
How Polyomaviruses Exploit the ERAD Machinery to Cause Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
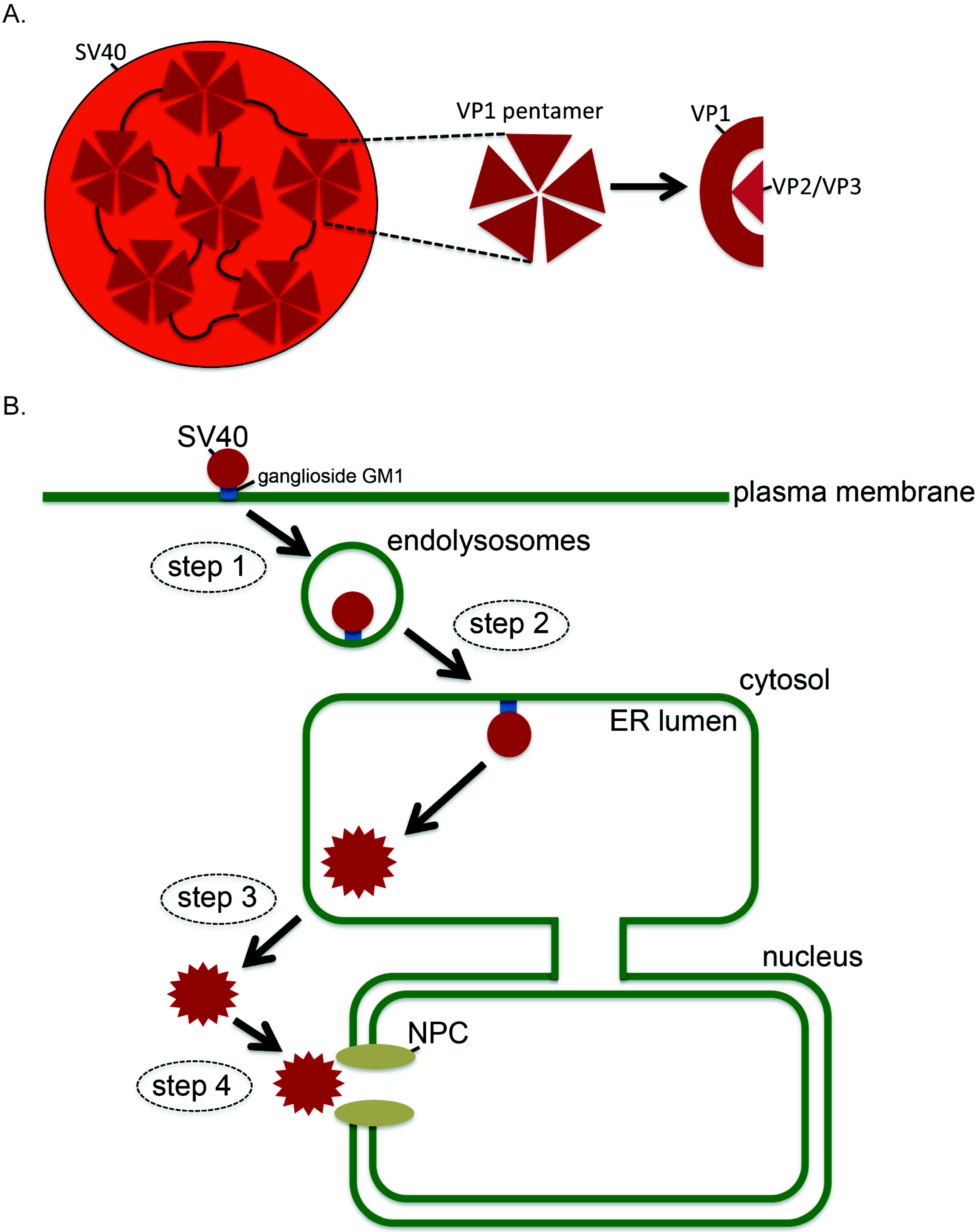
:1. Introduction
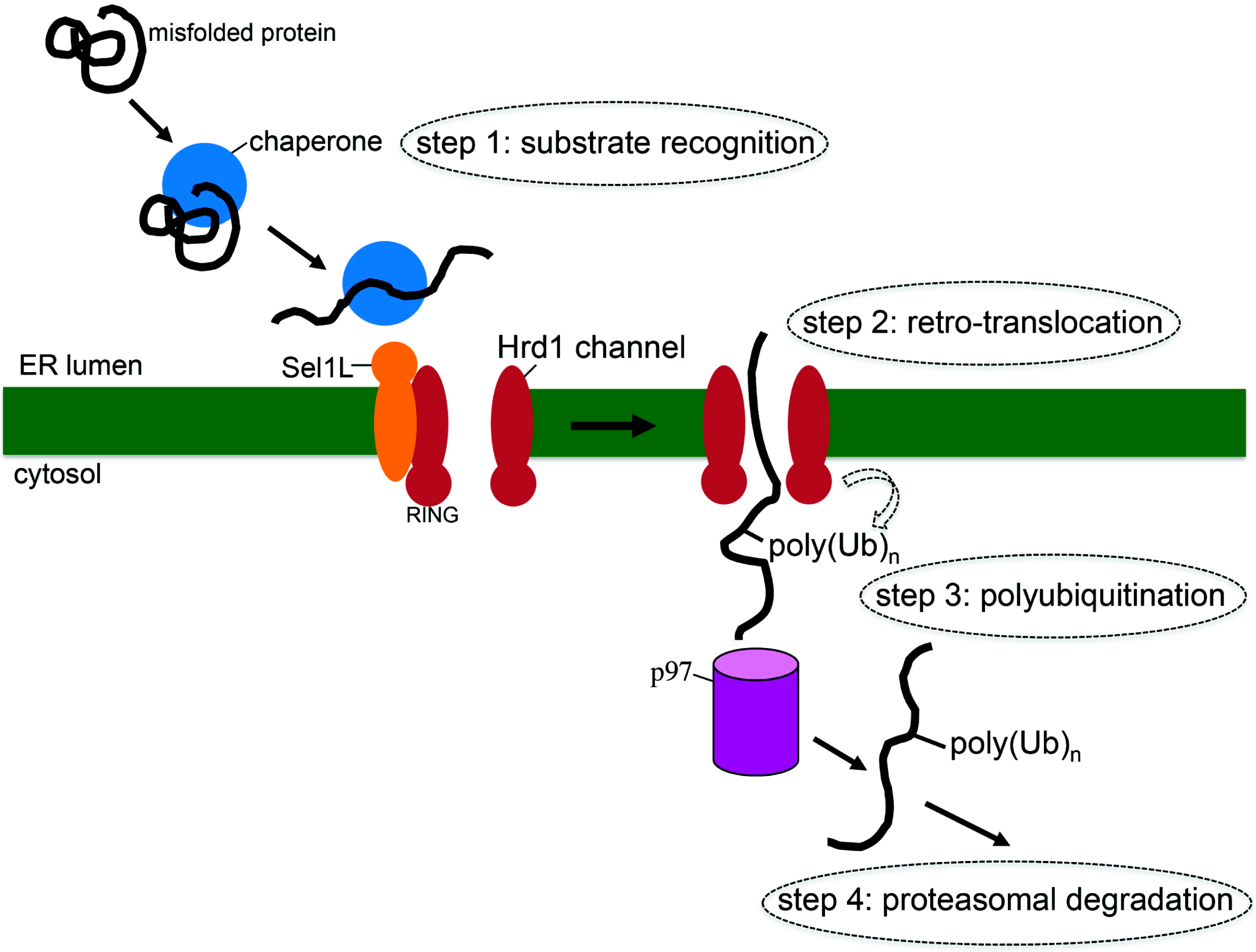
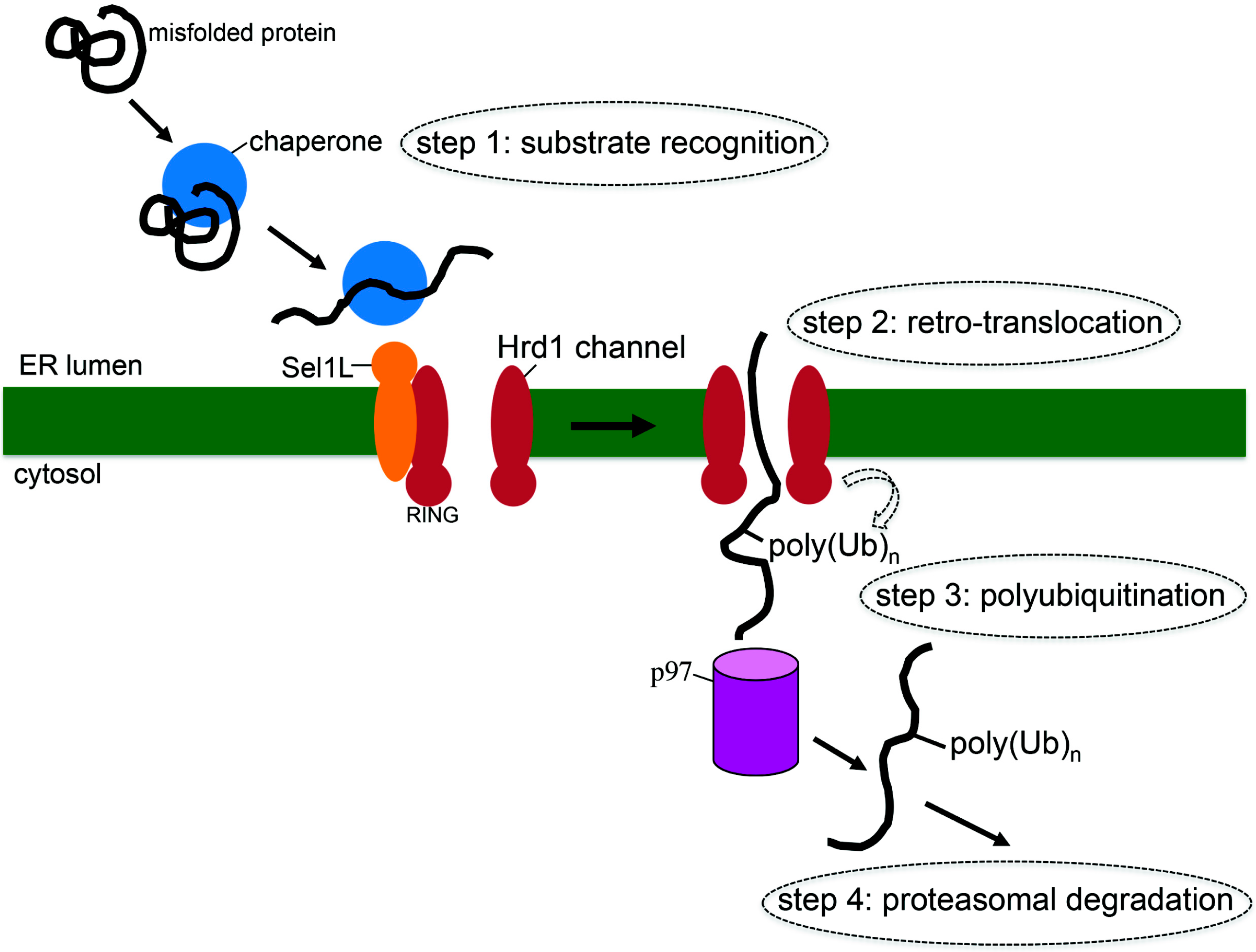
2. What is ERAD?
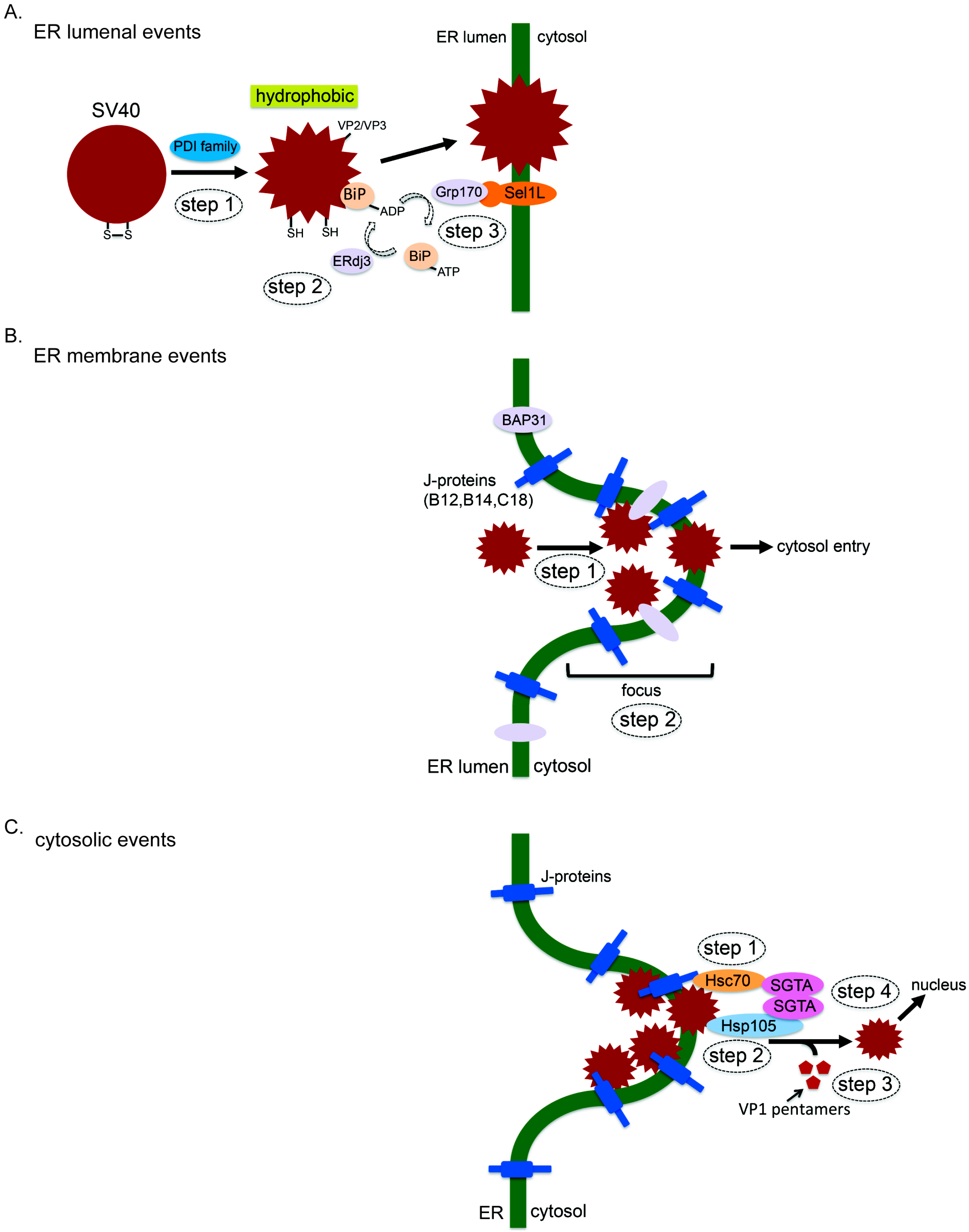
3. How SV40 Hijacks Elements of ERAD during ER Membrane Transport
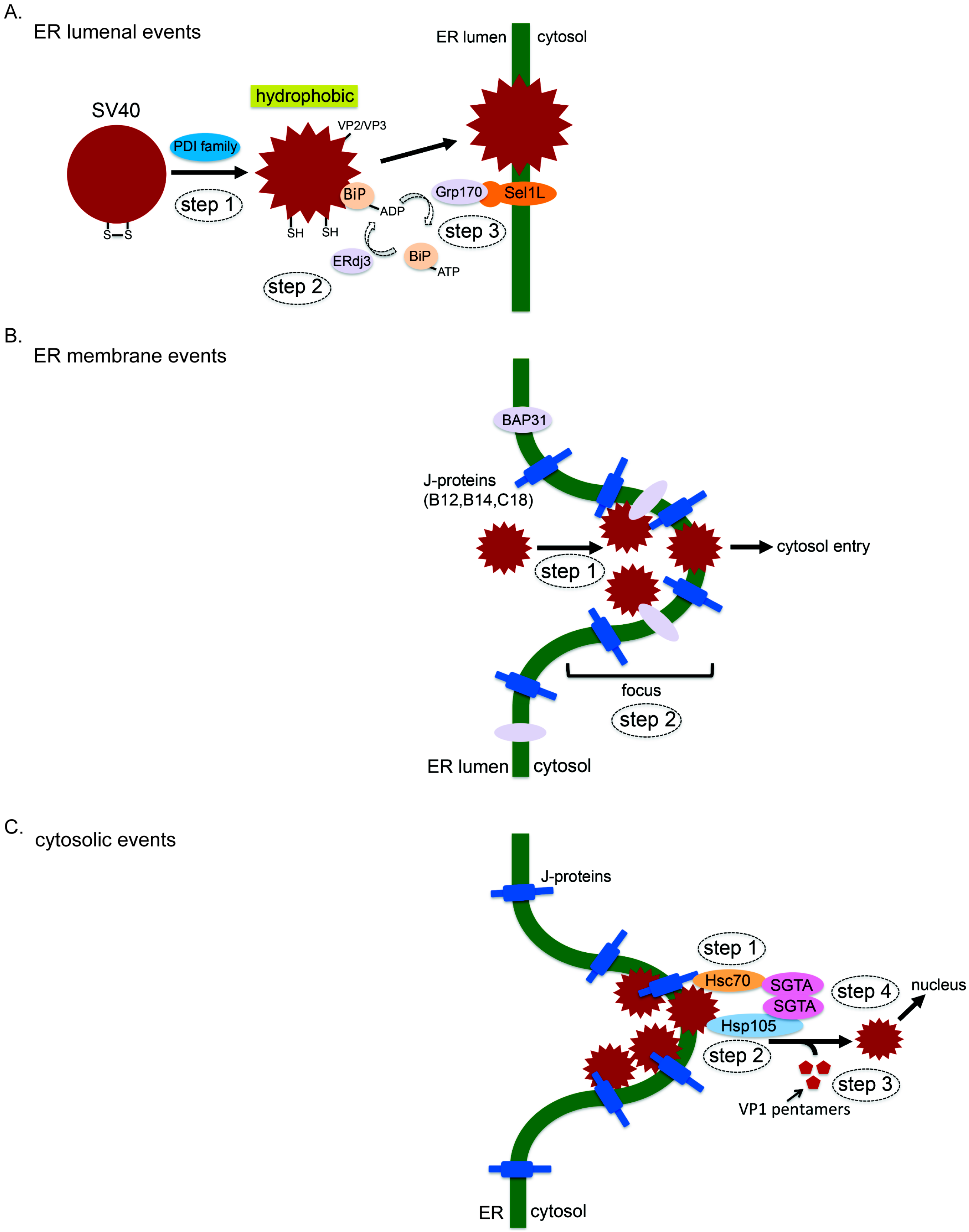
3.1. ER Luminal Events
3.2. ER Membrane Events
3.3. Cytosolic Events
4. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 1, 1253–1257. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Moens, U.; Johannessen, M. Human polyomaviruses and cancer: Expanding repertoire. J. Dtsch. Dermatol. Ges. 2008, 6, 704–708. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Gordon, J.; Khalili, K. The rapidly expanding family of human polyomaviruses: Recent developments in understanding their life cycle and role in human pathology. PLoS Pathog. 2013, 9, e1003206. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Chang, Y.; Moore, P.S. MCV and Merkel cell carcinoma: A molecular success story. Curr. Opin. Virol. 2012, 2, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.M.; Broekema, N.M.; Imperiale, M.J. BK polyomavirus: Emerging pathogen. Microbes Infect. 2012, 14, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Dalianis, T.; Hirsch, H.H. Human polyomaviruses in disease and cancer. Virology 2013, 437, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S.; Nelson, C.D.; Atwood, W.J. JC polyomavirus attachment, entry, and trafficking: Unlocking the keys to a fatal infection. J. Neurovirol. 2015, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.; Sadowicz, D.; Hebert, D.N. A very late viral protein triggers the lytic release of SV40. PLoS Pathog. 2007, 3, e98. [Google Scholar] [CrossRef] [PubMed]
- Liddington, R.C.; Yan, Y.; Moulai, J.; Sahli, R.; Benjamin, T.L.; Harrison, S.C. Structure of simian virus 40 at 3.8-A resolution. Nature 1991, 354, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Stehle, T.; Gamblin, S.J.; Yan, Y.; Harrison, S.C. The structure of simian virus 40 refined at 3.1 A resolution. Structure 1996, 4, 165–182. [Google Scholar] [CrossRef]
- Chen, X.S.; Stehle, T.; Harrison, S.C. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 1998, 17, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Gilbert, J.M.; Stehle, T.; Lencer, W.; Benjamin, T.L.; Rapoport, T.A. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 2003, 22, 4346–4355. [Google Scholar] [CrossRef] [PubMed]
- Campanero-Rhodes, M.A.; Smith, A.; Chai, W.; Sonnino, S.; Mauri, L.; Childs, R.A.; Zhang, Y.; Ewers, H.; Helenius, A.; Imberty, A.; et al. N-glycolyl GM1 ganglioside as a receptor for simian virus 40. J. Virol. 2007, 81, 12846–12858. [Google Scholar] [CrossRef] [PubMed]
- Ewers, H.; Romer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell Biol. 2010, 12, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.; Heger, T.; Mancini, R.; Herzog, F.; Kartenbeck, J.; Hayer, A.; Helenius, A. Role of endosomes in simian virus 40 entry and infection. J. Virol. 2011, 85, 4198–4211. [Google Scholar] [CrossRef] [PubMed]
- Kartenbeck, J.; Stukenbrok, H.; Helenius, A. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell Biol. 1989, 109, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Cai, D.; Verhey, K.J.; Tsai, B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Clever, J.; Yamada, M.; Li, P.P.; Kasamatsu, H. Association with capsid proteins promotes nuclear targeting of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1996, 93, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Clever, J.; Yamada, M.; Kasamatsu, H. Import of simian virus 40 virions through nuclear pore complexes. Proc. Natl. Acad. Sci. USA 1991, 88, 7333–7337. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Bagchi, P.; Cunningham, C.N.; Tsai, B. Opportunistic intruders: How viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol. 2016, 14, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rapoport, T.A. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 2012, 41, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.I.; Fewell, S.W.; Kato, Y.; Brodsky, J.L.; Endo, T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 2001, 153, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Christian, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar]
- Hosokawa, N.; Wada, I.; Nagasawa, K.; Moriyama, T.; Okawa, K.; Nagata, K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008, 283, 209014–209024. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Ruggiano, A.; Carvalho, P.; Rapoport, T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014, 158, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.G.; Swarbrick, G.M.; Bays, N.W.; Cronin, S.R.; Wilhovsky, S.; Seelig, L.; Kim, C.; Hampton, R.Y. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 2000, 151, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Mariano, J.; Tsai, Y.C.; Chan, A.H.; Cohen, M.; Weissman, A.M. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; van den Boomen, D.J.; Bye, H.; Antrobus, R.; Wiertz, E.J.; Lehner, P.J. MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proc. Natl. Acad. Sci. USA 2013, 110, 14290–14295. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Okuda-Shimizu, Y.; Hendershot, L.M. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol. Cell 2010, 40, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Soetandyo, N.; Baek, K.; Hegde, R.; Ye, Y. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 2011, 42, 758–770. [Google Scholar] [CrossRef]
- Xu, Y.; Cai, M.; Yang, Y.; Huang, L.; Ye, Y. SGTA recognizes a noncanonical ubiquitin-like domain in the Bag6-Ubl4A-Trc35 complex to promote endoplasmic reticulum-associated degradation. Cell Rep. 2012, 2, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Malmstrom, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grunewald, K.; Helenius, A. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.P.; Tsai, B. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J. Virol. 2011, 85, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Dosey, A.; Herbstman, J.F.; Ravindran, M.S.; Skiniotis, G.; Tsai, B. ERdj5 Reductase Cooperates with Protein Disulfide Isomerase To Promote Simian Virus 40 Endoplasmic Reticulum Membrane Translocation. J. Virol. 2015, 89, 8897–8908. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Rainey, E.K.; Benjamin, T.; Baryshev, M.; Mkrtchian, S.; Tsai, B. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 2005, 20, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Barger, E.K.; Mkrtchian, S.; Tsai, B. The C-terminal domain of ERp29 mediates polyomavirus binding, unfolding, and infection. J. Virol. 2009, 83, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Ou, W.; Silver, J.; Benjamin, T. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 2006, 80, 10868–10870. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.; Rusan, N.M.; Wadsworth, P.; Hebert, D.N. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: Implications for DNA translocation out of the ER. Mol. Cell 2006, 24, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Kuksin, D.; Norkin, L.C. Disassembly of simian virus 40 during passage through the endoplasmic reticulum and in the cytoplasm. J. Virol. 2012, 86, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.D.; Derdowski, A.; Maginnis, M.S.; O’Hara, B.A.; Atwood, W.J. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 2012, 428, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.; Van Goor, K.E.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio 2011, 2, e00101–e00111. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. A nucleotide exchange factor promotes endoplasmic reticulum-to-cytosol membrane penetration of the nonenveloped virus simian virus 40. J. Virol. 2015, 89, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. The Grp170 nucleotide exchange factor executes a key role during ERAD of cellular misfolded clients. Mol. Biol. Cell 2016, 27, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Gilbert, J.M.; Ploegh, H.L.; Benjamin, T.L. Murine polyomavirus requires the endoplasmic reticulum protein Derlin-2 to initiate infection. J. Virol. 2006, 80, 8739–8744. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Abend, J.R.; Tsai, B.; Imperiale, M.J. Early events during BK virus entry and disassembly. J. Virol. 2009, 83, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.H.; Kimura, T.; Momohara, S.; Takeuchi, M.; Tani, T.; Kimata, Y.; Kadokura, H.; Kohno, K. A novel ER J-protein DNAJB12 accelerates ER-associated degradation of membrane proteins including CFTR. Cell Struct. Funct. 2010, 35, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Grove, D.E.; Fan, C.Y.; Ren, H.Y.; Cyr, D.M. The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol. Biol. Cell 2011, 22, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Sopha, P.; Kadokura, H.; Yamamoto, Y.H.; Takeuchi, M.; Saito, M.; Tsuru, A.; Kohno, K. A novel mammalian ER-located J-protein, DNAJB14, can accelerate ERAD of misfolded membrane proteins. Cell Struct. Funct. 2012, 37, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.P.; Ravindran, M.S.; Inoue, T.; Tsai, B. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog. 2014, 10, e1004007. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Walczak, C.P.; Tsai, B. The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration. J. Virol. 2015, 89, 4058–4068. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Bagchi, P.; Inoue, T.; Tsai, B. A Non-enveloped Virus Hijacks Host Disaggregation Machinery to Translocate across the Endoplasmic Reticulum Membrane. PLoS Pathog. 2015, 11, e1005086. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011, 7, e1002037. [Google Scholar] [CrossRef] [PubMed]
- Hrizo, S.L.; Gusarova, V.; Habiel, D.M.; Goeckeler, J.L.; Fisher, E.A.; Brodsky, J.L. The Hsp110 molecular chaperone stabilizes apolipoprotein B from endoplasmic reticulum-associated degradation (ERAD). J. Biol. Chem. 2007, 282, 32665–32675. [Google Scholar] [CrossRef] [PubMed]
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell 2008, 133, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Bracher, A.; Verghese, J. GrpE, Hsp110/Grp170, HspBP1/Sil1 and BAG domain proteins: Nucleotide exchange factors for Hsp70 molecular chaperones. Subcell. Biochem. 2015, 78, 1–33. [Google Scholar] [PubMed]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, R.U.; Sharma, S.K.; Priya, S.; Finka, A.; Goloubinoff, P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 2013, 288, 21399–21411. [Google Scholar] [CrossRef] [PubMed]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Houck, S.A.; Ren, H.Y.; Madden, V.J.; Bonner, J.N.; Conlin, M.P.; Janovick, J.A.; Conn, P.M.; Cyr, D.M. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol. Cell 2014, 54, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Thapaliya, A.; Martinez-Lumbreras, S.; Krysztofinska, E.M.; Isaacson, R.L. Structural and Functional Insights into Small, Glutamine-Rich, Tetratricopeptide Repeat Protein Alpha. Front. Mol. Biosci. 2015, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Chartron, J.W.; Clemons, W.M., Jr.; Suloway, C.J. The complex process of GETting tail-anchored membrane proteins to the ER. Curr. Opin. Struct. Biol. 2012, 22, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Denic, V.; Dotsch, V.; Sinning, I. Endoplasmic reticulum targeting and insertion of tail-anchored membrane proteins by the GET pathway. Cold Spring Harb. Perspect. Biol. 2013, 5, a013334. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, P.C.; Walker, D.; Panganiban, A.T. Small glutamine-rich protein/viral protein U-binding protein is a novel cochaperone that affects heat shock protein 70 activity. Cell Stress Chaperones 2002, 7, 258–268. [Google Scholar] [CrossRef]
- Philp, L.K.; Butler, M.S.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Day, T.K. SGTA: A new player in the molecular co-chaperone game. Horm. Cancer 2013, 4, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Chartron, J.W.; VanderVelde, D.G.; Clemons, W.M., Jr. Structures of the Sgt2/SGTA dimerization domain with the Get5/UBL4A UBL domain reveal an interaction that forms a conserved dynamic interface. Cell Rep. 2012, 2, 1620–1632. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, K.; Morimatsu, M.; Kato, Y.; Ishiguro-Oonuma, T.; Udagawa, C.; Rungsuriyawiboon, O.; Azakami, D.; Michishita, M.; Ariyoshi, Y.; Ueki, H.; et al. Tumor suppressor REIC/DKK-3 and co-chaperone SGTA: Their interaction and roles in the androgen sensitivity. Oncotarget 2016, 7, 3283–3296. [Google Scholar] [PubMed]
- Yin, H.; Wang, H.; Zong, H.; Chen, X.; Wang, Y.; Yun, X.; Wu, Y.; Wang, J.; Gu, J. SGT, a Hsp90beta binding partner, is accumulated in the nucleus during cell apoptosis. Biochem. Biophys. Res. Commun. 2006, 343, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.A.; Handley, M.A.; Lee, Y.H.; Talbot, K.J.; Harper, J.W.; Panganiban, A.T. Functional interaction of human immunodeficiency virus type 1 Vpu and Gag with a novel member of the tetratricopeptide repeat protein family. J. Virol. 1998, 72, 5189–5197. [Google Scholar] [PubMed]
- Cziepluch, C.; Kordes, E.; Poirey, R.; Grewenig, A.; Rommelaere, J.; Jauniaux, J.C. Identification of a novel cellular TPR-containing protein, SGT, that interacts with the nonstructural protein NS1 of parvovirus H-1. J. Virol. 1998, 72, 4149–4156. [Google Scholar] [PubMed]
- Fielding, B.C.; Gunalan, V.; Tan, T.H.; Chou, C.F.; Shen, S.; Khan, S.; Lim, S.G.; Hong, W.; Tan, Y.J. Severe acute respiratory syndrome coronavirus protein 7a interacts with hSGT. Biochem. Biophys. Res. Commun. 2006, 343, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Liou, S.T.; Wang, C. Small glutamine-rich tetratricopeptide repeat-containing protein is composed of three structural units with distinct functions. Arch. Biochem. Biophys. 2005, 435, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Taguwa, S.; Maringer, K.; Li, X.; Bernal-Rubio, D.; Rauch, J.N.; Gestwicki, J.E.; Andino, R.; Fernandez-Sesma, A.; Frydman, J. Defining Hsp70 Subnetworks in Dengue Virus Replication Reveals Key Vulnerability in Flavivirus Infection. Cell 2015, 163, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Limjindaporn, T.; Wongwiwat, W.; Noisakran, S.; Srisawat, C.; Netsawang, J.; Puttikhunt, C.; Kasinrerk, W.; Avirutnan, P.; Thiemmeca, S.; Sriburi, R.; et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem. Biophys. Res. Commun. 2009, 379, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Suzuki, R.; Watanabe, N.; Masaki, T.; Tomonaga, M.; Muhammad, A.; Kato, T.; Matsuura, Y.; Watanabe, H.; Wakita, T.; et al. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 2011, 286, 37264–37273. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. MBio 2014, 5, e01777–e01714. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.K.; Chapman, J.A.; Deymier, M.J.; Bronnimann, M.P.; Ozbun, M.A. Opposing effects of bacitracin on human papillomavirus type 16 infection: Enhancement of binding and entry and inhibition of endosomal penetration. J. Virol. 2012, 86, 4169–4181. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupzyk, A.; Tsai, B. How Polyomaviruses Exploit the ERAD Machinery to Cause Infection. Viruses 2016, 8, 242. https://doi.org/10.3390/v8090242
Dupzyk A, Tsai B. How Polyomaviruses Exploit the ERAD Machinery to Cause Infection. Viruses. 2016; 8(9):242. https://doi.org/10.3390/v8090242
Chicago/Turabian StyleDupzyk, Allison, and Billy Tsai. 2016. "How Polyomaviruses Exploit the ERAD Machinery to Cause Infection" Viruses 8, no. 9: 242. https://doi.org/10.3390/v8090242
APA StyleDupzyk, A., & Tsai, B. (2016). How Polyomaviruses Exploit the ERAD Machinery to Cause Infection. Viruses, 8(9), 242. https://doi.org/10.3390/v8090242