
On the Selective Packaging of Genomic RNA by HIV-1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of RNA in Particle Assembly
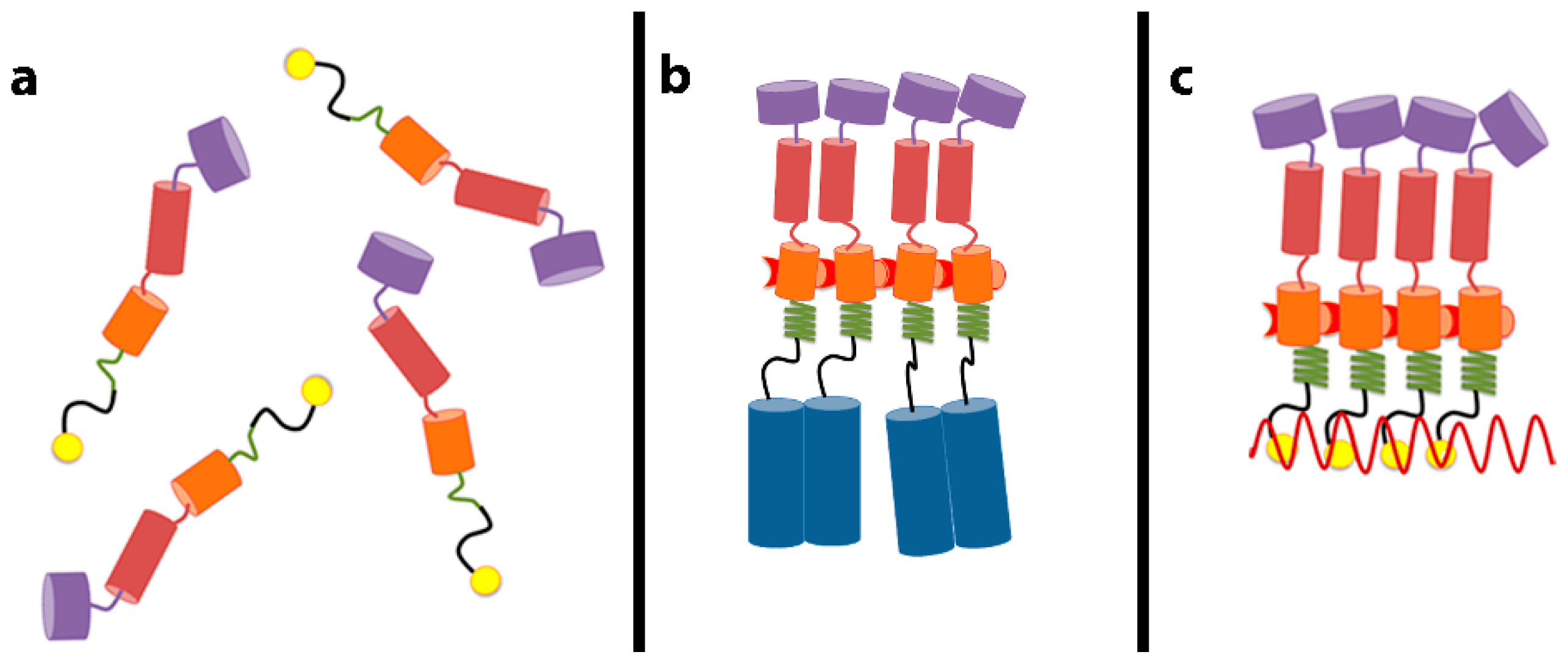
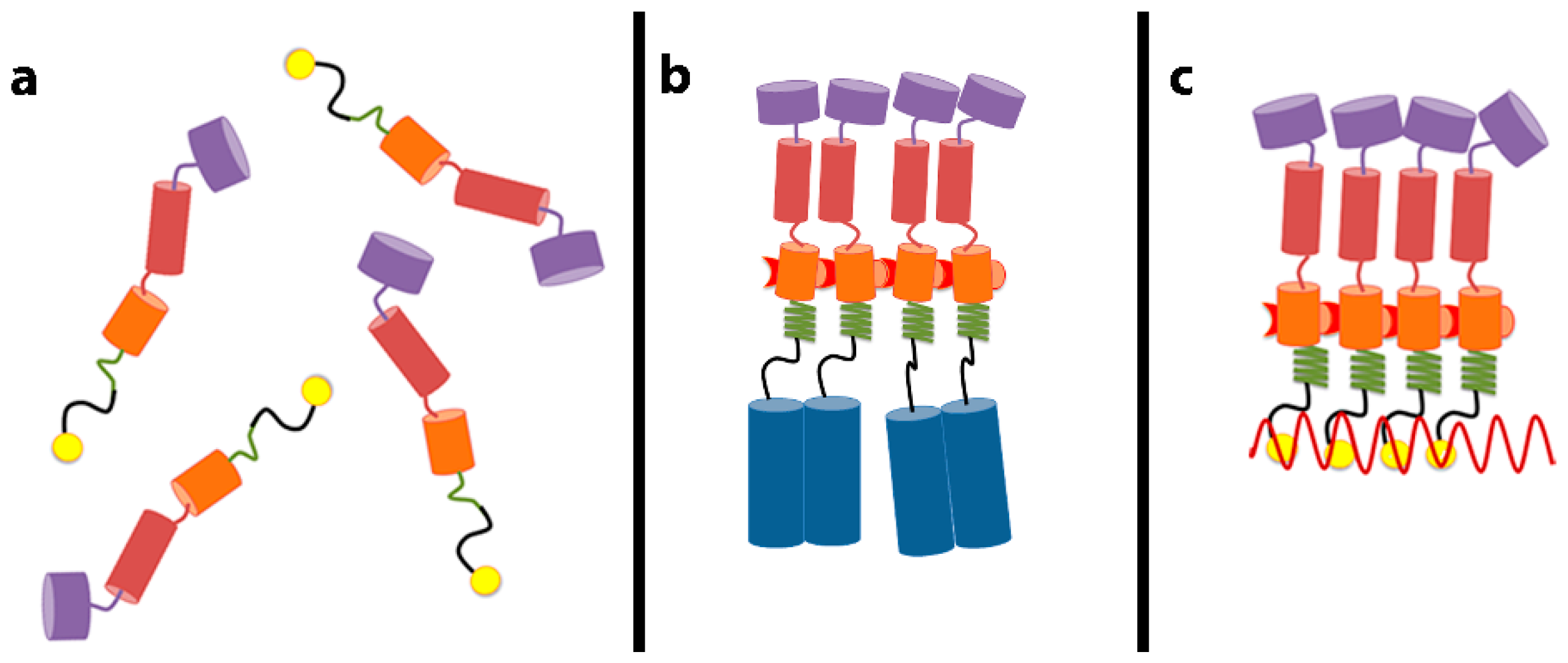
3. Gag–RNA Interactions Involve Multiple Domains of Gag
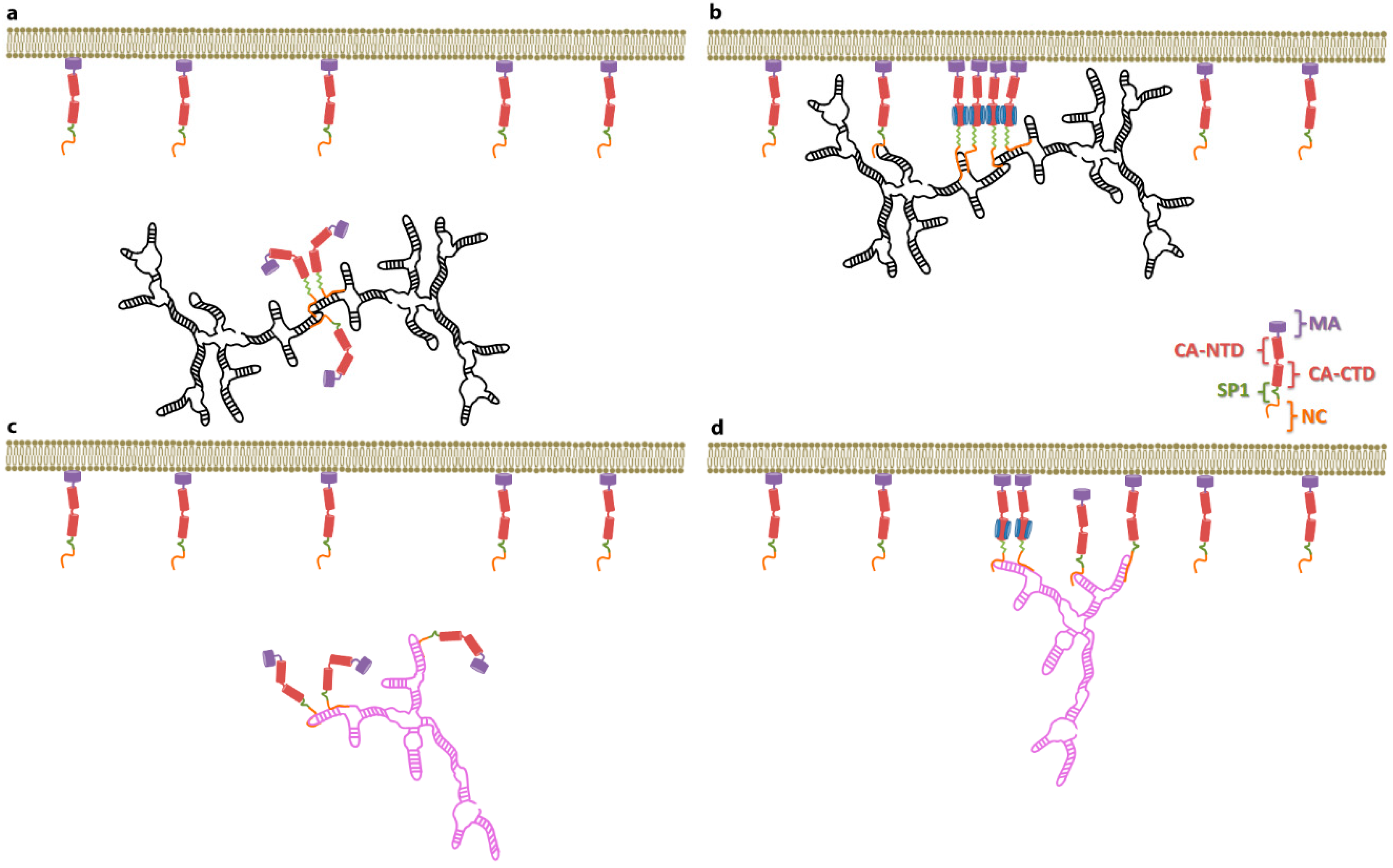
4. A Switch in Gag: The Decision to Assemble
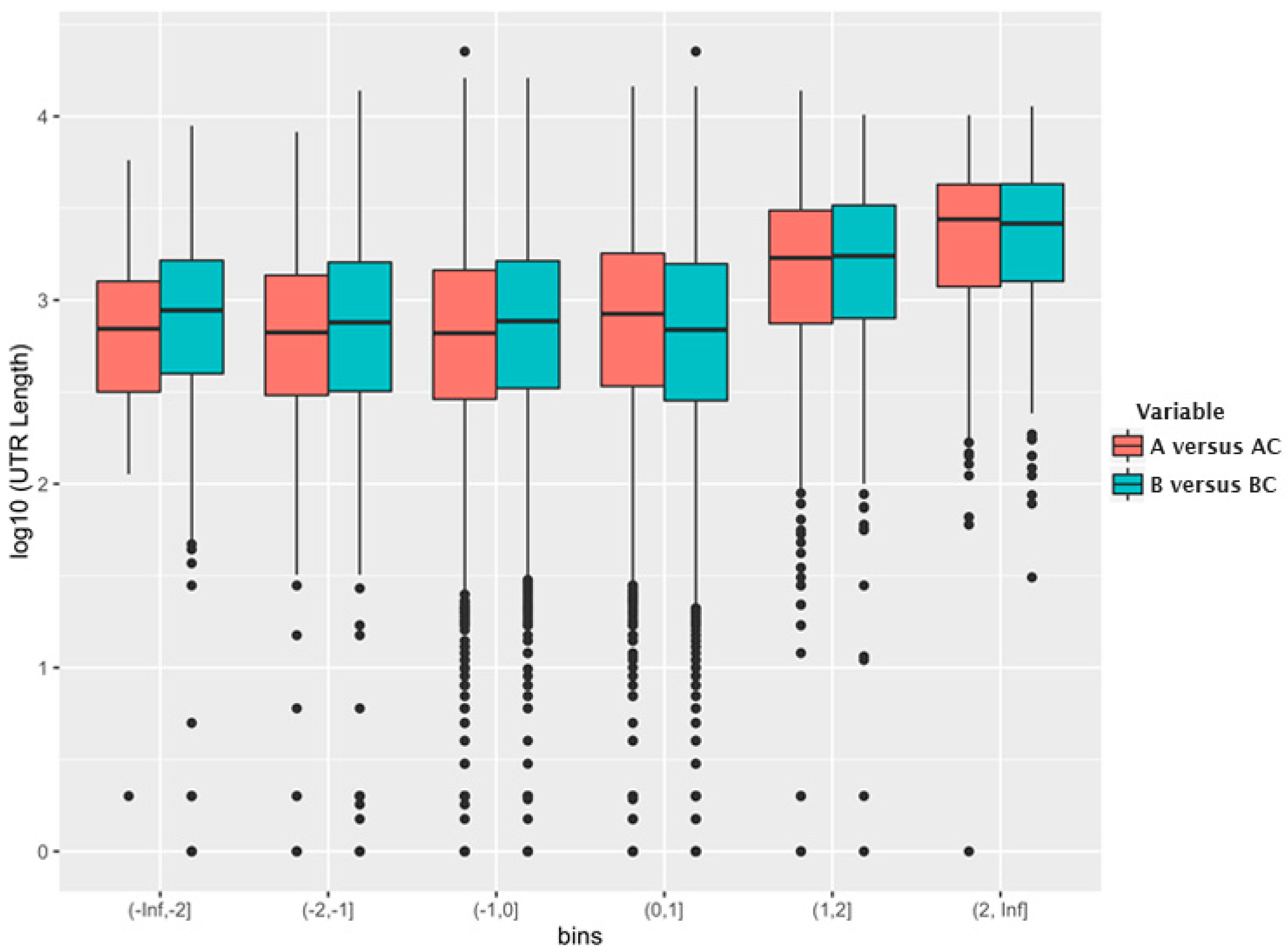
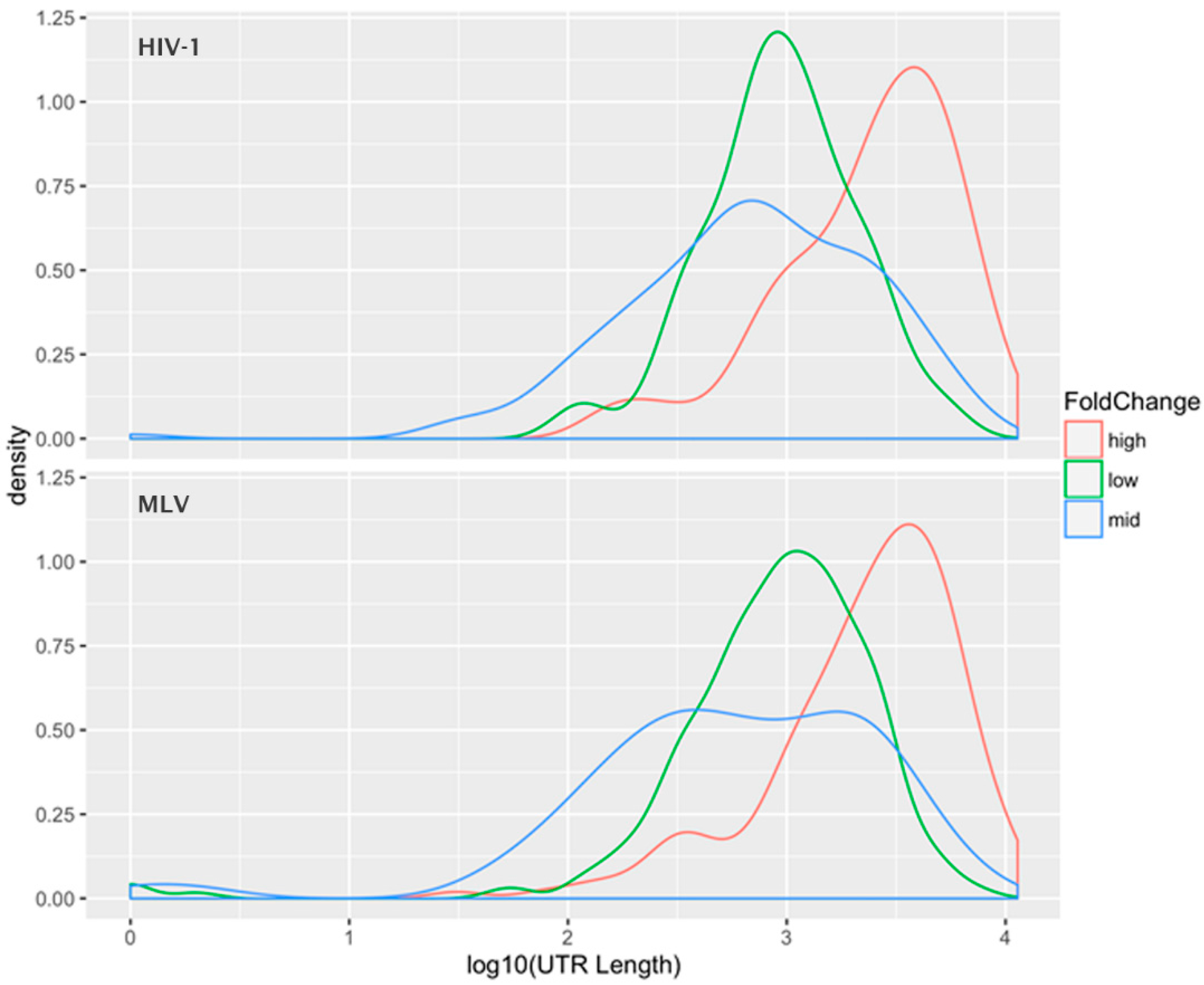
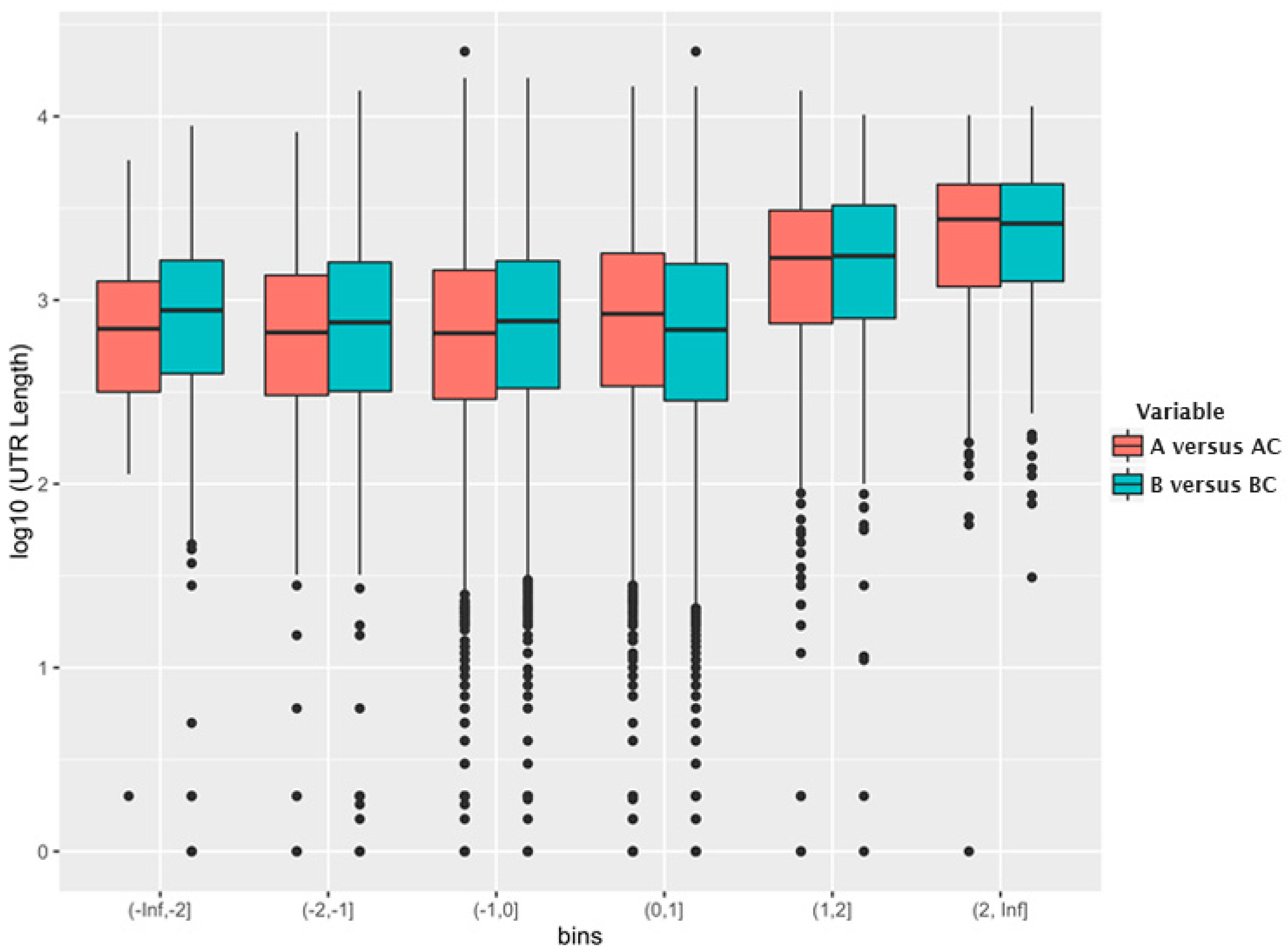
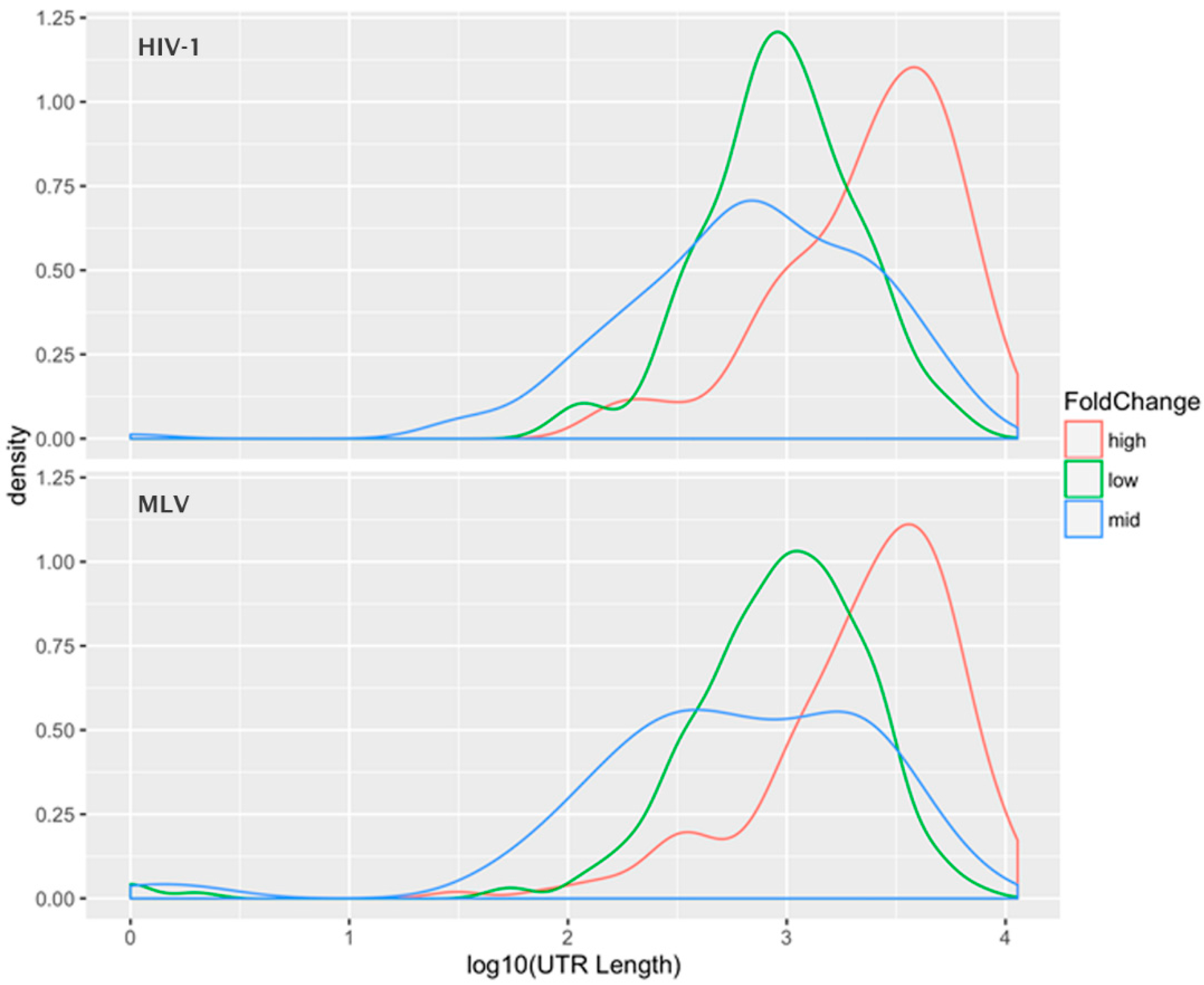
5. Selective Packaging and Ψ
6. Packaging Non-Viral RNAs
7. Dimeric RNA as the Right Substrate
8. Consequences of a Nucleation Mechanism
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moore, M.D.; Hu, W.S. HIV-1 RNA dimerization: It takes two to tango. AIDS Rev. 2009, 11, 91–102. [Google Scholar] [PubMed]
- Paillart, J.C.; Shehu-Xhilaga, M.; Marquet, R.; Mak, J. Dimerization of retroviral RNA genomes: An inseparable pair. Nat. Rev. Microbiol. 2004, 2, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Aldovini, A.; Young, R.A. Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J. Virol. 1990, 64, 1920–1926. [Google Scholar] [PubMed]
- Lever, A.; Gottlinger, H.; Haseltine, W.; Sodroski, J. Identification of a sequence required for efficient packaging of human immunodeficiency virus type 1 RNA into virions. J. Virol. 1989, 63, 4085–4087. [Google Scholar] [PubMed]
- Gorelick, R.J.; Nigida, S.M., Jr.; Bess, J.W., Jr.; Arthur, L.O.; Henderson, L.E.; Rein, A. Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J. Virol. 1990, 64, 3207–3211. [Google Scholar] [PubMed]
- Perlmutter, J.D.; Hagan, M.F. The role of packaging sites in efficient and specific virus assembly. J. Mol. Biol. 2015, 427, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.G.; Grimley, P.M.; Ramseur, J.M.; Berezesky, I.K. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J. Virol. 1974, 14, 152–161. [Google Scholar] [PubMed]
- Muriaux, D.; Mirro, J.; Harvin, D.; Rein, A. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 2001, 98, 5246–5251. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Rein, A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 1999, 73, 2270–2279. [Google Scholar] [PubMed]
- Zhang, Y.; Qian, H.; Love, Z.; Barklis, E. Analysis of the assembly function of the human immunodeficiency virus type 1 Gag protein nucleocapsid domain. J. Virol. 1998, 72, 1782–1789. [Google Scholar] [PubMed]
- Crist, R.M.; Datta, S.A.; Stephen, A.G.; Soheilian, F.; Mirro, J.; Fisher, R.J.; Nagashima, K.; Rein, A. Assembly properties of human immunodeficiency virus type 1 Gag-leucine zipper chimeras: Implications for retrovirus assembly. J. Virol. 2009, 83, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, V.; Oh, S.J.; Ono, A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. USA 2010, 107, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.P.; Datta, S.A.; Rein, A.; Rouzina, I.; Musier-Forsyth, K. Matrix domain modulates HIV-1 Gag’s nucleic acid chaperone activity via inositol phosphate binding. J. Virol. 2011, 85, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global changes in the RNA binding specificity of HIV-1 Gag regulate virion genesis. Cell 2014, 159, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Fisher, R.J.; Towler, E.M.; Fox, S.; Issaq, H.J.; Wolfe, T.; Phillips, L.R.; Rein, A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. USA 2001, 98, 10875–10879. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Curtis, J.E.; Ratcliff, W.; Clark, P.K.; Crist, R.M.; Lebowitz, J.; Krueger, S.; Rein, A. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 2007, 365, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Nath, A.; Farber, M.; Datta, S.A.; Rein, A.; Rhoades, E.; Mothes, W. A conformational transition observed in single HIV-1 Gag molecules during in vitro assembly of virus-like particles. J. Virol. 2014, 88, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.; Heinrich, F.; Raghunandan, S.; Krueger, S.; Curtis, J.E.; Rein, A.; Nanda, H. HIV-1 Gag extension: Conformational changes require simultaneous interaction with membrane and nucleic acid. J. Mol. Biol. 2011, 406, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Kempf, N.; Postupalenko, V.; Bora, S.; Didier, P.; Arntz, Y.; de Rocquigny, H.; Mely, Y. The HIV-1 nucleocapsid protein recruits negatively charged lipids to ensure its optimal binding to lipid membranes. J. Virol. 2015, 89, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Kutluay, S.B.; Bieniasz, P.D. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 2010, 6, e1001200. [Google Scholar] [CrossRef] [PubMed]
- Comas-Garcia, M.; Rein, A. manuscript in preparation. 2016.
- Datta, S.A.; Temeselew, L.G.; Crist, R.M.; Soheilian, F.; Kamata, A.; Mirro, J.; Harvin, D.; Nagashima, K.; Cachau, R.E.; Rein, A. On the role of the SP1 domain in HIV-1 particle assembly: A molecular switch? J. Virol. 2011, 85, 4111–4121. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.K.; Clark, P.K.; Fan, L.; Ma, B.; Harvin, D.P.; Sowder, R.C.; Nussinov, R.; Wang, Y.-X.; Rein, A. Dimerization of the SP1 region of HIV-1 Gag induces a helical conformation and association into helical bundles: Implications for particle assembly. J. Virol. 2016, 90, 1773–1787. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K.; Obr, M.; Hagen, W.J.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Krausslich, H.G.; Briggs, J.A. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Zadrozny, K.K.; Chrustowicz, J.; Purdy, M.D.; Yeager, M.; Ganser-Pornillos, B.K.; Pornillos, O. Crystal structure of an HIV assembly and maturation switch. Elife 2016, 5, e17063. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, R.D.; Hammarskjold, M.L.; Helga-Maria, C.; Rekosh, D.; Goff, S.P. 5′ regions of HIV-1 RNAs are not sufficient for encapsidation: Implications for the HIV-1 packaging signal. Virology 1995, 212, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.A.; Miller, A.D. Identification of a signal in a murine retrovirus that is sufficient for packaging of nonretroviral RNA into virions. J. Virol. 1988, 62, 3802–3806. [Google Scholar] [PubMed]
- Banks, J.D.; Yeo, A.; Green, K.; Cepeda, F.; Linial, M.L. A minimal avian retroviral packaging sequence has a complex structure. J. Virol. 1998, 72, 6190–6194. [Google Scholar] [PubMed]
- Hibbert, C.S.; Mirro, J.; Rein, A. mRNA molecules containing murine leukemia virus packaging signals are encapsidated as dimers. J. Virol. 2004, 78, 10927–10938. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, L.; Racine, P.J.; Houzet, L.; Chamontin, C.; Berkhout, B.; Mougel, M. Role of HIV-1 RNA and protein determinants for the selective packaging of spliced and unspliced viral RNA and host U6 and 7SL RNA in virus particles. Nucleic Acids Res. 2011, 39, 8915–8927. [Google Scholar] [CrossRef] [PubMed]
- Houzet, L.; Paillart, J.C.; Smagulova, F.; Maurel, S.; Morichaud, Z.; Marquet, R.; Mougel, M. HIV controls the selective packaging of genomic, spliced viral and cellular RNAs into virions through different mechanisms. Nucleic Acids Res. 2007, 35, 2695–2704. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Wahab, E.W.; Smyth, R.P.; Mailler, E.; Bernacchi, S.; Vivet-Boudou, V.; Hijnen, M.; Jossinet, F.; Mak, J.; Paillart, J.C.; Marquet, R. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat. Commun. 2014, 5, 4304. [Google Scholar] [CrossRef] [PubMed]
- Clever, J.; Sassetti, C.; Parslow, T.G. RNA secondary structure and binding sites for Gag gene products in the 5′ packaging signal of human immunodeficiency virus type 1. J. Virol. 1995, 69, 2101–2109. [Google Scholar] [PubMed]
- Smyth, R.P.; Despons, L.; Huili, G.; Bernacchi, S.; Hijnen, M.; Mak, J.; Jossinet, F.; Weixi, L.; Paillart, J.C.; von Kleist, M.; et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat. Methods 2015, 12, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Rulli, S.J., Jr.; Hibbert, C.S.; Mirro, J.; Pederson, T.; Biswal, S.; Rein, A. Selective and nonselective packaging of cellular RNAs in retrovirus particles. J. Virol. 2007, 81, 6623–6631. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, R.D.; Luban, J.; Goff, S.P. Specific binding of human immunodeficiency virus type 1 Gag polyprotein and nucleocapsid protein to viral RNAs detected by RNA mobility shift assays. J. Virol. 1993, 67, 7190–7200. [Google Scholar] [PubMed]
- Webb, J.A.; Jones, C.P.; Parent, L.J.; Rouzina, I.; Musier-Forsyth, K. Distinct binding interactions of HIV-1 Gag to Psi and non-Psi RNAs: Implications for viral genomic RNA packaging. RNA 2013, 19, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of affymetrix genechip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Dewan, V.; Wei, M.; Kleiman, L.; Musier-Forsyth, K. Dual role for motif 1 residues of human lysyl-tRNA synthetase in dimerization and packaging into HIV-1. J. Biol. Chem. 2012, 287, 41955–41962. [Google Scholar] [CrossRef] [PubMed]
- Eckwahl, M.J.; Arnion, H.; Kharytonchyk, S.; Zang, T.; Bieniasz, P.D.; Telesnitsky, A.; Wolin, S.L. Analysis of the human immunodeficiency virus-1 RNA packageome. RNA 2016, 22, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Eckwahl, M.J.; Telesnitsky, A.; Wolin, S.L. Host RNA packaging by retroviruses: A newly synthesized story. MBio 2016, 7, e02025–e02015. [Google Scholar] [CrossRef] [PubMed]
- Keane, S.C.; Heng, X.; Lu, K.; Kharytonchyk, S.; Ramakrishnan, V.; Carter, G.; Barton, S.; Hosic, A.; Florwick, A.; Santos, J.; et al. RNA structure. Structure of the HIV-1 RNA packaging signal. Science 2015, 348, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, J.C.; Prestwood, L.J.; Lever, A.M. A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci. Rep. 2015, 5, 14369. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.; Summers, M.F. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005, 3, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Gherghe, C.; Lombo, T.; Leonard, C.W.; Datta, S.A.; Bess, J.W., Jr.; Gorelick, R.J.; Rein, A.; Weeks, K.M. Definition of a high-affinity Gag recognition structure mediating packaging of a retroviral RNA genome. Proc. Natl. Acad. Sci. USA 2010, 107, 19248–19253. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rahman, S.A.; Nikolaitchik, O.A.; Grunwald, D.; Sardo, L.; Burdick, R.C.; Plisov, S.; Liang, E.; Tai, S.; Pathak, V.K.; et al. HIV-1 RNA genome dimerizes on the plasma membrane in the presence of Gag protein. Proc. Natl. Acad. Sci. USA 2016, 113, E201–E208. [Google Scholar] [CrossRef] [PubMed]
- Rein, A. Nucleic acid chaperone activity of retroviral Gag proteins. RNA Biol. 2010, 7, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Rein, A.; Henderson, L.E.; Levin, J.G. Nucleic acid chaperone activity of retroviral nucleocapsid proteins: Significance for viral replication. Trends Biochem. Sci. 1998, 23, 297–301. [Google Scholar] [CrossRef]
- Nikolaitchik, O.A.; Dilley, K.A.; Fu, W.; Gorelick, R.J.; Tai, S.H.; Soheilian, F.; Ptak, R.G.; Nagashima, K.; Pathak, V.K.; Hu, W.S. Dimeric RNA recognition regulates HIV-1 genome packaging. PLoS Pathog. 2013, 9, e1003249. [Google Scholar] [CrossRef] [PubMed]
- Sakuragi, J.; Sakuragi, S.; Shioda, T. Minimal region sufficient for genome dimerization in the human immunodeficiency virus type 1 virion and its potential roles in the early stages of viral replication. J. Virol. 2007, 81, 7985–7992. [Google Scholar] [CrossRef] [PubMed]
- Sakuragi, J.-I.; Shioda, T.; Panganiban, A.T. Duplication of the primary encapsidation and dimer linkage region of human immunodeficiency virus type 1 RNA results in the appearance of monomeric RNA in virions. J. Virol. 2001, 75, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comas-Garcia, M.; Davis, S.R.; Rein, A. On the Selective Packaging of Genomic RNA by HIV-1. Viruses 2016, 8, 246. https://doi.org/10.3390/v8090246
Comas-Garcia M, Davis SR, Rein A. On the Selective Packaging of Genomic RNA by HIV-1. Viruses. 2016; 8(9):246. https://doi.org/10.3390/v8090246
Chicago/Turabian StyleComas-Garcia, Mauricio, Sean R. Davis, and Alan Rein. 2016. "On the Selective Packaging of Genomic RNA by HIV-1" Viruses 8, no. 9: 246. https://doi.org/10.3390/v8090246
APA StyleComas-Garcia, M., Davis, S. R., & Rein, A. (2016). On the Selective Packaging of Genomic RNA by HIV-1. Viruses, 8(9), 246. https://doi.org/10.3390/v8090246