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Grimbacher, B.; Holland, S.M.; Puck, J.M. Hyper-IgE syndromes. Immunol. Rev. 2005, 203, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkurst, C.N.; Muratet, M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, K.J.; Weinmann, A.S. Master regulators or lineage-specifying? Changing views on CD4+ T cell transcription factors. Nat. Rev. Immunol. 2012, 12, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Christensen, J.; O’Garra, A.; Stockinger, B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 2009, 206, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Suto, A.; Iwamoto, T.; Kashiwakuma, D.; Kagami, S.; Suzuki, K.; Takatori, H.; Tamachi, T.; Hirose, K.; Onodera, A.; et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORgammat induction as downstream targets of Stat3. J. Exp. Med. 2014, 211, 1857–1874. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.K.; Lahesmaa, R. Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol. Rev. 2014, 261, 62–83. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Zhang, H.H.; Tsang, H.; Gardina, P.J.; Myers, T.G.; Nagarajan, V.; Lee, C.H.; Farber, J.M. PLZF regulates CCR6 and is critical for the acquisition and maintenance of the Th17 phenotype in human cells. J. Immunol. 2015, 194, 4350–4361. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, Y.; Suto, A.; Kashiwakuma, D.; Kanari, H.; Kagami, S.; Ikeda, K.; Hirose, K.; Watanabe, N.; Grusby, M.J.; Iwamoto, I.; et al. c-Maf activates the promoter and enhancer of the IL-21 gene, and TGF-β inhibits c-Maf-induced IL-21 production in CD4+ T cells. J. Leukocyte Biol. 2010, 87, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Jin, H.; Burns, E.J.; Nadeau, M.; Yeste, A.; Kumar, D.; Rangachari, M.; Zhu, C.; Xiao, S.; Seavitt, J.; et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat. Immunol. 2012, 13, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; Tato, C.M.; Davidson, T.S.; Kanno, Y.; Chen, Z.; Yao, Z.; Blank, R.B.; Meylan, F.; Siegel, R.; Hennighausen, L.; et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007, 26, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wei, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.Y.; Hatfield, J.K.; Brown, M.A. Ikaros sets the potential for Th17 lineage gene expression through effects on chromatin state in early T cell development. J. Biol. Chem. 2013, 288, 35170–35179. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Iwai, Y.; Oh-Hora, M.; Yamamoto, M.; Morio, T.; Aoki, K.; Ohya, K.; Jetten, A.M.; Akira, S.; Muta, T.; et al. I κ B[zeta] regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 2010, 464, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ruan, Q.; Hilliard, B.; Devirgiliis, J.; Karin, M.; Chen, Y.H. Transcriptional regulation of the Th17 immune response by IKK(α). J. Exp. Med. 2011, 208, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Meng, G.; Strober, W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol. 2008, 9, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnolzer, M.; et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Gokmen, M.R.; Dong, R.; Kanhere, A.; Powell, N.; Perucha, E.; Jackson, I.; Howard, J.K.; Hernandez-Fuentes, M.; Jenner, R.G.; Lord, G.M. Genome-wide regulatory analysis reveals that T-bet controls Th17 lineage differentiation through direct suppression of IRF4. J. Immunol. 2013, 191, 5925–5932. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Chen, X.; Shim, J.H.; Hwang, E.S.; Jang, E.; Bolm, A.N.; Oukka, M.; Kuchroo, V.K.; Glimcher, L.H. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat. Immunol. 2011, 12, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Sharma, A.; Ghosh, A.; Sen, J.M. T cell factor-1 negatively regulates expression of IL-17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. J. Immunol. 2011, 186, 3946–3952. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Davidson, T.S.; Wei, G.; Jankovic, D.; Cui, K.; Schones, D.E.; Guo, L.; Zhao, K.; Shevach, E.M.; Paul, W.E. Down-regulation of Gfi-1 expression by TGF-β is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J. Exp. Med. 2009, 206, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhang, R.; Yang, J.; Li, Q.; Qin, L.; Zhu, C.; Liu, J.; Ning, H.; Shin, M.S.; Gupta, M.; et al. Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat. Commun. 2011, 2, 314. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.; Walline, C.C.; Hollister, K.; Dent, A.L.; Blum, J.S.; Firulli, A.B.; Kaplan, M.H. The transcription factor Twist1 limits T helper 17 and T follicular helper cell development by repressing the gene encoding the interleukin-6 receptor α chain. J. Biol. Chem. 2013, 288, 27423–27433. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Nowak, N.; Beyer, M.; Mayer, G.; et al. The nuclear receptor PPAR γ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Moisan, J.; Grenningloh, R.; Bettelli, E.; Oukka, M.; Ho, I.C. Ets-1 is a negative regulator of Th17 differentiation. J. Exp. Med. 2007, 204, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Puppi, M.; Schluns, K.S.; Yu-Lee, L.Y.; Dong, C.; Lacorazza, H.D. The transcription factor E74-like factor 4 suppresses differentiation of proliferating CD4+ T cells to the Th17 lineage. J. Immunol. 2014, 192, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Li, J.; Vaque, J.P.; Konkel, J.E.; Wang, W.; Zhang, B.; Zhang, P.; Zamarron, B.F.; Yu, D.; Wu, Y.; et al. Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nat. Immunol. 2011, 12, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Miao, T.; Raymond, M.; Bhullar, P.; Ghaffari, E.; Symonds, A.L.; Meier, U.C.; Giovannoni, G.; Li, S.; Wang, P. Early growth response gene-2 controls IL-17 expression and Th17 differentiation by negatively regulating Batf. J. Immunol. 2013, 190, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Yu, J.; Zufferey, C.; Brustle, A.; Wirtz, S.; Weigmann, B.; Hoffman, A.; Schenk, M.; Galle, P.R.; Lehr, H.A.; et al. IRF4 regulates IL-17A promoter activity and controls RORγt-dependent Th17 colitis in vivo. Inflamm. Bowel Dis. 2011, 17, 1343–1358. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. Regulation of IL-17 in autoimmune diseases by transcriptional factors and microRNAs. Front. Genet. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Stritesky, G.L.; Yeh, N.; Kaplan, M.H. IL-23 promotes maintenance but not commitment to the Th17 lineage. J. Immunol. 2008, 181, 5948–5955. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Flavell, R.A.; Stockinger, B. Signals mediated by transforming growth factor-β initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol. 2006, 7, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Flavell, R.A. T cell-produced transforming growth factor-β1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 2007, 26, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Gutcher, I.; Donkor, M.K.; Ma, Q.; Rudensky, A.Y.; Flavell, R.A.; Li, M.O. Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation. Immunity 2011, 34, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ren, G.; Zhang, L.; Roberts, A.I.; Zhao, X.; Bothwell, A.L.; van Kaer, L.; Shi, Y.; Das, G. Transforming growth factor β is dispensable for the molecular orchestration of Th17 cell differentiation. J. Exp. Med. 2009, 206, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; O’Shea, J.J. T(H)-17 differentiation: Of mice and men. Nat. Immunol. 2007, 8, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Defining the human T helper 17 cell phenotype. Trends Immunol. 2012, 33, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Annunziato, F. Th17 plasticity: Pathophysiology and treatment of chronic inflammatory disorders. Curr. Opin. Pharmacol. 2014, 17, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T(H)-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupe, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-β are required for differentiation of human T(H)17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tato, C.M.; Muul, L.; Laurence, A.; O’Shea, J.J. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheumatol. 2007, 56, 2936–2946. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.G.; Williams, C.B. Generation and function of induced regulatory T cells. Front. Immunol. 2013, 4, 152. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Chen, Y.; Tato, C.M.; Laurence, A.; Joyce-Shaikh, B.; Blumenschein, W.M.; McClanahan, T.K.; O’Shea, J.J.; Cua, D.J. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 2009, 10, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Haines, C.J.; Chen, Y.; Blumenschein, W.M.; Jain, R.; Chang, C.; Joyce-Shaikh, B.; Porth, K.; Boniface, K.; Mattson, J.; Basham, B.; et al. Autoimmune memory T helper 17 cell function and expansion are dependent on interleukin-23. Cell Rep. 2013, 3, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Kang, Z.; Bulek, K.; Youzhong, W.; Kim, T.W.; Chen, Y.; Altuntas, C.Z.; Sass Bak-Jensen, K.; McGeachy, M.J.; Do, J.S.; et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity 2010, 32, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, F.; Basso, C.; Preite, S.; Reboldi, A.; Baumjohann, D.; Perlini, L.; Lanzavecchia, A.; Sallusto, F. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1β production by myeloid cells. Nat. Commun. 2016, 7, 11541. [Google Scholar] [CrossRef] [PubMed]
- Micci, L.; Cervasi, B.; Ende, Z.S.; Iriele, R.I.; Reyes-Aviles, E.; Vinton, C.; Else, J.; Silvestri, G.; Ansari, A.A.; Villinger, F.; et al. Paucity of IL-21-producing CD4(+) T cells is associated with Th17 cell depletion in SIV infection of rhesus macaques. Blood 2012, 120, 3925–3935. [Google Scholar] [CrossRef] [PubMed]
- Santarlasci, V.; Maggi, L.; Capone, M.; Querci, V.; Beltrame, L.; Cavalieri, D.; D’Aiuto, E.; Cimaz, R.; Nebbioso, A.; Liotta, F.; et al. Rarity of human T helper 17 cells is due to retinoic acid orphan receptor-dependent mechanisms that limit their expansion. Immunity 2012, 36, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Pallikkuth, S.; Micci, L.; Ende, Z.S.; Iriele, R.I.; Cervasi, B.; Lawson, B.; McGary, C.S.; Rogers, K.A.; Else, J.G.; Silvestri, G.; et al. Maintenance of intestinal Th17 cells and reduced microbial translocation in SIV-infected rhesus macaques treated with interleukin (IL)-21. PLoS Pathog. 2013, 9, e1003471. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Schmitt, N.; Bentebibel, S.E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Djuretic, I.; Tanasa, B.; Rao, A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu. Rev. Immunol. 2006, 24, 607–656. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Carr, E.J.; Linterman, M.A.; Veldhoen, M. Cellular plasticity of CD4+ T cells in the intestine. Front. Immunol. 2014, 5, 488. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Cox, C.A.; Vistica, B.P.; Tan, C.; Wawrousek, E.F.; Gery, I. Phenotype switching by inflammation-inducing polarized Th17 cells, but not by Th1 cells. J. Immunol. 2008, 181, 7205–7213. [Google Scholar] [CrossRef] [PubMed]
- Abromson-Leeman, S.; Bronson, R.T.; Dorf, M.E. Encephalitogenic T cells that stably express both T-bet and ROR γ t consistently produce IFNβ but have a spectrum of IL-17 profiles. J. Neuroimmunol. 2009, 215, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Fili, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Bending, D.; Newland, S.; Krejci, A.; Phillips, J.M.; Bray, S.; Cooke, A. Epigenetic changes at Il12rb2 and Tbx21 in relation to plasticity behavior of Th17 cells. J. Immunol. 2011, 186, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Blumenschein, W.M.; Brovont-Porth, K.; McGeachy, M.J.; Basham, B.; Desai, B.; Pierce, R.; McClanahan, T.K.; Sadekova, S.; de Waal Malefyt, R. Human Th17 cells comprise heterogeneous subsets including IFN-γ-producing cells with distinct properties from the Th1 lineage. J. Immunol. 2010, 185, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Latorre, D.; Mele, F.; Foglierini, M.; de Gregorio, C.; Cassotta, A.; Fernandez, B.; Kelderman, S.; Schumacher, T.N.; Corti, D.; et al. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 2015, 347, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C.; Tolaini, M.; Menzel, U.; et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, A.; Santarlasci, V.; Maggi, L.; Capone, M.; Rossi, M.C.; Querci, V.; de Palma, R.; Chang, H.D.; Thiel, A.; Cimaz, R.; et al. Demethylation of the RORC2 and IL17A in human CD4+ T lymphocytes defines Th17 origin of nonclassic Th1 cells. J. Immunol. 2015, 194, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Cimaz, R.; Capone, M.; Santarlasci, V.; Querci, V.; Simonini, G.; Nencini, F.; Liotta, F.; Romagnani, S.; Maggi, E.; et al. Brief report: Etanercept inhibits the tumor necrosis factor α-driven shift of Th17 lymphocytes toward a nonclassic Th1 phenotype in juvenile idiopathic arthritis. Arthritis Rheumatol. 2014, 66, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Capone, M.; Cardilicchia, E.; Frosali, F.; Querci, V.; Angeli, R.; Matucci, A.; Fambrini, M.; et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J. Allergy Clin. Immunol. 2010, 125, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Turner, J.E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat. Immunol. 2013, 14, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Gamrekelashvili, J.; Manns, M.P.; Greten, T.F.; Korangy, F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011, 117, 6532–6541. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Koenen, H.J.; Smeets, R.L.; Vink, P.M.; van Rijssen, E.; Boots, A.M.; Joosten, I. Human CD25 high Foxp3 pos regulatory T cells differentiate into IL-17-producing cells. Blood 2008, 112, 2340–2352. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Mackay, C.R.; Lanzavecchia, A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science 1997, 277, 2005–2007. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Butcher, E.C. Chemokines in tissue-specific and microenvironment-specific lymphocyte homing. Curr. Opin. Immunol. 2000, 12, 336–341. [Google Scholar] [CrossRef]
- Altin, J.G.; Sloan, E.K. The role of CD45 and CD45-associated molecules in T cell activation. Immunol. Cell Biol. 1997, 75, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Rivino, L.; Messi, M.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F.; Geginat, J. Chemokine receptor expression identifies Pre-T helper (Th)1, Pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J. Exp. Med. 2004, 200, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Woodland, D.L.; Kohlmeier, J.E. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat. Rev. Immunol. 2009, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Reiss, Y.; Proudfoot, A.E.; Power, C.A.; Campbell, J.J.; Butcher, E.C. CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J. Exp. Med. 2001, 194, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Zhang, H.H.; Foley, J.F.; Hedrick, M.N.; Farber, J.M. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J. Immunol. 2008, 180, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kang, S.G.; Lee, J.; Sun, Z.; Kim, C.H. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2009, 2, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Impellizzieri, D.; Basso, C.; Laroni, A.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B. T-cell trafficking in the central nervous system. Immunol. Rev. 2012, 248, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Campbell, D.J. IL-1β promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J. Immunol. 2014, 193, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Seigel, B.; Flecken, T.; Wolanski, J.; Blum, H.E.; Thimme, R. Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J. Immunol. 2012, 188, 5438–5447. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; De Palma, R.; Santarlasci, V.; Maggi, L.; Capone, M.; Frosali, F.; Rodolico, G.; Querci, V.; Abbate, G.; Angeli, R.; et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J. Exp. Med. 2008, 205, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Main features of human T helper 17 cells. Ann. N. Y. Acad. Sci. 2013, 1284, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Wacleche, V.S.; Goulet, J.P.; Gosselin, A.; Monteiro, P.; Soudeyns, H.; Fromentin, R.; Jenabian, M.A.; Vartanian, S.; Deeks, S.G.; Chomont, N.; et al. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Kozhaya, L.; ElHed, A.; Ramesh, R.; Carlson, T.J.; Djuretic, I.M.; Sundrud, M.S.; Unutmaz, D. Cytokine signals through PI-3 kinase pathway modulate Th17 cytokine production by CCR6+ human memory T cells. J. Exp. Med. 2011, 208, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Dunay, G.A.; Toth, I.; Eberhard, J.M.; Degen, O.; Tolosa, E.; van Lunzen, J.; Hauber, J.; Schulze Zur Wiesch, J. Parallel assessment of Th17 cell frequencies by surface marker co-expression versus ex vivo IL-17 production in HIV-1 infection. Cytometry B Clin. Cytom. 2016, 90, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.M.; Lee, E.J.; Donovan, A.M.; Guo, K.; Harper, M.S.; Frank, D.N.; McCarter, M.D.; Santiago, M.L.; Wilson, C.C. Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology 2016, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.C.; Guo, K.; Dillon, S.M.; Phang, T.; Lee, E.J.; Harper, M.S.; Helm, K.; Kappes, J.C.; Ochsenbauer, C.; McCarter, M.D.; et al. The transcriptome of HIV-1 infected intestinal CD4+ T cells exposed to enteric bacteria. PLoS Pathog. 2017, 13, e1006226. [Google Scholar] [CrossRef] [PubMed]
- Macal, M.; Sankaran, S.; Chun, T.W.; Reay, E.; Flamm, J.; Prindiville, T.J.; Dandekar, S. Effective CD4+ T-cell restoration in gut-associated lymphoid tissue of HIV-infected patients is associated with enhanced Th17 cells and polyfunctional HIV-specific T-cell responses. Mucosal Immunol. 2008, 1, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.M.; Manuzak, J.A.; Leone, A.K.; Lee, E.J.; Rogers, L.M.; McCarter, M.D.; Wilson, C.C. HIV-1 infection of human intestinal lamina propria CD4+ T cells in vitro is enhanced by exposure to commensal Escherichia coli. J. Immunol. 2012, 189, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; McKinnon, L.R.; Kovacs, C.; Kandel, G.; Huibner, S.; Chege, D.; Shahabi, K.; Benko, E.; Loutfy, M.; Ostrowski, M.; et al. Mucosal Th17 cell function is altered during HIV infection and is an independent predictor of systemic immune activation. J. Immunol. 2013, 191, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Chege, D.; Sheth, P.M.; Kain, T.; Kim, C.J.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Kandel, G.; Chun, T.W.; Ostrowski, M.; et al. Sigmoid Th17 populations, the HIV latent reservoir, and microbial translocation in men on long-term antiretroviral therapy. AIDS 2011, 25, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, A.; Deleage, C.; Sereti, I.; Rerknimitr, R.; Phanuphak, N.; Phuang-Ngern, Y.; Estes, J.D.; Sandler, N.G.; Sukhumvittaya, S.; Marovich, M.; et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014, 10, e1004543. [Google Scholar] [CrossRef] [PubMed]
- Boily-Larouche, G.; Omollo, K.; Cheruiyot, J.; Njoki, J.; Kimani, M.; Kimani, J.; Oyugi, J.; Lajoie, J.; Fowke, K.R. CD161 identifies polyfunctional Th1/Th17 cells in the genital mucosa that are depleted in HIV-infected female sex workers from Nairobi, Kenya. Sci. Rep. 2017, 7, 11123. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Diveu, C.; McGeachy, M.J.; Boniface, K.; Stumhofer, J.S.; Sathe, M.; Joyce-Shaikh, B.; Chen, Y.; Tato, C.M.; McClanahan, T.K.; de Waal Malefyt, R.; et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J. Immunol. 2009, 182, 5748–5756. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Lanzavecchia, A.; Sallusto, F.; Campbell, D.J. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 2012, 119, 4430–4440. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.R.; Nowyhed, H.N.; Choi, J.Y.; Poholek, A.C.; Odegard, J.M.; Flavell, R.A.; Craft, J. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat. Immunol. 2009, 10, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, L.A.; Jain, R.; Haines, C.; Cua, D.J. Th17 cell development: From the cradle to the grave. Immunol. Rev. 2013, 252, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernandez-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Massot, B.; Michel, M.L.; Diem, S.; Ohnmacht, C.; Latour, S.; Dy, M.; Eberl, G.; Leite-de-Moraes, M.C. TLR-induced cytokines promote effective proinflammatory natural Th17 cell responses. J. Immunol. 2014, 192, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.S.; Huang, S.T.; Tsai, M.H.; Yen, C.C.; Lai, Y.G.; Liou, Y.H.; Lin, C.K.; Liao, N.S. The interleukin-15 system suppresses T cell-mediated autoimmunity by regulating negative selection and nT(H)17 cell homeostasis in the thymus. J. Autoimmun. 2015, 56, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Sundrud, M.S.; Trivigno, C. Identity crisis of Th17 cells: Many forms, many functions, many questions. Semin. Immunol. 2013, 25, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in human immunity: Lessons from genetics and therapeutic interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Lochner, M.; Peduto, L.; Cherrier, M.; Sawa, S.; Langa, F.; Varona, R.; Riethmacher, D.; Si-Tahar, M.; Di Santo, J.P.; Eberl, G. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORβt+ T cells. J. Exp. Med. 2008, 205, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Nikoopour, E.; Schwartz, J.A.; Huszarik, K.; Sandrock, C.; Krougly, O.; Lee-Chan, E.; Singh, B. Th17 polarized cells from nonobese diabetic mice following mycobacterial adjuvant immunotherapy delay type 1 diabetes. J. Immunol. 2010, 184, 4779–4788. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weiner, J.; Liu, Y.; Smith, A.J.; Huss, D.J.; Winger, R.; Peng, H.; Cravens, P.D.; Racke, M.K.; Lovett-Racke, A.E. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J. Exp. Med. 2009, 206, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.R.; Nelson, M.H.; Himes, R.A.; Li, Z.; Mehrotra, S.; Paulos, C.M. Th17 cells in cancer: The ultimate identity crisis. Front. Immunol. 2014, 5, 276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Hoechst, B.; Gamrekelashvili, J.; Ormandy, L.A.; Voigtlander, T.; Wedemeyer, H.; Ylaya, K.; Wang, X.W.; Hewitt, S.M.; Manns, M.P.; et al. Human CCR4+ CCR6+ Th17 cells suppress autologous CD8+ T cell responses. J. Immunol. 2012, 188, 6055–6062. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Saito, H.; Ikeguchi, M. Prevalence and clinical relevance of Th17 cells in patients with gastric cancer. J. Surg. Res. 2012, 178, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, Y.; Odunsi, K.; Chen, W.; Peng, G.; Matsuzaki, J.; Wang, R.F. Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 15505–15510. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kono, K.; Mizukami, Y.; Kawaguchi, Y.; Mimura, K.; Watanabe, M.; Izawa, S.; Fujii, H. Distribution of Th17 cells and FoxP3(+) regulatory T cells in tumor-infiltrating lymphocytes, tumor-draining lymph nodes and peripheral blood lymphocytes in patients with gastric cancer. Cancer Sci. 2010, 101, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Ye, J.; Hsueh, E.C.; Zhang, Y.; Hoft, D.F.; Peng, G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J. Immunol. 2010, 184, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Zhao, E.; Liu, Y.; Wang, Y.; Vatan, L.; Szeliga, W.; Moyer, J.; Klimczak, A.; Lange, A.; Zou, W. Human TH17 cells are long-lived effector memory cells. Sci. Transl. Med. 2011, 3, 104ra100. [Google Scholar] [CrossRef] [PubMed]
- Muranski, P.; Borman, Z.A.; Kerkar, S.P.; Klebanoff, C.A.; Ji, Y.; Sanchez-Perez, L.; Sukumar, M.; Reger, R.N.; Yu, Z.; Kern, S.J.; et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 2011, 35, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat. Med. 2005, 11, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Swanson, H.M.; Fujii, N.; Estey, E.H.; Riddell, S.R. A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity 2009, 31, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Luckey, C.J.; Weaver, C.T. Stem-cell-like qualities of immune memory; CD4+ T cells join the party. Cell Stem Cell 2012, 10, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Ji, Y.; Restifo, N.P. Wnt/β-catenin signaling in T-cell immunity and cancer immunotherapy. Clin. Cancer Res. 2010, 16, 4695–4701. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Schacker, T.W.; Ruff, L.E.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Douek, D.C.; Picker, L.J.; Koup, R.A. T cell dynamics in HIV-1 infection. Annu. Rev. Immunol. 2003, 21, 265–304. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Mattapallil, J.J. Loss and dysregulation of Th17 cells during HIV infection. Clin. Dev. Immunol. 2013, 2013, 852418. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Trindade, C.J.; Laurence, A.; Heraud, J.M.; Brenchley, J.M.; Ferrari, M.G.; Zaffiri, L.; Tryniszewska, E.; Tsai, W.P.; Vaccari, M.; et al. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 2008, 1, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Douek, D.C. HIV disease: Fallout from a mucosal catastrophe? Nat. Immunol. 2006, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Ancuta, P.; Kamat, A.; Kunstman, K.J.; Kim, E.Y.; Autissier, P.; Wurcel, A.; Zaman, T.; Stone, D.; Mefford, M.; Morgello, S.; et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE 2008, 3, e2516. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Funderburg, N.T.; Brenchley, J.M. Microbial translocation, immune activation, and HIV disease. Trends Microbiol. 2013, 21, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 2008, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Paiardini, M.; Knox, K.S.; Asher, A.I.; Cervasi, B.; Asher, T.E.; Scheinberg, P.; Price, D.A.; Hage, C.A.; Kholi, L.M.; et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 2008, 112, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, E.J.; Greenwald, J.H.; Lee, P.I.; Biancotto, A.; Read, S.W.; Yao, M.A.; Hodge, J.N.; Thompson, W.L.; Kovacs, S.B.; Chairez, C.L.; et al. CD4+ T cells, including Th17 and cycling subsets, are intact in the gut mucosa of HIV-1-infected long-term nonprogressors. J. Virol. 2011, 85, 5880–5888. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, S.; George, M.D.; Baumler, A.J. Th17 cells, HIV and the gut mucosal barrier. Curr. Opin. HIV AIDS 2010, 5, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.R.; Routy, J.P.; et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J. Immunol. 2010, 184, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- El Hed, A.; Khaitan, A.; Kozhaya, L.; Manel, N.; Daskalakis, D.; Borkowsky, W.; Valentine, F.; Littman, D.R.; Unutmaz, D. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J. Infect. Dis. 2010, 201, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, Y.; Tuen, M.; Shen, G.; Nawaz, F.; Arthos, J.; Wolff, M.J.; Poles, M.A.; Hioe, C.E. Preferential HIV infection of CCR6+ Th17 cells is associated with higher levels of virus receptor expression and lack of CCR5 ligands. J. Virol. 2013, 87, 10843–10854. [Google Scholar] [CrossRef] [PubMed]
- Mavigner, M.; Cazabat, M.; Dubois, M.; L’Faqihi, F.E.; Requena, M.; Pasquier, C.; Klopp, P.; Amar, J.; Alric, L.; Barange, K.; et al. Altered CD4+ T cell homing to the gut impairs mucosal immune reconstitution in treated HIV-infected individuals. J. Clin. Investig. 2012, 122, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, C.; Requena, M.; Mavigner, M.; Cazabat, M.; Carrere, N.; Suc, B.; Barange, K.; Alric, L.; Marchou, B.; Massip, P.; et al. CCR6(−) regulatory T cells blunt the restoration of gut Th17 cells along the CCR6-CCL20 axis in treated HIV-1-infected individuals. Mucosal Immunol. 2016, 9, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Favre, D.; Lederer, S.; Kanwar, B.; Ma, Z.M.; Proll, S.; Kasakow, Z.; Mold, J.; Swainson, L.; Barbour, J.D.; Baskin, C.R.; et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009, 5, e1000295. [Google Scholar] [CrossRef] [PubMed]
- Favre, D.; Mold, J.; Hunt, P.W.; Kanwar, B.; Loke, P.; Seu, L.; Barbour, J.D.; Lowe, M.M.; Jayawardene, A.; Aweeka, F.; et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2010, 2, 32ra36. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Sandler, N.G.; Douek, D.C.; Mattapallil, J.J. Suppressed Th17 levels correlate with elevated PIAS3, SHP2, and SOCS3 expression in CD4 T cells during acute simian immunodeficiency virus infection. J. Virol. 2013, 87, 7093–7101. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Estes, J.D.; Sun, X.; Ortiz, A.M.; Barber, J.S.; Harris, L.D.; Cervasi, B.; Yokomizo, L.K.; Pan, L.; Vinton, C.L.; et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 2012, 5, 646–657. [Google Scholar] [CrossRef] [PubMed]
- DaFonseca, S.; Niessl, J.; Pouvreau, S.; Wacleche, V.S.; Gosselin, A.; Cleret-Buhot, A.; Bernard, N.; Tremblay, C.; Jenabian, M.A.; Routy, J.P.; et al. Impaired Th17 polarization of phenotypically naive CD4(+) T-cells during chronic HIV-1 infection and potential restoration with early ART. Retrovirology 2015, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Mercer, F.; Khaitan, A.; Kozhaya, L.; Aberg, J.A.; Unutmaz, D. Differentiation of IL-17-producing effector and regulatory human T cells from lineage-committed naive precursors. J. Immunol. 2014, 193, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Zhang, Y.; Monteiro, P.; Goulet, J.P.; Gosselin, A.; Grandvaux, N.; Hope, T.J.; Fassati, A.; Routy, J.P.; Ancuta, P. HIV-1 selectively targets gut-homing CCR6+CD4+ T cells via mTOR-dependent mechanisms. JCI Insight 2017, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Chapman, J.M.; Jha, A.R.; Snyder-Cappione, J.E.; Pagan, M.; Leal, F.E.; Boland, B.S.; Norris, P.J.; Rosenberg, M.G.; Nixon, D.F. Suppression of HIV-1 plasma viral load below detection preserves IL-17 producing T cells in HIV-1 infection. AIDS 2008, 22, 990–992. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, A.; Prado, J.G.; Kang, Y.H.; Chen, F.; Riddell, L.A.; Luzzi, G.; Goulder, P.; Klenerman, P. HIV-1 infection is characterized by profound depletion of CD161+ Th17 cells and gradual decline in regulatory T cells. AIDS 2010, 24, 491–502. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.S.; Alvarez, X.; Harrington, S.; Cervasi, B.; Ryan, E.S.; Iriele, R.I.; Paganini, S.; Harper, J.L.; Easley, K.; Silvestri, G.; et al. The loss of CCR6+ and CD161+ CD4+ T-cell homeostasis contributes to disease progression in SIV-infected rhesus macaques. Mucosal Immunol. 2017, 10, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Gosselin, A.; Wacleche, V.S.; El-Far, M.; Said, E.A.; Kared, H.; Grandvaux, N.; Boulassel, M.R.; Routy, J.P.; Ancuta, P. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin β7. J. Immunol. 2011, 186, 4618–4630. [Google Scholar] [CrossRef] [PubMed]
- Kader, M.; Wang, X.; Piatak, M.; Lifson, J.; Roederer, M.; Veazey, R.; Mattapallil, J.J. α4(+)α7(hi)CD4(+) memory T cells harbor most Th-17 cells and are preferentially infected during acute SIV infection. Mucosal Immunol. 2009, 2, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Stieh, D.J.; Matias, E.; Xu, H.; Fought, A.J.; Blanchard, J.L.; Marx, P.A.; Veazey, R.S.; Hope, T.J. Th17 cells are preferentially infected very early after vaginal transmission of SIV in macaques. Cell Host Microbe 2016, 19, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.; Wiche Salinas, T.R.; Planas, D.; Wacleche, V.S.; Zhang, Y.; Fromentin, R.; Chomont, N.; Cohen, E.A.; Shacklett, B.; Mehraj, V.; et al. HIV persists in CCR6+CD4+ T cells from colon and blood during antiretroviral therapy. AIDS 2017, 31, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Cleret-Buhot, A.; Zhang, Y.; Planas, D.; Goulet, J.P.; Monteiro, P.; Gosselin, A.; Wacleche, V.S.; Tremblay, C.L.; Jenabian, M.A.; Routy, J.P.; et al. Identification of novel HIV-1 dependency factors in primary CCR4(+)CCR6(+)Th17 cells via a genome-wide transcriptional approach. Retrovirology 2015, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Bernier, A.; Cleret-Buhot, A.; Zhang, Y.; Goulet, J.P.; Monteiro, P.; Gosselin, A.; DaFonseca, S.; Wacleche, V.S.; Jenabian, M.A.; Routy, J.P.; et al. Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor γ as an intrinsic negative regulator of viral replication. Retrovirology 2013, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Christensen-Quick, A.; Lafferty, M.; Sun, L.; Marchionni, L.; DeVico, A.; Garzino-Demo, A. Human Th17 cells lack HIV-inhibitory RNases and are highly permissive to productive HIV infection. J. Virol. 2016, 90, 7833–7847. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, M.; Barr, F.D.; Crist, S.G.; Fahey, J.V.; Wira, C.R. Phenotype and susceptibility to HIV infection of CD4+ Th17 cells in the human female reproductive tract. Mucosal Immunol. 2014, 7, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Jambo, K.C.; Banda, D.H.; Kankwatira, A.M.; Sukumar, N.; Allain, T.J.; Heyderman, R.S.; Russell, D.G.; Mwandumba, H.C. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal Immunol. 2014, 7, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. Macrophages sustain HIV replication in vivo independently of T cells. J. Clin. Investig. 2016, 126, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Costiniuk, C.T.; Jenabian, M.A. The lungs as anatomical reservoirs of HIV infection. Rev. Med. Virol. 2014, 24, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Tremblay, C.; Routy, J.P.; Boulassel, M.R.; Toma, E.; Ahmad, A. Decreased levels of circulating IL-21 in HIV-infected AIDS patients: Correlation with CD4+ T-cell counts. Viral Immunol. 2008, 21, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.M.; Klase, Z.A.; DiNapoli, S.R.; Vujkovic-Cvijin, I.; Carmack, K.; Perkins, M.R.; Calantone, N.; Vinton, C.L.; Riddick, N.E.; Gallagher, J.; et al. IL-21 and probiotic therapy improve Th17 frequencies, microbial translocation, and microbiome in ARV-treated, SIV-infected macaques. Mucosal Immunol. 2016, 9, 458. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Bernasconi, E.; Speck, R.F.; Proietti, M.; Sauermann, U.; D’Agostino, G.; Danelon, G.; Rezzonico Jost, T.; Grassi, F.; Raeli, L.; et al. Impairment of CCR6+ and CXCR3+ Th Cell migration in HIV-1 infection is rescued by modulating actin polymerization. J. Immunol. 2017, 198, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, B.; Favre, D.; McCune, J.M. Th17 and regulatory T cells: Implications for AIDS pathogenesis. Curr. Opin. HIV AIDS 2010, 5, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Montagnoli, C.; Zelante, T.; Bonifazi, P.; Bozza, S.; Moretti, S.; D’Angelo, C.; Vacca, C.; Boon, L.; Bistoni, F.; et al. Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J. Immunol. 2007, 179, 5999–6008. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Fallarino, F.; De Luca, A.; Montagnoli, C.; D’Angelo, C.; Zelante, T.; Vacca, C.; Bistoni, F.; Fioretti, M.C.; Grohmann, U.; et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 2008, 451, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Toledo, C.M.; Wietgrefe, S.W.; Duan, L.; Schacker, T.W.; Reilly, C.S.; Haase, A.T. The immunosuppressive role of IL-32 in lymphatic tissue during HIV-1 infection. J. Immunol. 2011, 186, 6576–6584. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.; Benfield, T.; Mens, H.; Clausen, L.N.; Katzenstein, T.L.; Fomsgaard, A.; Karlsson, I. Low level of regulatory T cells and maintenance of balance between regulatory T cells and TH17 cells in HIV-1-infected elite controllers. J. Acquir. Immune Defic. Syndr. 2011, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, N.M.; Vazquez, N.; Greenwell-Wild, T.; Ecevit, I.; Horn, J.; Orenstein, J.; Wahl, S.M. Regulation of the tonsil cytokine milieu favors HIV susceptibility. J. Leukocyte Biol. 2006, 80, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Rousseau, R.; Huibner, S.; Kovacs, C.; Benko, E.; Shahabi, K.; Kandel, G.; Ostrowski, M.; Kaul, R. Impact of intensified antiretroviral therapy during early HIV infection on gut immunology and inflammatory blood biomarkers. AIDS 2017, 31, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Gaardbo, J.C.; Hartling, H.J.; Gerstoft, J.; Nielsen, S.D. Incomplete immune recovery in HIV infection: Mechanisms, relevance for clinical care, and possible solutions. Clin. Dev. Immunol. 2012, 2012, 670957. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, J.; Zheng, Y.; Luo, Y.; Zhou, H.; Yao, Y.; Chen, X.; Chen, Z.; He, M. A randomized case-control study of dynamic changes in peripheral blood Th17/Treg cell balance and interleukin-17 levels in highly active antiretroviral-treated HIV type 1/AIDS patients. AIDS Res. Hum. Retrovirus. 2012, 28, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hernandez, L.A.; Jave-Suarez, L.F.; Fafutis-Morris, M.; Montes-Salcedo, K.E.; Valle-Gutierrez, L.G.; Campos-Loza, A.E.; Enciso-Gomez, L.F.; Andrade-Villanueva, J.F. Synbiotic therapy decreases microbial translocation and inflammation and improves immunological status in HIV-infected patients: A double-blind randomized controlled pilot trial. Nutr. J. 2012, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- De Repentigny, L.; Goupil, M.; Jolicoeur, P. Oropharyngeal candidiasis in HIV infection: Analysis of impaired mucosal immune response to Candida albicans in mice expressing the HIV-1 transgene. Pathogens 2015, 4, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.; Anderson, J.L.; Fromentin, R.; Hartogenesis, W.; Smith, M.Z.; Bacchetti, P.; Hecht, F.M.; Chomont, N.; Cameron, P.U.; Deeks, S.G.; et al. Persistence of integrated HIV DNA in CXCR3 + CCR6 + memory CD4+ T cells in HIV-infected individuals on antiretroviral therapy. AIDS 2016, 30, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Cianfriglia, M.; Dupuis, M.L.; Molinari, A.; Verdoliva, A.; Costi, R.; Galluzzo, C.M.; Andreotti, M.; Cara, A.; di Santo, R.; Palmisano, L. HIV-1 integrase inhibitors are substrates for the multidrug transporter MDR1-P-glycoprotein. Retrovirology 2007, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, R.V.; Middlemas, D.; Flynn, P.; Fridland, A. Human immunodeficiency virus protease inhibitors serve as substrates for multidrug transporter proteins MDR1 and MRP1 but retain antiviral efficacy in cell lines expressing these transporters. Antimicrob. Agents Chemother. 1998, 42, 3157–3162. [Google Scholar] [PubMed]
- Sun, H.; Kim, D.; Li, X.; Kiselinova, M.; Ouyang, Z.; Vandekerckhove, L.; Shang, H.; Rosenberg, E.S.; Yu, X.G.; Lichterfeld, M. Th1/17 polarization of CD4 T cells supports HIV-1 persistence during antiretroviral therapy. J. Virol. 2015, 89, 11284–11293. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.M.; de Leval, L.; Pantaleo, G.; Perreau, M. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016, 22, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wacleche, V.S.; Landay, A.; Routy, J.-P.; Ancuta, P. The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses 2017, 9, 303. https://doi.org/10.3390/v9100303
Wacleche VS, Landay A, Routy J-P, Ancuta P. The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses. 2017; 9(10):303. https://doi.org/10.3390/v9100303
Chicago/Turabian StyleWacleche, Vanessa Sue, Alan Landay, Jean-Pierre Routy, and Petronela Ancuta. 2017. "The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis" Viruses 9, no. 10: 303. https://doi.org/10.3390/v9100303
APA StyleWacleche, V. S., Landay, A., Routy, J.-P., & Ancuta, P. (2017). The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses, 9(10), 303. https://doi.org/10.3390/v9100303