Expression Profiles of Ligands for Activating Natural Killer Cell Receptors on HIV Infected and Uninfected CD4+ T Cells


Abstract
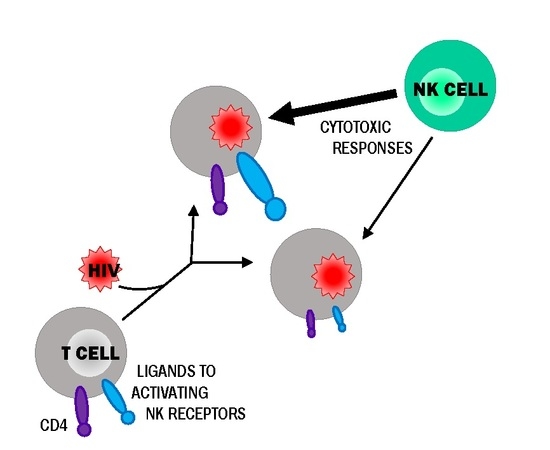
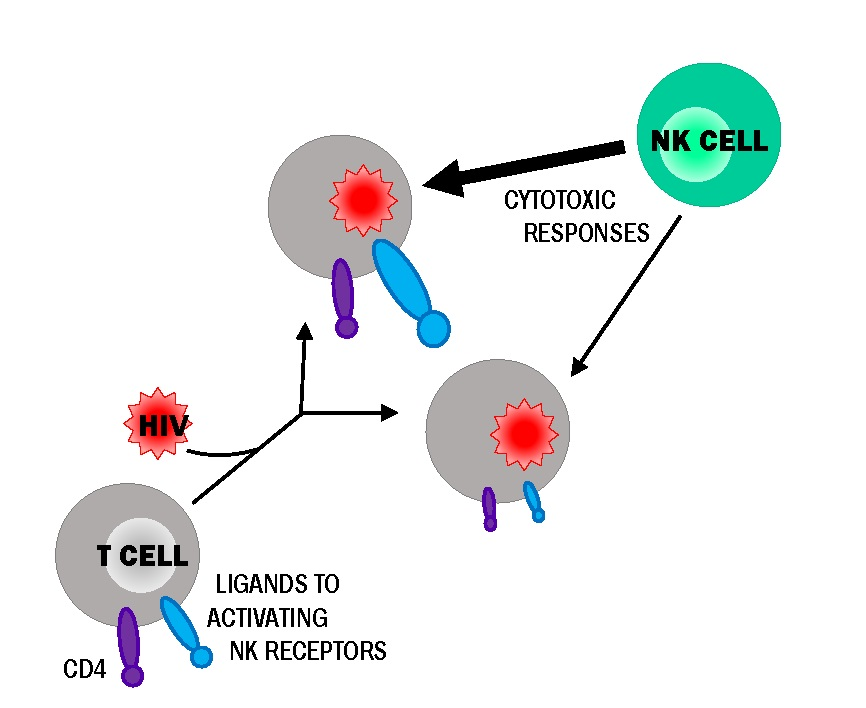
:
{kind=link}
{kind=link}
{kind=link}
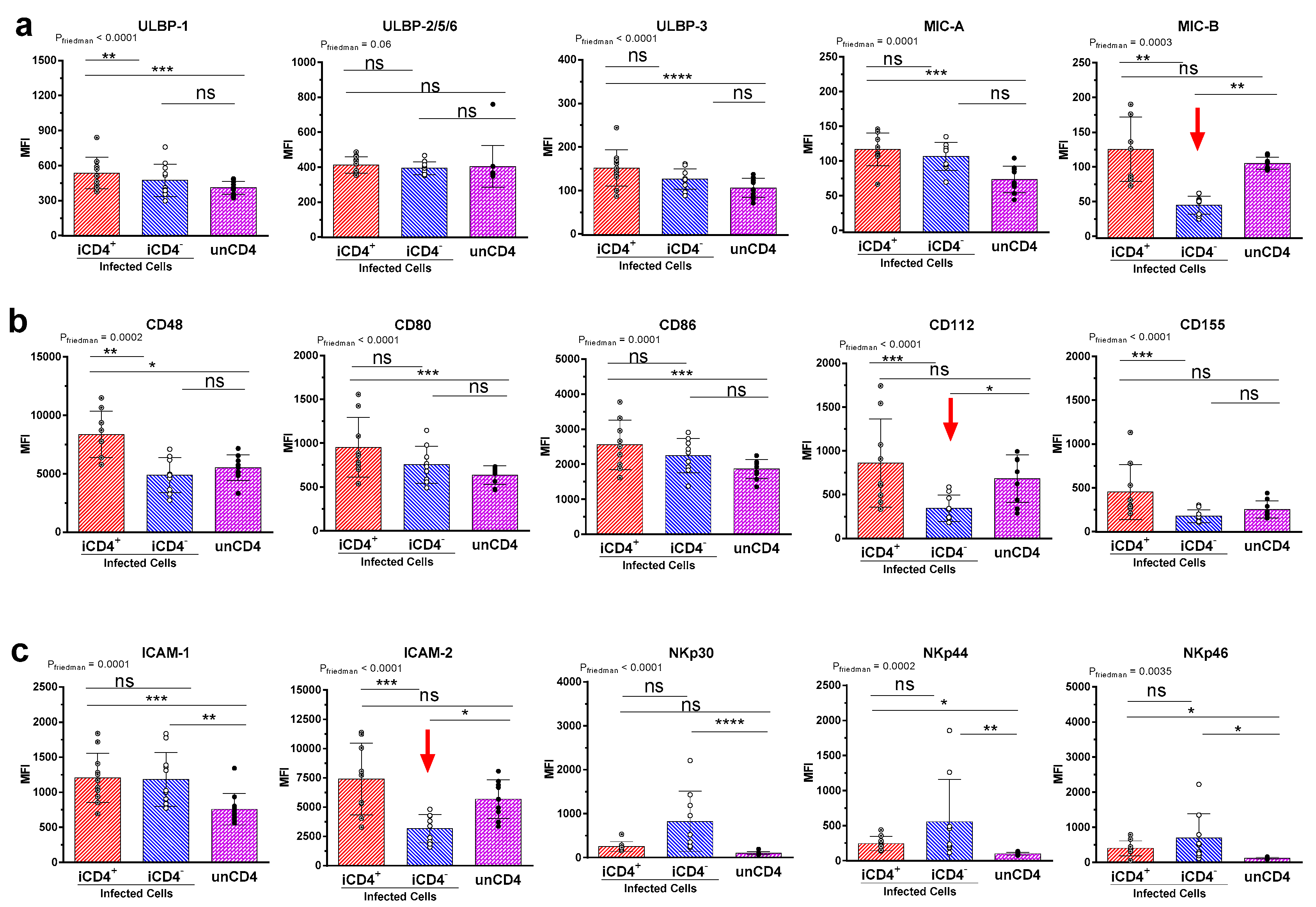{kind=link}
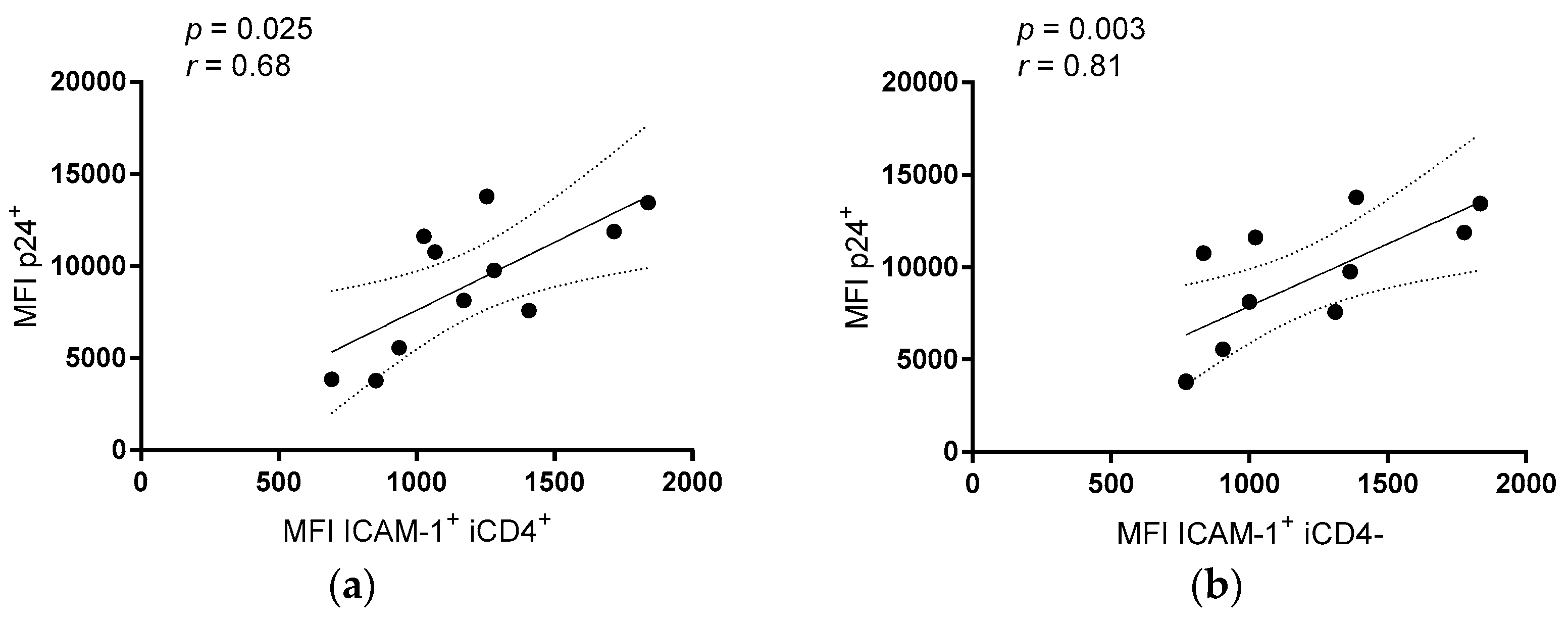{kind=link}
{kind=link}
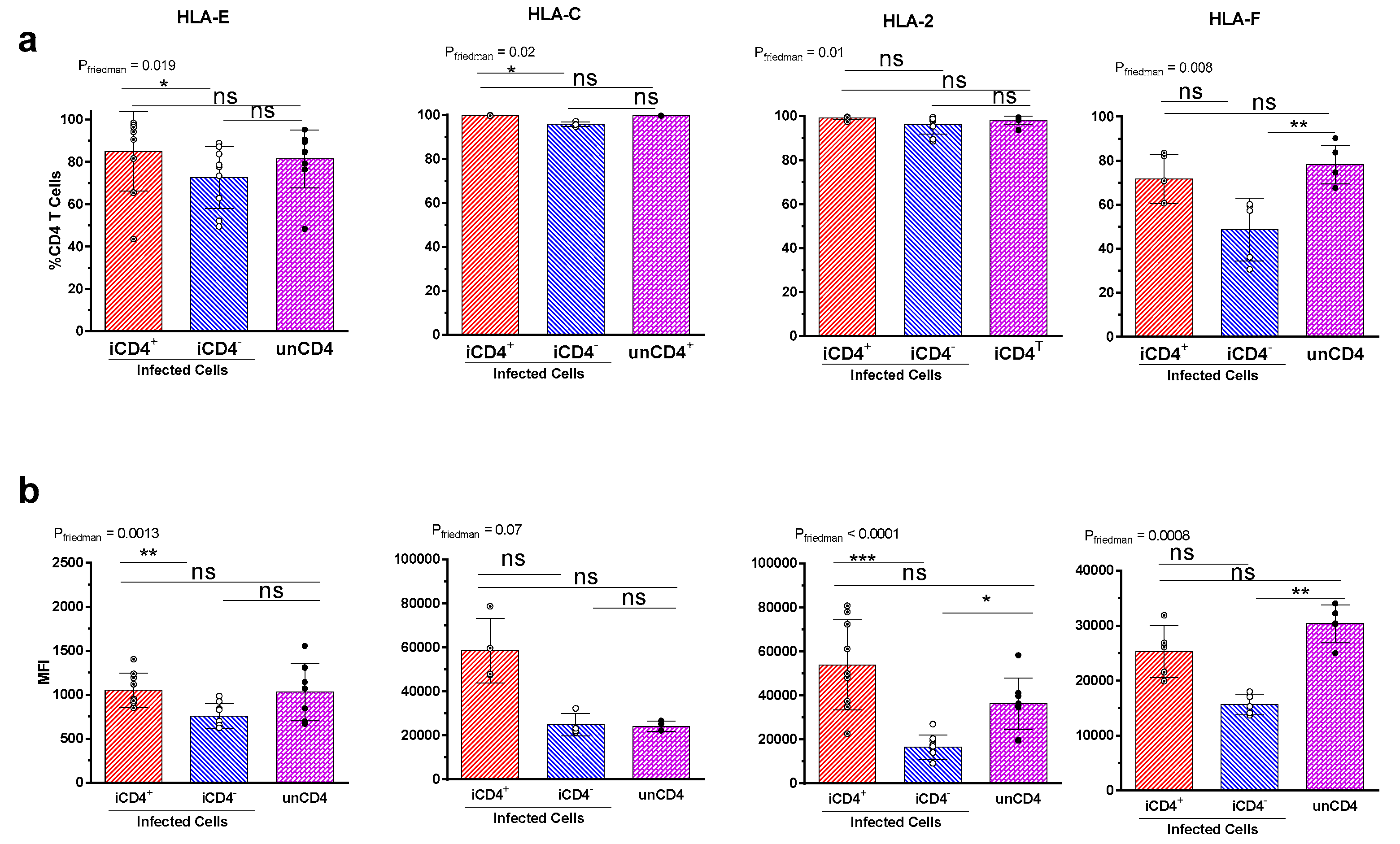{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Population
2.3. CD4+ T Cell Isolation
2.4. HIV Infection
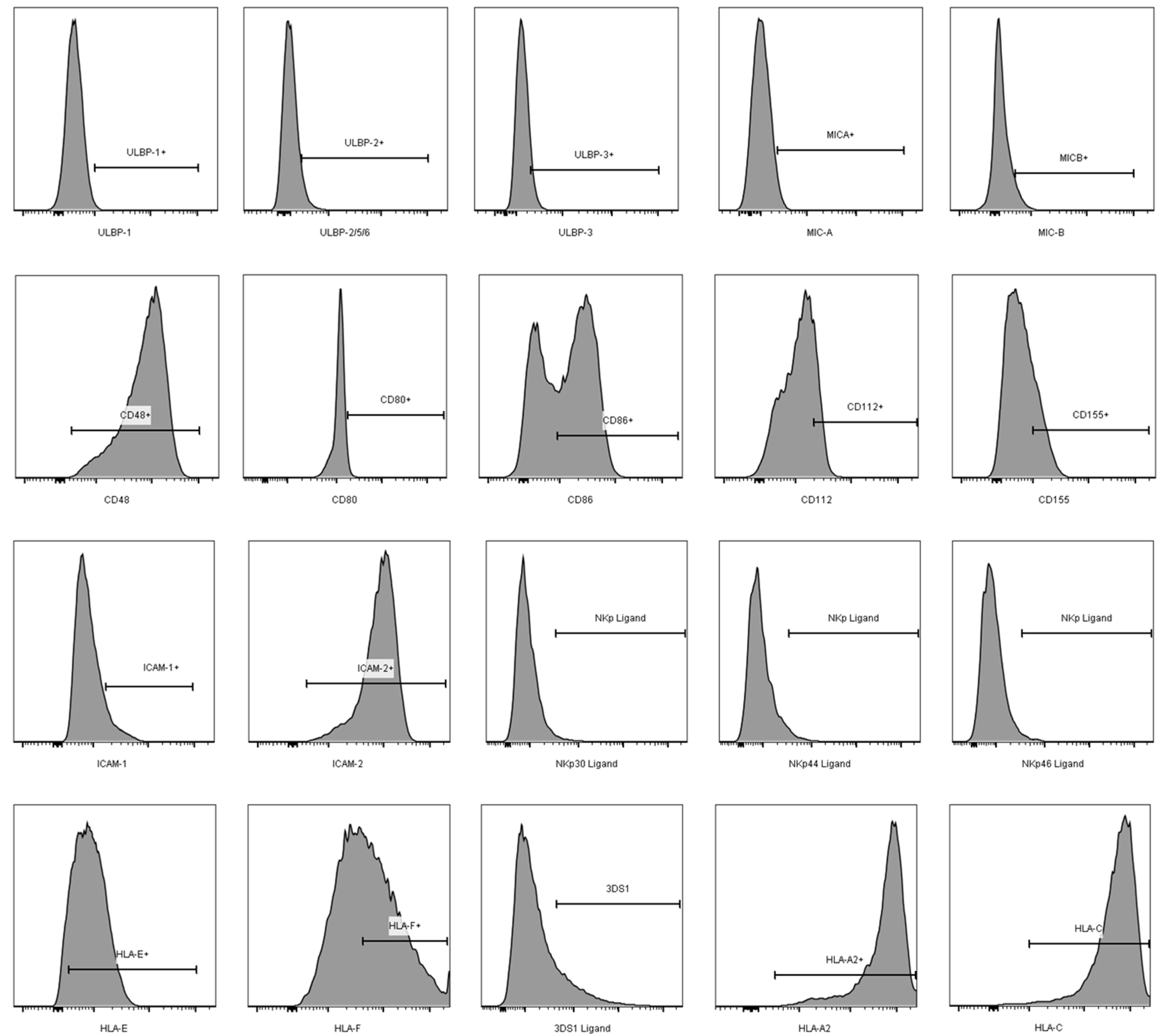
2.5. Antibody Staining and Acquisition
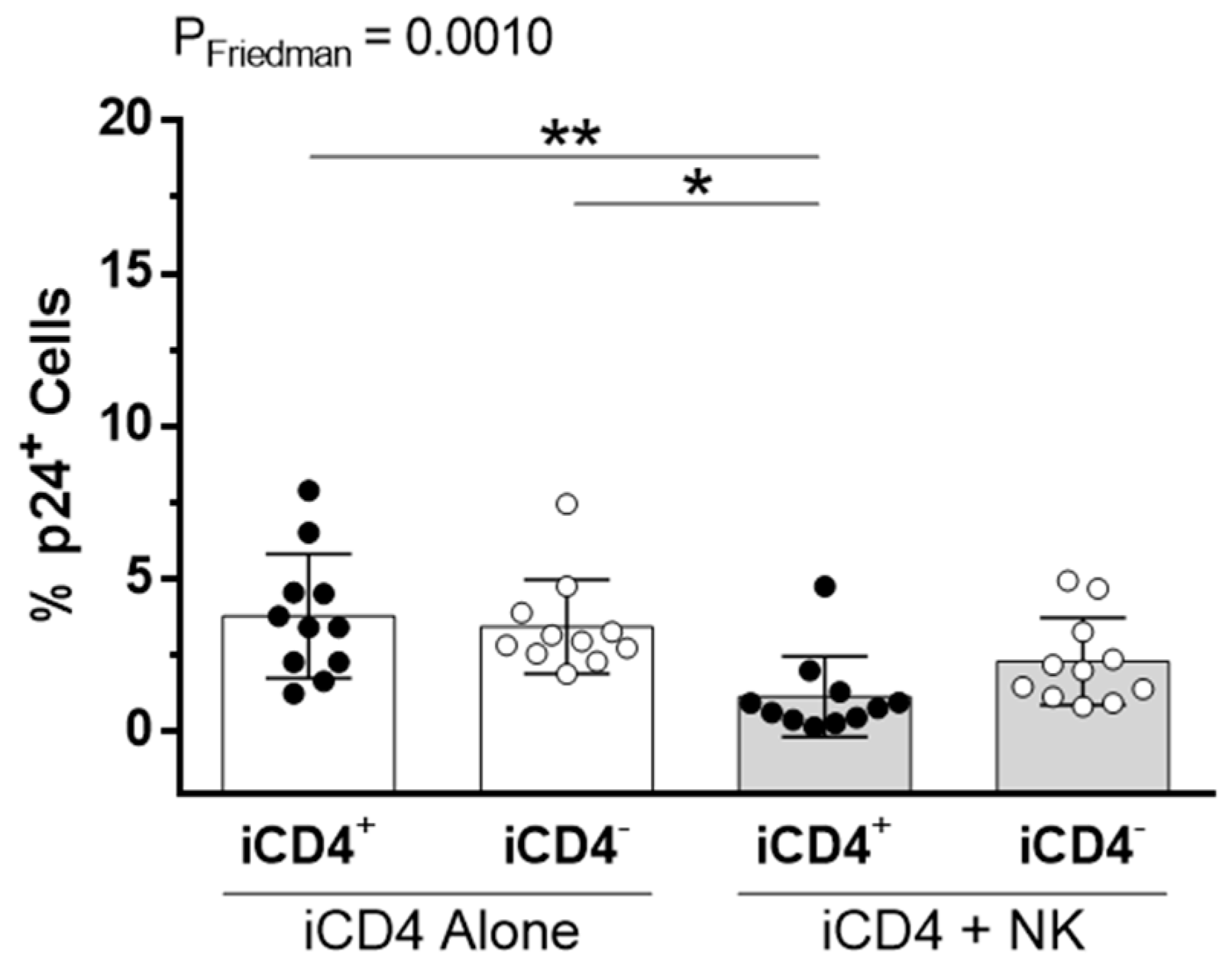
2.6. Inhibition of Viral Replication Assay
2.7. Statistical Analysis
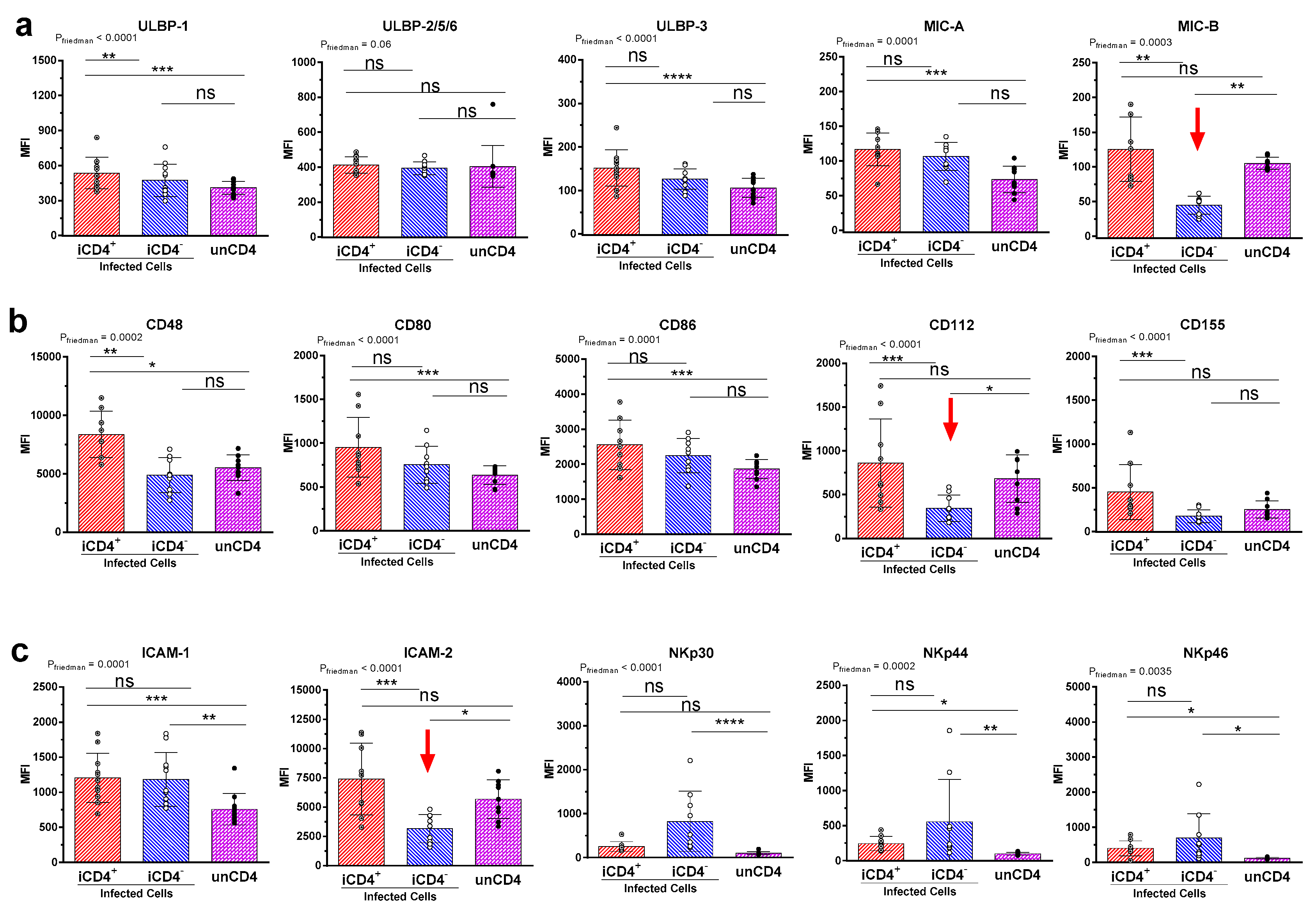
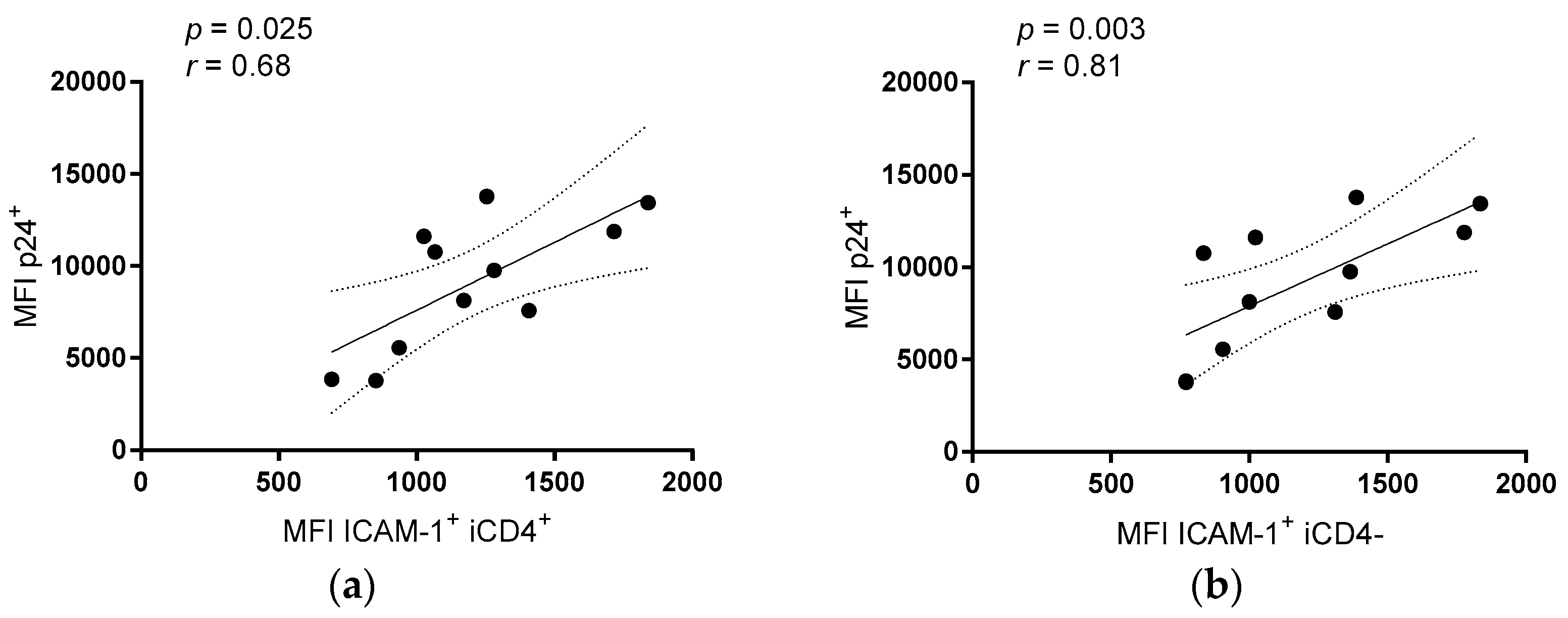
3. Results
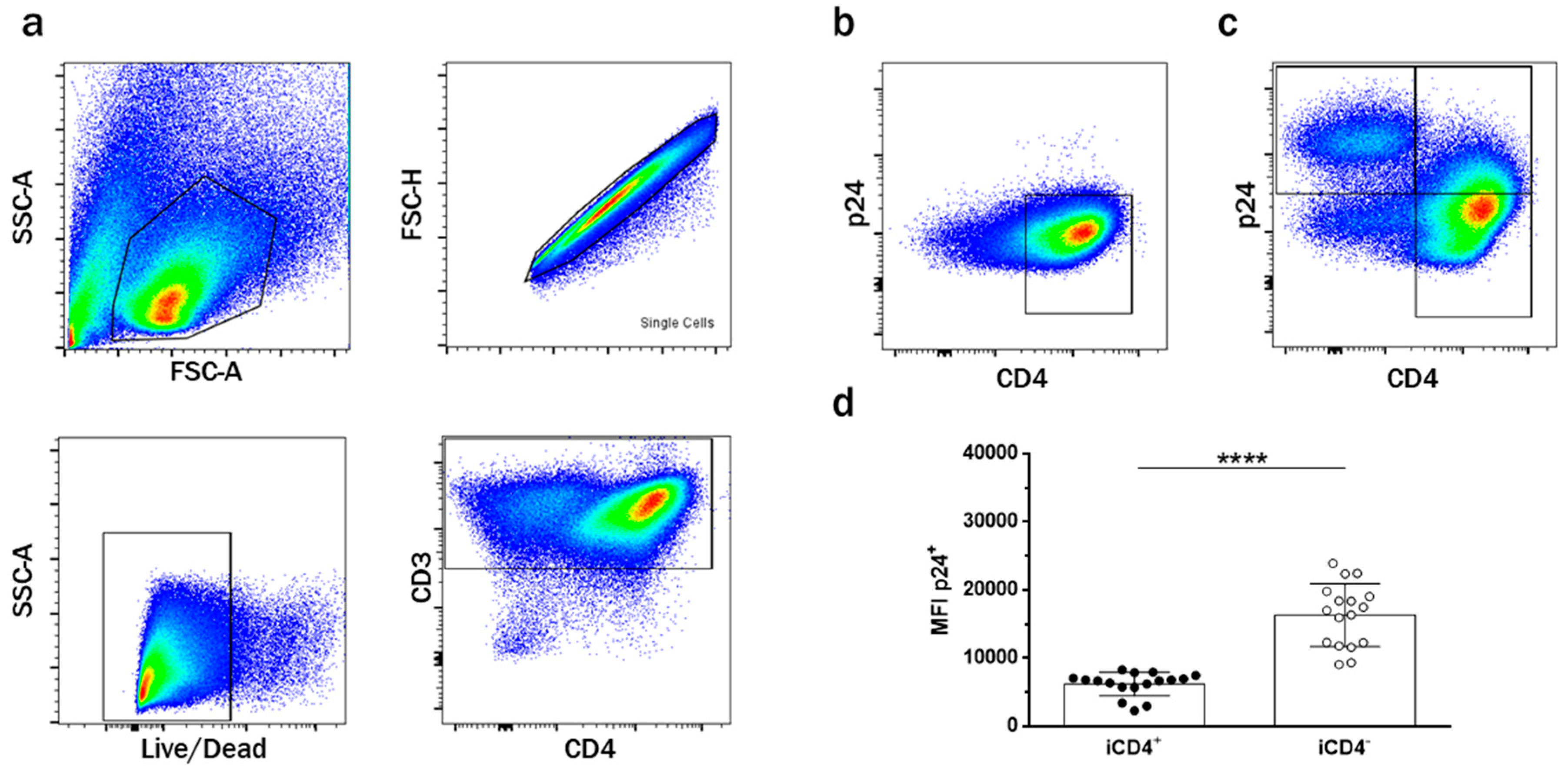
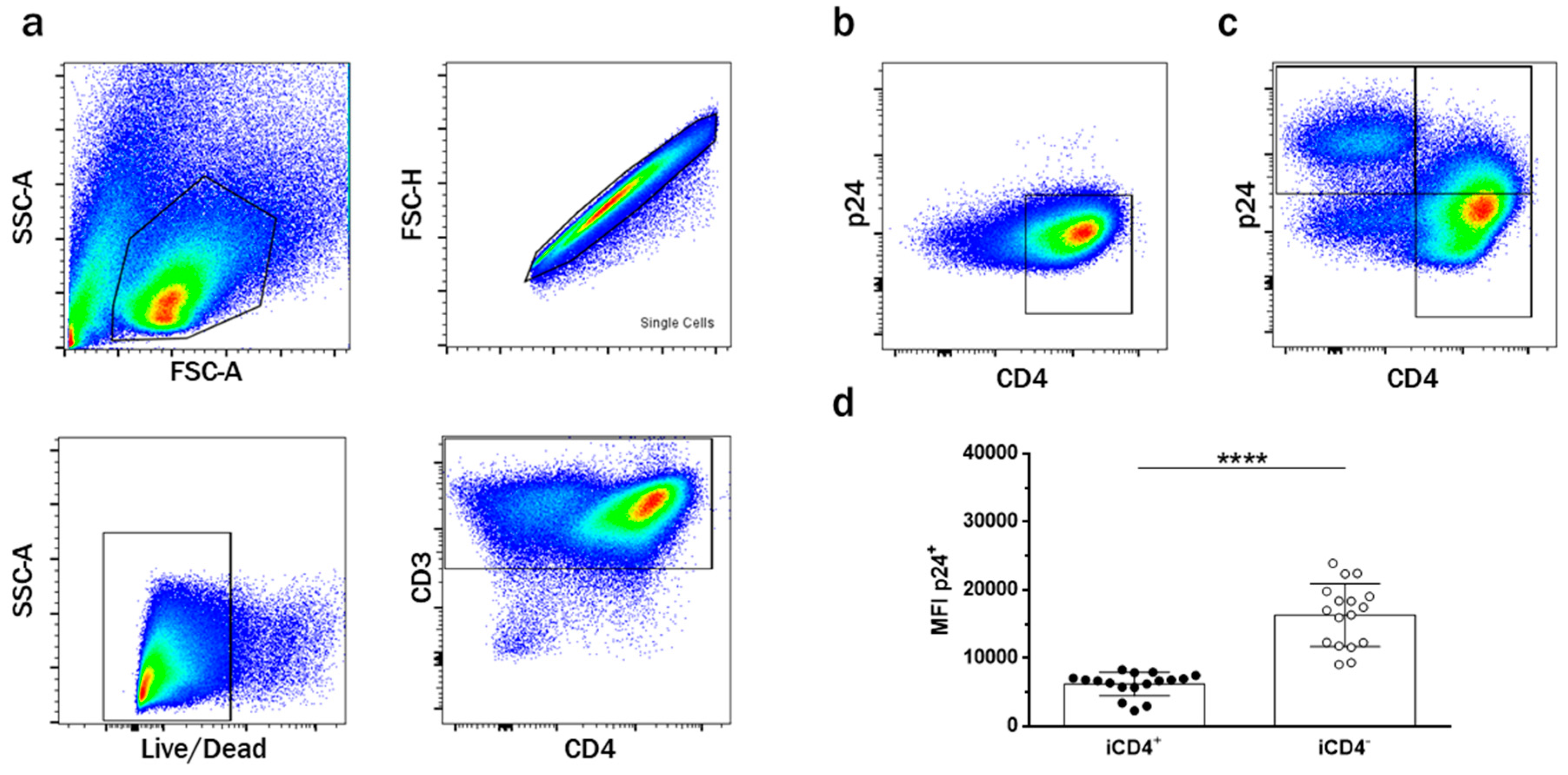
3.1. CD4+ T Cell Infection Characteristics
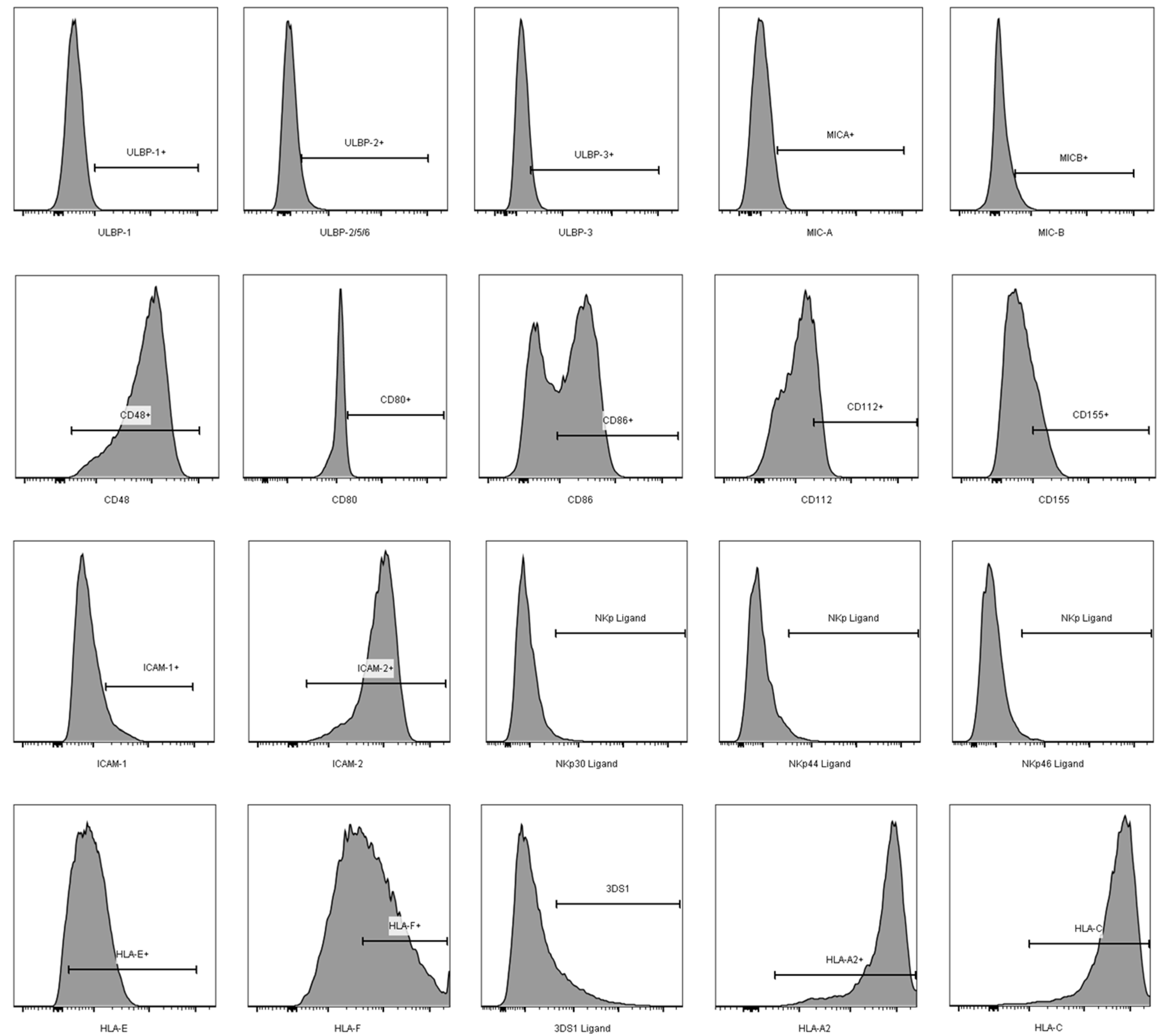
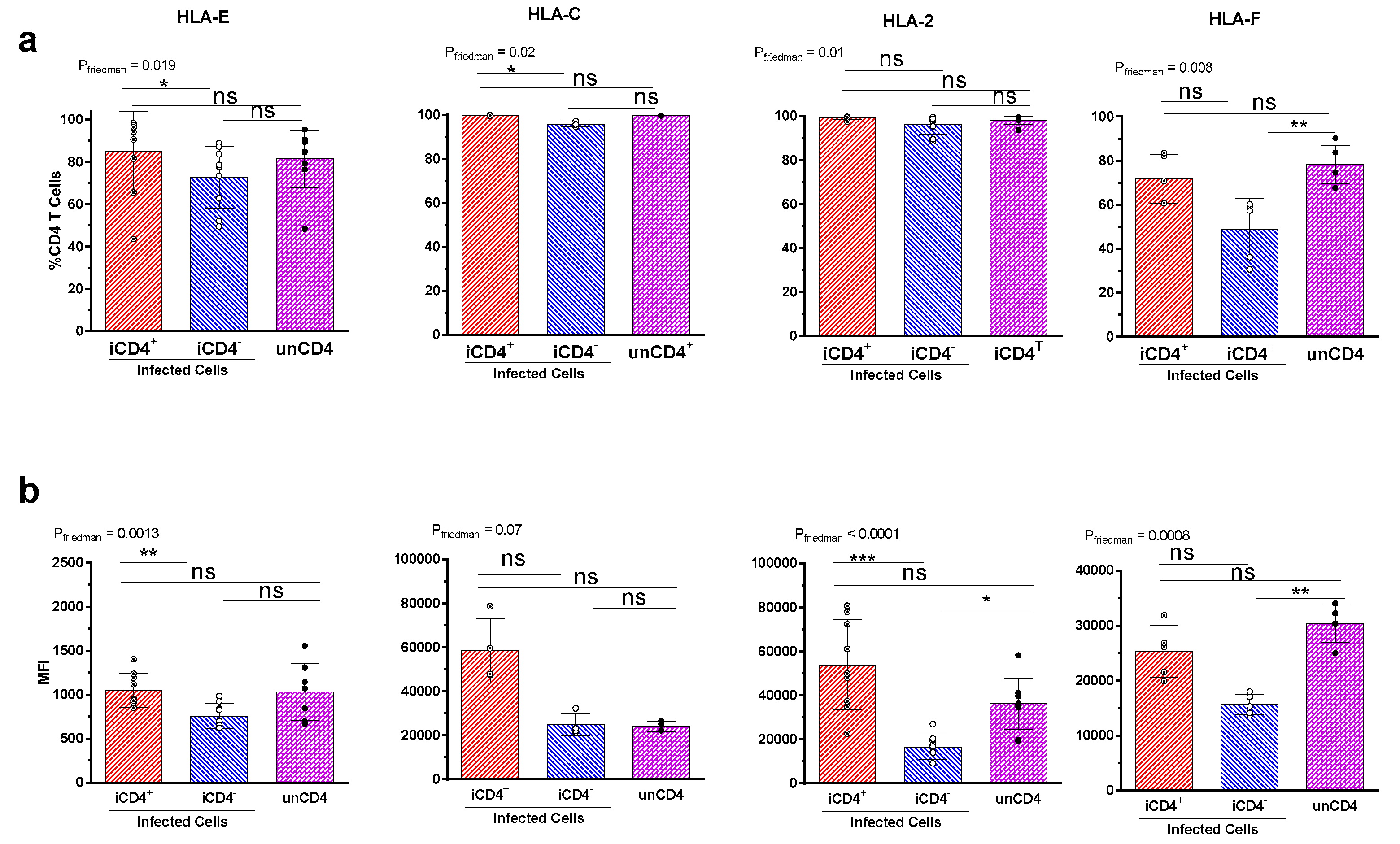
3.2. Comparison of the aNKR Ligand Expression Intensity on iCD4 and unCD4 T Cells
3.3. Expression Profiles of HLA-E, HLA-F, HLA-A2, and HLA-C on iCD4 and unCD4
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Fauriat, C.; Long, E.O.; Ljunggren, H.-G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Watzl, C.; Stebbins, C.C.; Long, E.O. NK cell inhibitory receptors prevent tyrosine phosphorylation of the activation receptor 2B4 (CD244). J. Immunol. 2000, 165, 3545–3548. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible mica. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vitale, M.; Morelli, L.; Sanseverino, L.; Augugliaro, R.; Bottino, C.; Moretta, L.; Moretta, A. P46, a novel natural killer cell—Specific surface molecule that mediates cell activation. J. Exp. Med. 1997, 186, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Bloushtain, N.; Qimron, U.; Bar-Ilan, A.; Hershkovitz, O.; Gazit, R.; Fima, E.; Korc, M.; Vlodavsky, I.; Bovin, N.V.; Porgador, A. Membrane-associated heparan sulfate proteoglycans are involved in the recognition of cellular targets by NKp30 and NKp46. J. Immunol. 2004, 173, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Arnon, T.I.; Lev, M.; Katz, G.; Chernobrov, Y.; Porgador, A.; Mandelboim, O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol. 2001, 31, 2680–2689. [Google Scholar] [CrossRef]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.L.; Rosental, B.; Horlacher, T.; Hershkovitz, O.; de Paz, J.L.; Noti, C.; Schauer, S.; Porgador, A.; Seeberger, P.H. Natural cytotoxicity receptors NKp30, NKp44 and NKp46 bind to different heparan sulfate/heparin sequences. J. Proteome Res. 2009, 8, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.H.; Boles, K.; van der Merwe, P.A.; Kumar, V.; Mathew, P.A.; Barclay, A.N. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Bottino, C.; Castriconi, R.; Cantoni, C.; Marcenaro, S.; Rivera, P.; Spaggiari, G.M.; Dondero, A.; Carnemolla, B.; Reymond, N.; et al. PVR (CD155) and Nectin-2 (CD112) as ligands of the human DNAM-1 (CD226) activating receptor: Involvement in tumor cell lysis. Mol. Immunol. 2005, 42, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.; Lanier, L.L.; Shibuya, A. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and Nectin-2 (PRR-2/CD112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef]
- Shibuya, K.; Lanier, L.L.; Phillips, J.H.; Ochs, H.D.; Shimizu, K.; Nakayama, E.; Nakauchi, H.; Shibuya, A. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity 1999, 11, 615–623. [Google Scholar] [CrossRef]
- Gross, C.C.; Brzostowski, J.A.; Liu, D.; Long, E.O. Tethering of intercellular adhesion molecule on target cells is required for LFA-1-dependent NK cell adhesion and granule polarization. J. Immunol. 2010, 185, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Schleinitz, N.; March, M.E.; Long, E.O. Recruitment of activation receptors at inhibitory NK cell immune synapses. PLoS ONE 2008, 3, e3278. [Google Scholar] [CrossRef] [PubMed]
- Culley, F.J.; Johnson, M.; Evans, J.H.; Kumar, S.; Crilly, R.; Casasbuenas, J.; Schnyder, T.; Mehrabi, M.; Deonarain, M.P.; Ushakov, D.S.; et al. Natural killer cell signal integration balances synapse symmetry and migration. PLoS Biol. 2009, 7, e1000159. [Google Scholar] [CrossRef] [PubMed]
- Luque, I.; Reyburn, H.; Strominger, J.L. Expression of the CD80 and CD86 molecules enhances cytotoxicity by human natural killer cells. Hum. Immunol. 2000, 61, 721–728. [Google Scholar] [CrossRef]
- Wilson, J.L.; Charo, J.; Martin-Fontecha, A.; Dellabona, P.; Casorati, G.; Chambers, B.J.; Kiessling, R.; Bejarano, M.T.; Ljunggren, H.G. NK cell triggering by the human costimulatory molecules CD80 and CD86. J. Immunol. 1999, 163, 4207–4212. [Google Scholar] [PubMed]
- Galea-Lauri, J.; Darling, D.; Gan, S.U.; Krivochtchapov, L.; Kuiper, M.; Gaken, J.; Souberbielle, B.; Farzaneh, F. Expression of a variant of CD28 on a subpopulation of human NK cells: Implications for B7-mediated stimulation of NK cells. J. Immunol. 1999, 163, 62–70. [Google Scholar] [PubMed]
- Costa, C.; Barber, D.F.; Fodor, W.L. Human NK cell-mediated cytotoxicity triggered by CD86 and gal α 1,3-gal is inhibited in genetically modified porcine cells. J. Immunol. 2002, 168, 3808–3816. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Mavilio, D.; Kottilil, S. NK cells in HIV infection: Paradigm for protection or targets for ambush. Nat. Rev. Immunol. 2005, 5, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Barahmand-Pour, F.; Paulsene, W.; Medley, S.; Geraghty, D.E.; Strong, R.K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J. Immunol. 2005, 174, 2878–2884. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Matsumoto, N.; Maenaka, K.; Suzuki, K.; Yamamoto, K. The inhibitory NK cell receptor CD94/NKG2A and the activating receptor CD94/NKG2C bind the top of HLA-E through mostly shared but partly distinct sets of HLA-E residues. Eur. J. Immunol. 2004, 34, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; López-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA 1998, 95, 5199–5204. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Holzemer, A.; Martrus, G.; Chung, A.W.; Pacheco, Y.; Simoneau, C.R.; Rucevic, M.; Lamothe-Molina, P.A.; Pertel, T.; Kim, T.E.; et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol. 2016, 17, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Burian, A.; Wang, K.L.; Finton, K.A.K.; Lee, N.; Ishitani, A.; Strong, R.K.; Geraghty, D.E. HLA-F and MHC-I open conformers bind natural killer cell Ig-like receptor KIR3DS1. PLoS ONE 2016, 11, e0163297. [Google Scholar] [CrossRef] [PubMed]
- Alter, G.; Martin, M.P.; Teigen, N.; Carr, W.H.; Suscovich, T.J.; Schneidewind, A.; Streeck, H.; Waring, M.; Meier, A.; Brander, C.; et al. Differential natural killer cell–mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J. Exp. Med. 2007, 204, 3027–3036. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Lisovsky, I.; Lebouche, B.; Routy, J.P.; Bruneau, J.; Bernard, N.F. HIV protective KIR3DL1/S1-HLA-B genotypes influence NK cell-mediated inhibition of HIV replication in autologous CD4 targets. PLoS Pathog. 2014, 10, e1003867. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.; Lindwasser, O.W.; Smith, W.J.; Hurley, J.H.; Bonifacino, J.S. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J. Virol. 2007, 81, 3877–3890. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, P.A.; Yonemoto, W.; Ferrell, S.; Williams-Herman, D.; Geleziunas, R.; Greene, W.C. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr. Biol. 1998, 8, 1235–1238. [Google Scholar] [CrossRef]
- Lindwasser, O.W.; Chaudhuri, R.; Bonifacino, J.S. Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr. Mol. Med. 2007, 7, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Fruh, K. A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur. J. Immunol. 2000, 30, 509–515. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Apps, R.; Del Prete, G.Q.; Chatterjee, P.; Lara, A.; Brumme, Z.L.; Brockman, M.A.; Neil, S.; Pickering, S.; Schneider, D.K.; Piechocka-Trocha, A.; et al. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe 2016, 19, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Ueno, T.; Nakahara, T.; Sasaki, K.; Ishimoto, A.; Sakai, H. Downregulation of CD4 is required for maintenance of viral infectivity of HIV-1. Virology 2003, 311, 316–325. [Google Scholar] [CrossRef]
- Lundquist, C.A.; Tobiume, M.; Zhou, J.; Unutmaz, D.; Aiken, C. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J. Virol. 2002, 76, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Nattermann, J.; Nischalke, H.D.; Hofmeister, V.; Kupfer, B.; Ahlenstiel, G.; Feldmann, G.; Rockstroh, J.; Weiss, E.H.; Sauerbruch, T.; Spengler, U. HIV-1 infection leads to increased HLA-E expression resulting in impaired function of natural killer cells. Antivir. Ther. 2005, 10, 95–107. [Google Scholar] [PubMed]
- Brown, A.; Gartner, S.; Kawano, T.; Benoit, N.; Cheng-Mayer, C. HLA-A2 down-regulation on primary human macrophages infected with an M-tropic EGFP-tagged HIV-1 reporter virus. J. Leukoc. Biol. 2005, 78, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Meng, Z.; Del Prete, G.Q.; Lifson, J.D.; Zhou, M.; Carrington, M. Relative expression levels of the HLA class-I proteins in normal and HIV-infected cells. J. Immunol. 2015, 194, 3594–3600. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Bonaparte, M.; Sacks, J.; Guterman, J.; Fogli, M.; Mavilio, D.; Barker, E. HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 2007, 110, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Fogli, M.; Mavilio, D.; Brunetta, E.; Varchetta, S.; Ata, K.; Roby, G.; Kovacs, C.; Follmann, D.; Pende, D.; Ward, J.; et al. Lysis of endogenously infected CD4+ T cell blasts by rIL-2 activated autologous natural killer cells from HIV-infected viremic individuals. PLoS Pathog. 2008, 4, e1000101. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Davis, Z.; DeHart, J.; Zimmerman, E.; Bosque, A.; Brunetta, E.; Mavilio, D.; Planelles, V.; Barker, E. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 2009, 5, e1000613. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Bolduan, S.; Reif, T.; Schindler, M.; Schubert, U. HIV-1 Vpu mediated downregulation of CD155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology 2014, 464–465, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.B.; Sowrirajan, B.; Cogswell, A.; Ward, J.P.; Planelles, V.; Barker, E. CD155 on HIV-infected cells is not modulated by HIV-1 Vpu and Nef but synergizes with NKG2D ligands to trigger NK cell lysis of autologous primary HIV-infected cells. AIDS Res. Hum. Retrovirus 2017, 33, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Das, S.R.; Hussain, A.; Mayor, S.; George, A.; Bal, V.; Jameel, S.; Rath, S. The Nef protein of HIV-1 induces loss of cell surface costimulatory molecules CD80 and CD86 in APCs. J. Immunol. 2005, 175, 4566–4574. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Giguère, K.; Ouellet, M.; Tremblay, M.J. Exon level transcriptomic profiling of HIV-1-infected CD4+ T cells reveals virus-induced genes and host environment favorable for viral replication. PLoS Pathog. 2012, 8, e1002861. [Google Scholar] [CrossRef] [PubMed]
- Geleziunas, R.; Bour, S.; Wainberg, M.A. Cell surface down-modulation of CD4 after infection by HIV-1. FASEB J. 1994, 8, 593–600. [Google Scholar] [PubMed]
- Piguet, V.; Schwartz, O.; Le Gall, S.; Trono, D. The downregulation of CD4 and MHC-I by primate lentiviruses: A paradigm for the modulation of cell surface receptors. Immunol. Rev. 1999, 168, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Trono, D. The Nef protein of primate lentiviruses. Rev. Med. Virol. 1999, 9, 111–120. [Google Scholar] [CrossRef]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.-M.; Schwartz, O. Nef interacts with the μ subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Swann, S.A.; Williams, M.; Story, C.M.; Bobbitt, K.R.; Fleis, R.; Collins, K.L. HIV-1 Nef blocks transport of mhc class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology 2001, 282, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Kwas, C.; Wu, L. Intercellular adhesion molecule 1 (ICAM-1), but not ICAM-2 and -3, is important for dendritic cell-mediated human immunodeficiency virus type 1 transmission. J. Virol. 2009, 83, 4195–4204. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.F.; Cantin, R.; Lamontagne, G.; Tremblay, M. Host-derived ICAM-1 glycoproteins incorporated on human immunodeficiency virus type 1 are biologically active and enhance viral infectivity. J. Virol. 1997, 71, 3588–3596. [Google Scholar] [PubMed]
- Petravic, J.; Ellenberg, P.; Chan, M.L.; Paukovics, G.; Smyth, R.P.; Mak, J.; Davenport, M.P. Intracellular dynamics of HIV infection. J. Virol. 2014, 88, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremblay-McLean, A.; Bruneau, J.; Lebouché, B.; Lisovsky, I.; Song, R.; Bernard, N.F. Expression Profiles of Ligands for Activating Natural Killer Cell Receptors on HIV Infected and Uninfected CD4+ T Cells. Viruses 2017, 9, 295. https://doi.org/10.3390/v9100295
Tremblay-McLean A, Bruneau J, Lebouché B, Lisovsky I, Song R, Bernard NF. Expression Profiles of Ligands for Activating Natural Killer Cell Receptors on HIV Infected and Uninfected CD4+ T Cells. Viruses. 2017; 9(10):295. https://doi.org/10.3390/v9100295
Chicago/Turabian StyleTremblay-McLean, Alexandra, Julie Bruneau, Bertrand Lebouché, Irene Lisovsky, Rujun Song, and Nicole F. Bernard. 2017. "Expression Profiles of Ligands for Activating Natural Killer Cell Receptors on HIV Infected and Uninfected CD4+ T Cells" Viruses 9, no. 10: 295. https://doi.org/10.3390/v9100295
APA StyleTremblay-McLean, A., Bruneau, J., Lebouché, B., Lisovsky, I., Song, R., & Bernard, N. F. (2017). Expression Profiles of Ligands for Activating Natural Killer Cell Receptors on HIV Infected and Uninfected CD4+ T Cells. Viruses, 9(10), 295. https://doi.org/10.3390/v9100295