
Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Virus Infections and Cellular mRNA Quality Controls
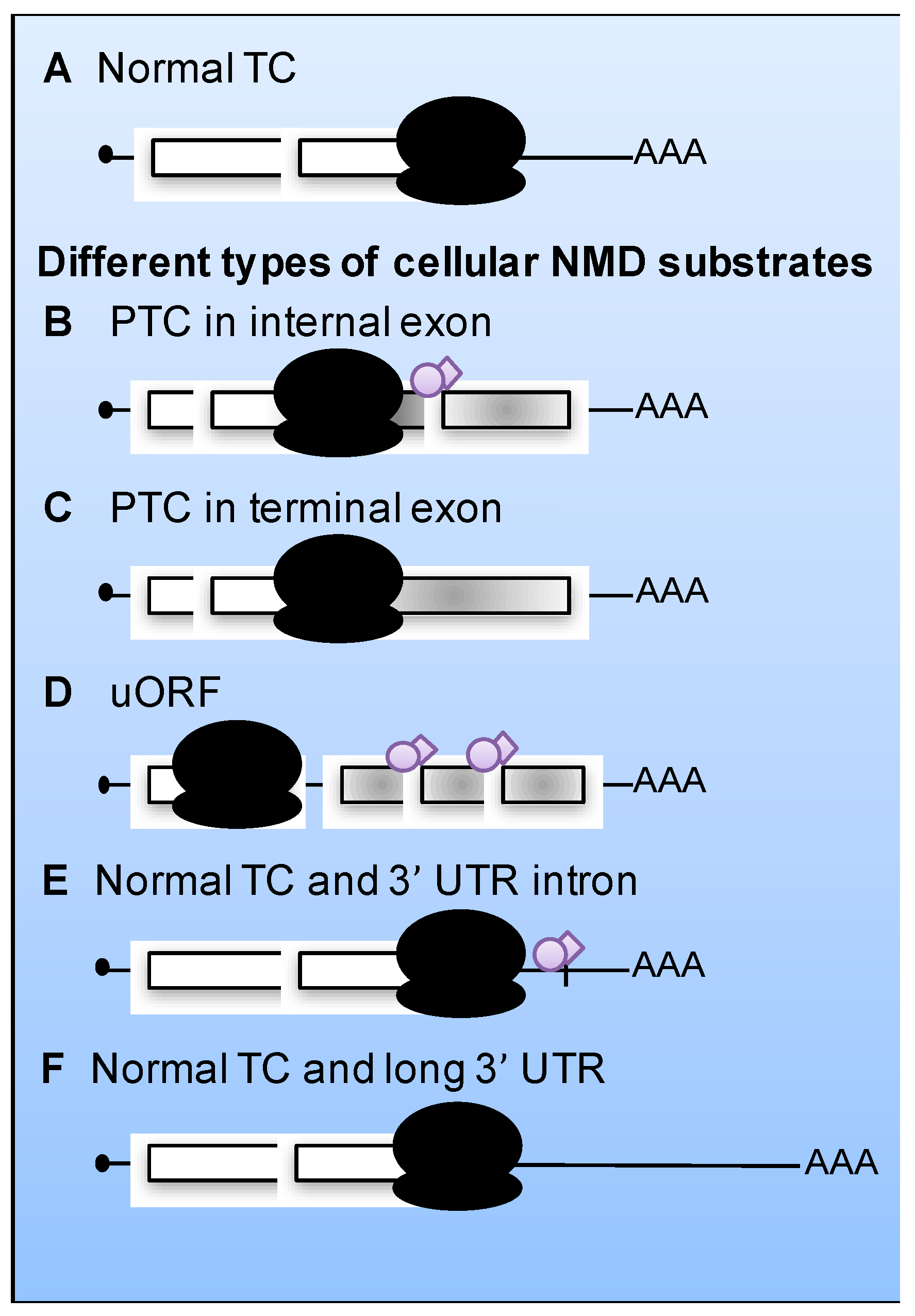
2. The Nonsense-Mediated mRNA Decay (NMD) Pathway
3. NMD Factors
4. Current NMD Model
Aberrant Translation Termination Activates NMD
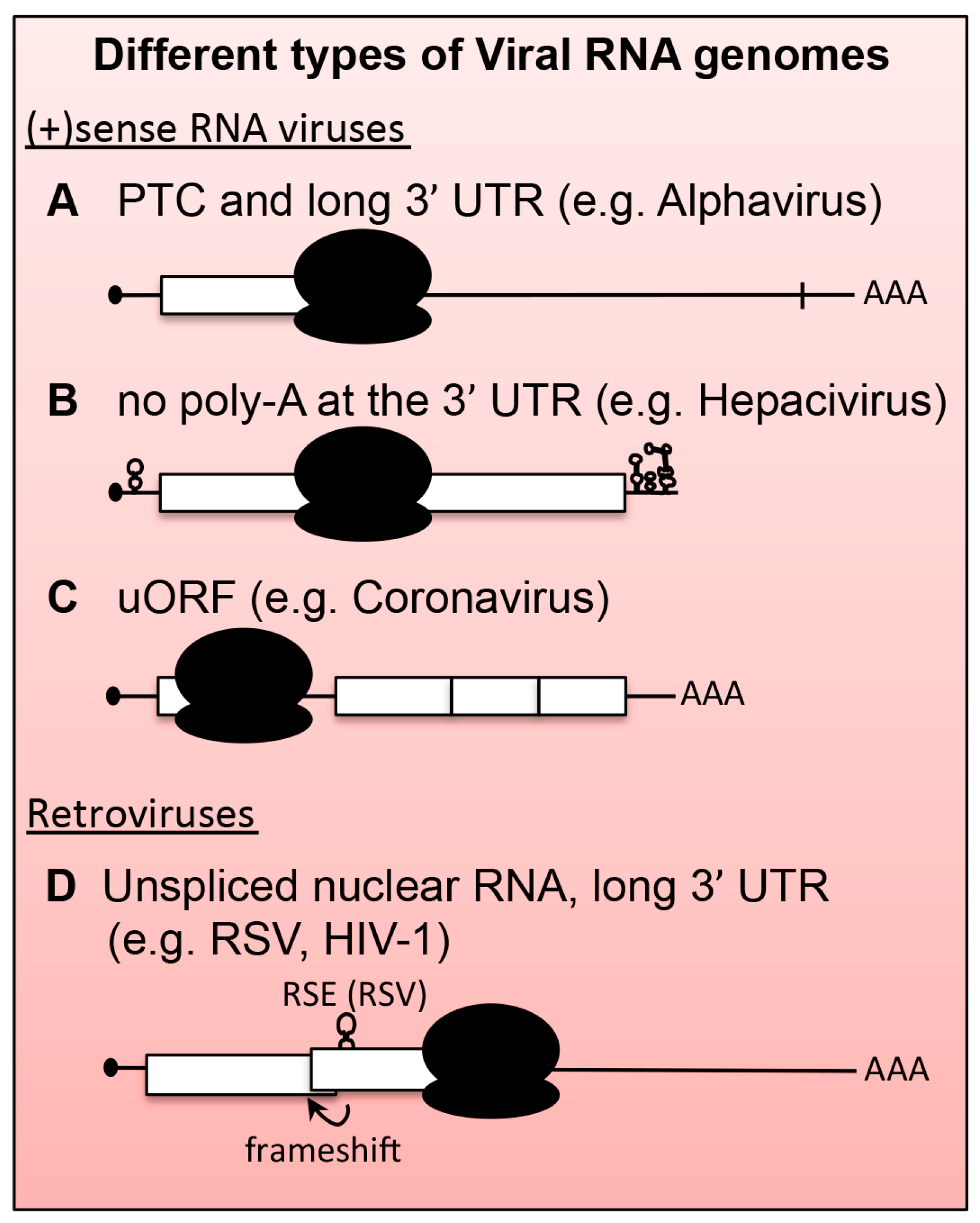
5. Viral mRNAs as Substrates for NMD
6. Documented Role of NMD and NMD Factors in Virus Infection
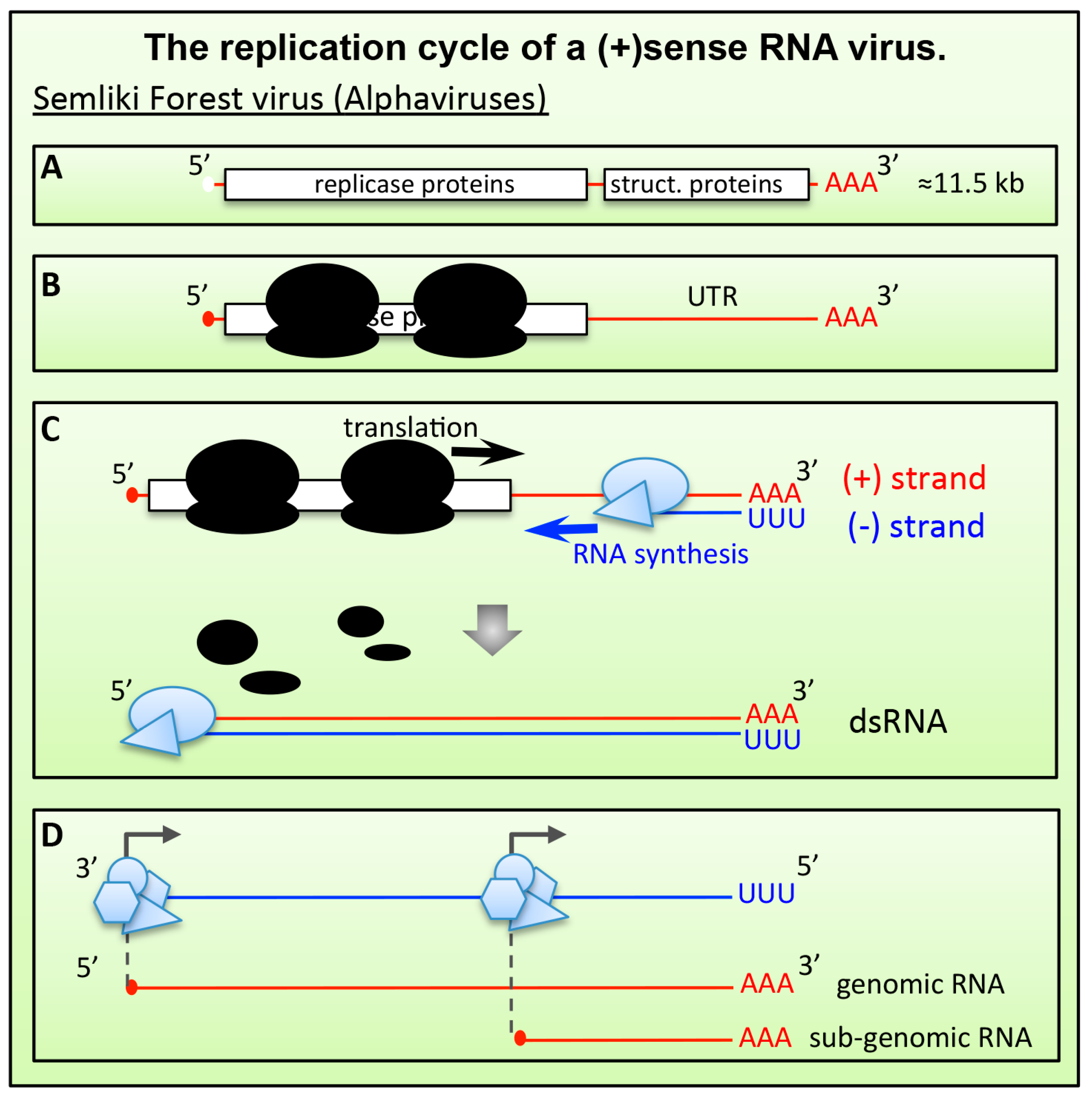
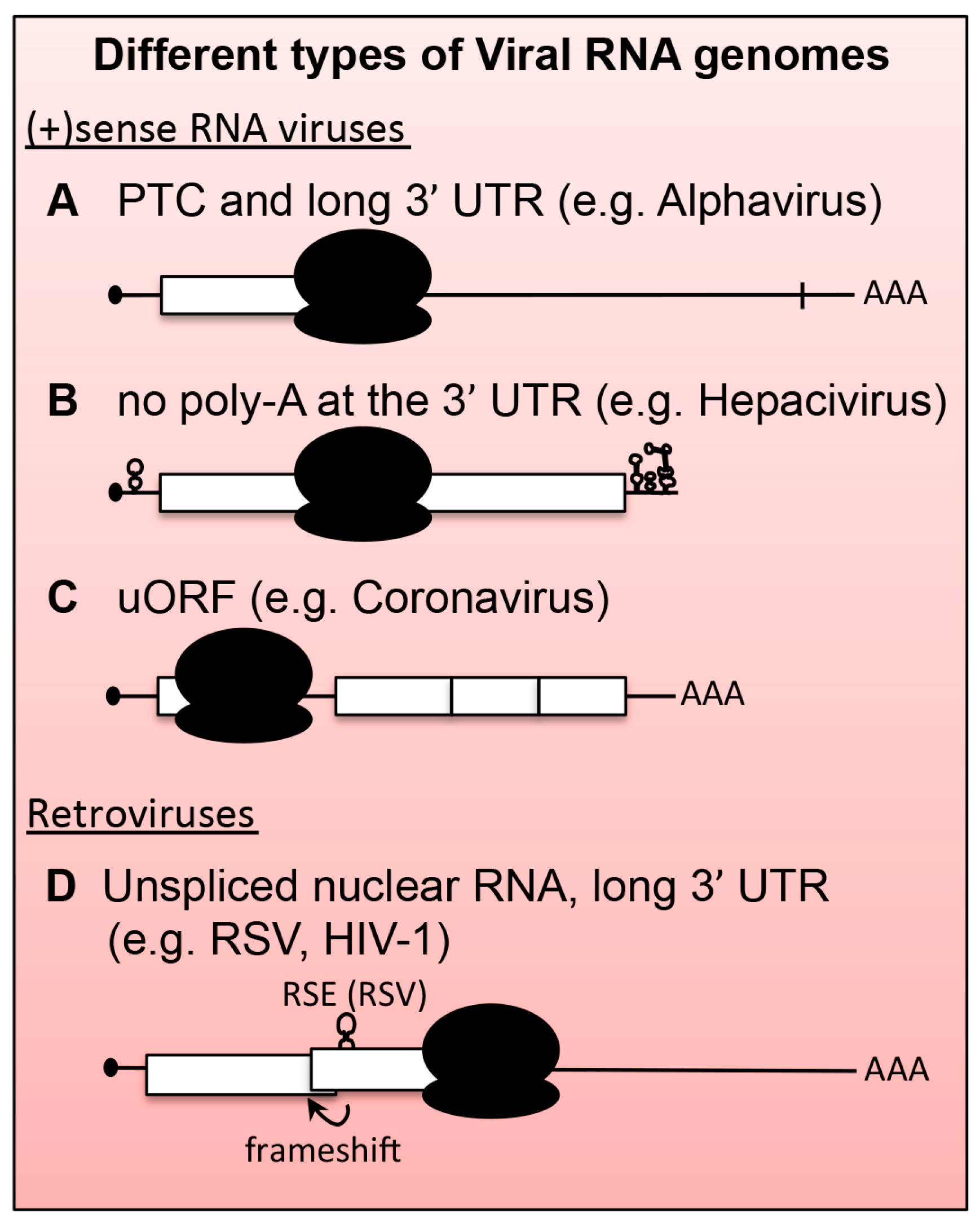
6.1. Positive-Stranded RNA ((+)RNA) Viruses
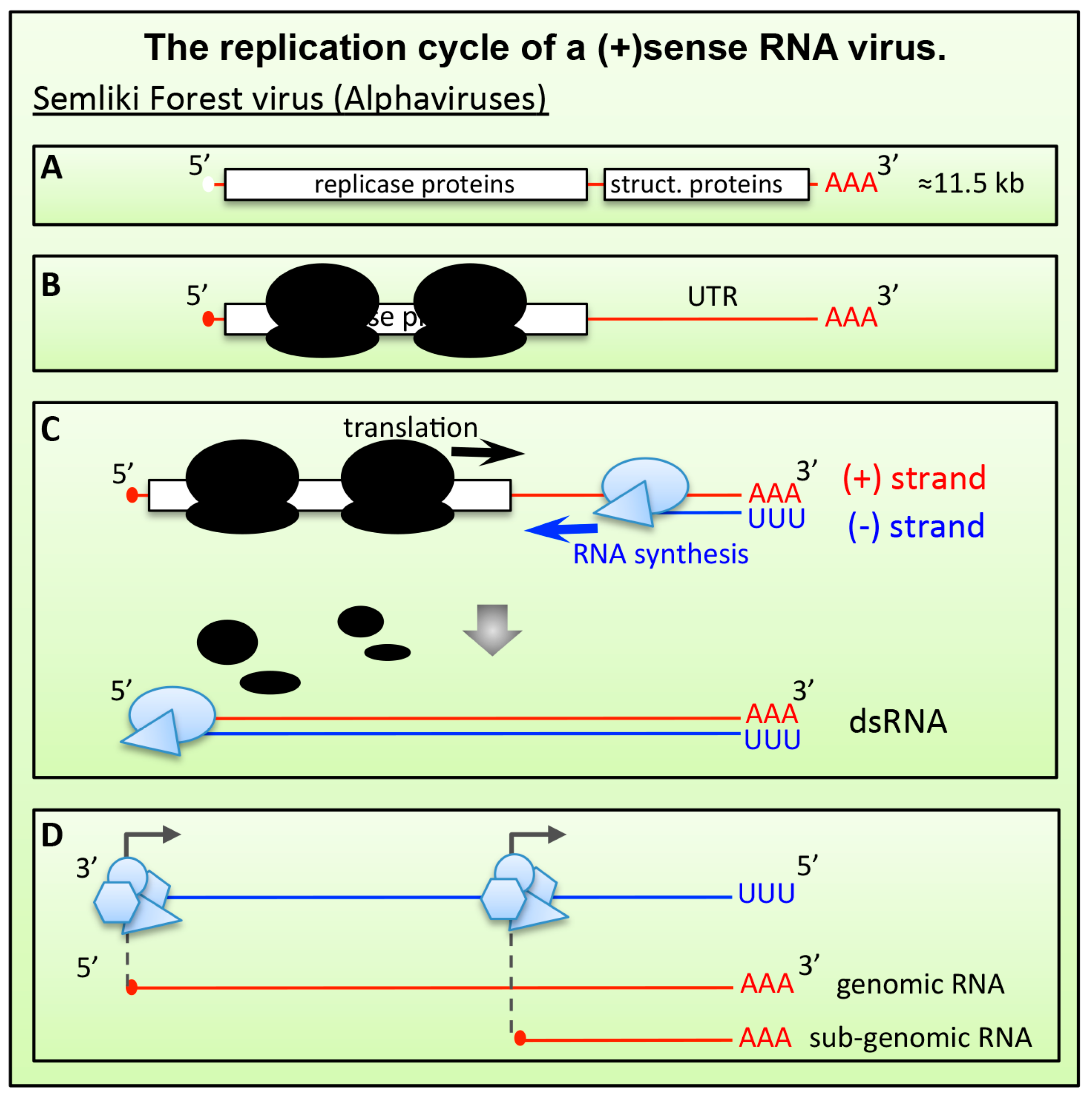
6.1.1. Alphaviruses and Plant (+)RNA Viruses
6.1.2. Hepatitis C Virus (HCV)
6.2. Retroviruses
6.2.1. Rous Sarcoma Virus (RSV)
6.2.2. Human T-lymphotropic Virus Type 1 (HTLV-1)
6.2.3. Human Immunodeficiency Virus Type 1 (HIV-1)
7. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lykke-Andersen, J.; Bennett, E.J. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol. 2014, 204, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Karousis, E.D.; Nasif, S.; Muhlemann, O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley interdiscip. Rev. RNA 2016, 7, 661–682. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, X.; Spatrick, P.; Casillo, R.; Dong, S.; Jacobson, A. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5' to 3' mRNA decay pathways in yeast. Mol. Cell 2003, 12, 1439–1452. [Google Scholar] [CrossRef]
- Mendell, J.T.; Sharifi, N.A.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat.Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Losson, R.; Lacroute, F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl. Acad. Sci. USA 1979, 76, 5134–5137. [Google Scholar] [CrossRef] [PubMed]
- Maquat, L.E.; Kinniburgh, A.J.; Rachmilewitz, E.A.; Ross, J. Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell 1981, 27, 543–553. [Google Scholar] [CrossRef]
- Culbertson, M.R.; Leeds, P.F. Looking at mRNA decay pathways through the window of molecular evolution. Curr. Opin. Genet. Dev. 2003, 13, 207–214. [Google Scholar] [CrossRef]
- Rebbapragada, I.; Lykke-Andersen, J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr. Opin. Cell Biol. 2009, 21, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, P.; Yepiskoposyan, H.; Metze, S.; Zamudio Orozco, R.; Kleinschmidt, N.; Mühlemann, O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol. Life Sci. 2010, 67, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Schweingruber, C.; Rufener, S.C.; Zund, D.; Yamashita, A.; Muhlemann, O. Nonsense-mediated mRNA decay - mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim. Biophys. Acta. 2013, 1829, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Puretzky, A.A.; Liang, L.; Li, X.; Xiao, K.; Wang, K.; Mahjouri-Samani, M.; Basile, L.; Idrobo, J.C.; Sumpter, B.G.; Meunier, V. Low-Frequency Raman Fingerprints of Two-Dimensional Metal Dichalcogenide Layer Stacking Configurations. ACS Nano 2015, 9, 6333–6342. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Bio. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Shaul, O. Unique Aspects of Plant Nonsense-Mediated mRNA Decay. Trends Plant Sci. 2015, 20, 767–779. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Brown, A.H.; Jacobson, A. Upf1p, Nmd2p, and Upf3p are interacting components of the yeast nonsense-mediated mRNA decay pathway. Mol. Cell. Biol. 1997, 17, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Min, E.E.; Roy, B.; Amrani, N.; He, F.; Jacobson, A. Yeast Upf1 CH domain interacts with Rps26 of the 40S ribosomal subunit. RNA 2013, 19, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Jacobson, A. Identification of a novel component of the nonsense-mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev. 1995, 9, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Deniaud, A.; Boehm, V.; Gehring, N.H.; Schaffitzel, C.; Cusack, S. Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res. 2014, 42, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell 41, 693–703. [CrossRef] [PubMed]
- Lykke-Andersen, J.; Shu, M.D.; Steitz, J.A. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 2000, 103, 1121–1131. [Google Scholar] [CrossRef]
- Le Hir, H.; Sauliere, J.; Wang, Z. The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 2016, 17, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.B.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E.; Gehring, N.H. Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA 2006, 12, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Bhalla, A.D.; Le Hir, H.; Nguyen, L.S.; Huang, L.; Gécz, J.; Wilkinson, M.F. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat. Struct. Mol. Biol. 2009, 16, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Shum, E.Y.; Jones, S.H.; Shao, A.; Dumdie, J.; Krause, M.D.; Chan, W.K.; Lou, C.H.; Espinoza, J.L.; Song, H.W.; Phan, M.H.; et al. The Antagonistic Gene Paralogs Upf3a and Upf3b Govern Nonsense-Mediated RNA Decay. Cell 2016, 165, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Denning, G.; Jamieson, L.; Maquat, L.E.; Thompson, E.A.; Fields, A.P. Cloning of a novel phosphatidylinositol kinase-related kinase: characterization of the human SMG-1 RNA surveillance protein. J. Biol. Chem. 2001, 276, 22709–22714. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Caceres, J.F. The RNA helicase DHX34 activates NMD by promoting a transition from the surveillance to the decay-inducing complex. Cell Rep. 2014, 8, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Izumi, N.; Kashima, I.; Ohnishi, T.; Saari, B.; Katsuhata, Y.; Muramatsu, R.; Morita, T.; Iwamatsu, A.; Hachiya, T.; et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009, 23, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Uchiyama, A.; Castaño, R.; Kataoka, N.; Kurosawa, H.; Ohno, S.; Yamashita, A.; Llorca, O. Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure 2014, 22, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol. Cells 2014, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Loh, B.; Jonas, S.; Izaurralde, E. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013, 27, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Bonneau, F.; Schussler, S.; Eppinger, E.; Conti, E. Phospho-dependent and phospho-independent interactions of the helicase UPF1 with the NMD factors SMG5-SMG7 and SMG6. Nucleic Acids Res 2014, 42, 9447–9460. [Google Scholar] [CrossRef] [PubMed]
- Eberle, A.B.; Lykke-Andersen, S.; Muhlemann, O.; Jensen, T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009, 16, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Okada-Katsuhata, Y.; Yamashita, A.; Kutsuzawa, K.; Izumi, N.; Hirahara, F.; Ohno, S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012, 40, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, P.; Josi, C.; Kurosawa, H.; Yamashita, A.; Muhlemann, O. A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res. 2014, 42, 9217–9235. [Google Scholar] [CrossRef] [PubMed]
- Longman, D.; Hug, N.; Keith, M.; Anastasaki, C.; Patton, E.E.; Grimes, G.; Cáceres, J.F. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013, 41, 8319–8331. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Longman, D.; Capper, A.; Patton, E.E.; Caceres, J.F. Dhx34 and Nbas function in the NMD pathway and are required for embryonic development in zebrafish. Nucleic Acids Res. 2011, 39, 3686–3694. [Google Scholar] [CrossRef] [PubMed]
- Casadio, A.; Longman, D.; Hug, N.; Delavaine, L.; Vallejos Baier, R.; Alonso, C.R.; Cáceres, J.F. Identification and characterization of novel factors that act in the nonsense-mediated mRNA decay pathway in nematodes, flies and mammals. EMBO Rep. 2015, 16, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ito, K.; Matsumura, K.; Kawazu, Y.; Ebihara, K. Regulation of translation termination: conserved structural motifs in bacterial and eukaryotic polypeptide release factors. Biochem. Cell Biol. 1995, 73, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Pisarev, A.V.; Skabkin, M.A.; Pisareva, V.P.; Skabkina, O.V.; Rakotondrafara, A.M.; Hentze, M.W.; Hellen, C.U.; Pestova, T.V. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell 2010, 37, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Barthelme, D.; Dinkelaker, S.; Albers, S.V.; Londei, P.; Ermler, U.; Tampé, R. Ribosome recycling depends on a mechanistic link between the FeS cluster domain and a conformational switch of the twin-ATPase ABCE1. Proc. Natl. Acad. Sci. USA 2011, 108, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Mikhailova, T.; Eliseev, B.; Yeramala, L.; Sokolova, E.; Susorov, D.; Shuvalov, A.; Schaffitzel, C.; Alkalaeva, E. PABP enhances release factor recruitment and stop codon recognition during translation termination. Nucleic Acids Research 2016, 44, 7766–7776. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Rebbapragada, I.; Lykke-Andersen, J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008, 6, e111. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Ribeiro, P.; Inacio, A.; Liebhaber, S.A.; Romao, L. Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated mRNA decay. RNA 2008, 14, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Muhlemann, O.; Jensen, T.H. mRNP quality control goes regulatory. Trends Genet. 2012, 28, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Maquat, L.E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004, 5, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Amrani, N.; Ganesan, R.; Kervestin, S.; Mangus, D.A.; Ghosh, S.; Jacobson, A. A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 2004, 432, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Peixeiro, I.; Silva, A.L.; Romao, L. Control of human beta-globin mRNA stability and its impact on beta-thalassemia phenotype. Haematologica 2011, 96, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Chen, Y.; Ardal, B.R.; Lilje, B.; Waage, J.; Sandelin, A.; Jensen, T.H. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014, 28, 2498–2517. [Google Scholar] [CrossRef] [PubMed]
- Boehm, V.; Haberman, N.; Ottens, F.; Ule, J.; Gehring, N.H. 3' UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep. 2014, 9, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 2002, 22, 8114–8121. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kim, K.M.; Kim, Y.K. Human proline-rich nuclear receptor coregulatory protein 2 mediates an interaction between mRNA surveillance machinery and decapping complex. Mol. Cell 2009, 33, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.; Cho, H.; Liu, Z.; Bowler, M.W.; Piao, S.; Parker, R.; Kim, Y.K.; Song, H. Structural basis of the PNRC2-mediated link between mrna surveillance and decapping. Structure 2012, 20, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Han, S.; Choe, J.; Park, S.G.; Choi, S.S.; Kim, Y.K. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res. 2013, 41, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Franks, T. M.; Singh, G.; Lykke-Andersen, J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell 2010, 143, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Yamashita, A.; Kashima, I.; Schell, T.; Anders, K.R.; Grimson, A.; Hachiya, T.; Hentze, M.W.; Anderson, P.; Ohno, S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell 2003, 12, 1187–1200. [Google Scholar] [CrossRef]
- Chiu, S.Y.; Serin, G.; Ohara, O.; Maquat, L.E. Characterization of human Smg5/7a: a protein with similarities to Caenorhabditis elegans SMG5 and SMG7 that functions in the dephosphorylation of Upf1. RNA 2003, 9, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Mühlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe 2014, 16, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Garcia, S.; Voinnet, O. Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe 2014, 16, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Quek, B.L.; Beemon, K. Retroviral strategy to stabilize viral RNA. Curr. Opin. Microbiol. 2014, 18, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Mocquet, V.; Durand, S.; Jalinot, P. How Retroviruses Escape the Nonsense-Mediated mRNA Decay. AIDS Res. Hum. Retroviruses 2015, 31, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Wernet, M.F.; Klovstad, M.; Clandinin, T.R. Generation of infectious virus particles from inducible transgenic genomes. Curr. Biol. 2014, 24, R107–R108. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.L.; Wilusz, J. Cytoplasmic viruses: rage against the (cellular RNA decay) machine. PLoS Pathog. 2013, 9, e1003762. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Caldentey, J.; Kaariainen, L.; Ahola, T. Enzymatic defects of the nsP2 proteins of Semliki Forest virus temperature-sensitive mutants. J. Virol 2007, 81, 2849–2860. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.R.; Kumar, G.R.; Verschueren, E.; Johnson, J.R.; Von Dollen, J.; Johnson, T.; Newton, B.; Shah, P.; Horner, J.; Krogan, N.J.; et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 2015, 57, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Barker, G.F.; Beemon, K. Rous sarcoma virus RNA stability requires an open reading frame in the gag gene and sequences downstream of the gag-pol junction. Mol. Cell. Biol. 1994, 14, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.J.; Beemon, K.L. Unspliced Rous sarcoma virus genomic RNAs are translated and subjected to nonsense-mediated mRNA decay before packaging. J. Virol 2004, 78, 5139–5146. [Google Scholar] [CrossRef] [PubMed]
- Weil, J.E.; Beemon, K.L. A 3' UTR sequence stabilizes termination codons in the unspliced RNA of Rous sarcoma virus. RNA 2006, 12, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Withers, J.B.; Beemon, K.L. Structural features in the Rous sarcoma virus RNA stability element are necessary for sensing the correct termination codon. Retrovirology 2012, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Withers, J.B.; Beemon, K.L. The structure and function of the rous sarcoma virus RNA stability element. J. Cell Biochem. 2011, 112, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Quek, B.L.; Beemon, K.L.; Hogg, J.R. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Mocquet, V.; Neusiedler, J.; Rende, F.; Cluet, D.; Robin, J.P.; Terme, J.M.; Duc Dodon, M.; Wittmann, J.; Morris, C.; Le Hir, H.; et al. The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J Virol 2012, 86, 7530–7543. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Ando, T.; Yamagishi, M.; Yokoyama, K.; Ishida, T.; Ohsugi, T.; Tanaka, Y.; Brighty, D.W.; Watanabe, T. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: implications for retroviral replication. Microbes Infect. 2013, 15, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, L.; Abrahamyan, L.; Milev, M.; Ivanov, P.V.; Kulozik, A.E.; Gehring, N.H.; Mouland, A.J. Unexpected roles for UPF1 in HIV-1 RNA metabolism and translation. RNA 2008, 14, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, L.; Abel, K.; Rao, S.; Vyboh, K.; García-de-Gracia, F.; Soto-Rifo, R.; Kulozik, A.E.; Gehring, N.H.; Mouland, A.J. HIV-1 Recruits UPF1 but Excludes UPF2 to Promote Nucleocytoplasmic Export of the Genomic RNA. Biomolecules 2015, 5, 2808–2839. [Google Scholar] [CrossRef] [PubMed]
- Serquina, A.K.; Das, S.R.; Popova, E.; Ojelabi, O.A.; Roy, C.K.; Göttlinger, H.G. UPF1 is crucial for the infectivity of human immunodeficiency virus type 1 progeny virions. J. Virol 2013, 87, 8853–8861. [Google Scholar] [CrossRef] [PubMed]
- Hamid, F.M.; Makeyev, E.V. Exaptive origins of regulated mRNA decay in eukaryotes. Bioessays 2016, 38, 830–838. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balistreri, G.; Bognanni, C.; Mühlemann, O. Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay. Viruses 2017, 9, 24. https://doi.org/10.3390/v9010024
Balistreri G, Bognanni C, Mühlemann O. Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay. Viruses. 2017; 9(1):24. https://doi.org/10.3390/v9010024
Chicago/Turabian StyleBalistreri, Giuseppe, Claudia Bognanni, and Oliver Mühlemann. 2017. "Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay" Viruses 9, no. 1: 24. https://doi.org/10.3390/v9010024
APA StyleBalistreri, G., Bognanni, C., & Mühlemann, O. (2017). Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay. Viruses, 9(1), 24. https://doi.org/10.3390/v9010024