
Experimental Approaches to Study Genome Packaging of Influenza A Viruses

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mapping of cis-Acting Packaging Signals on Individual vRNAs
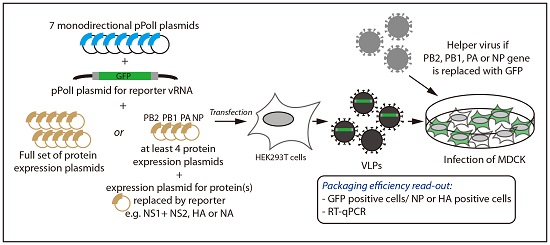
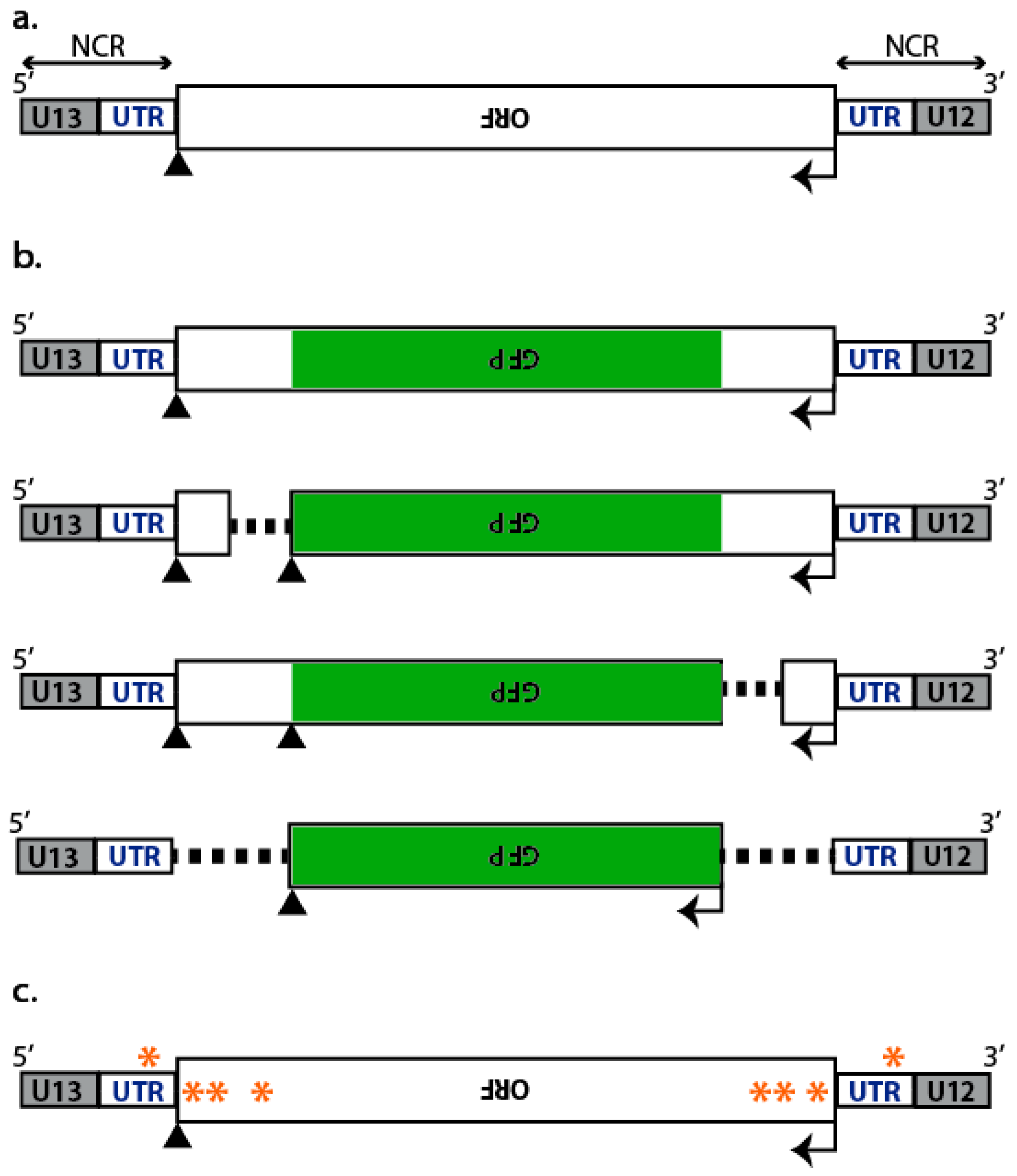
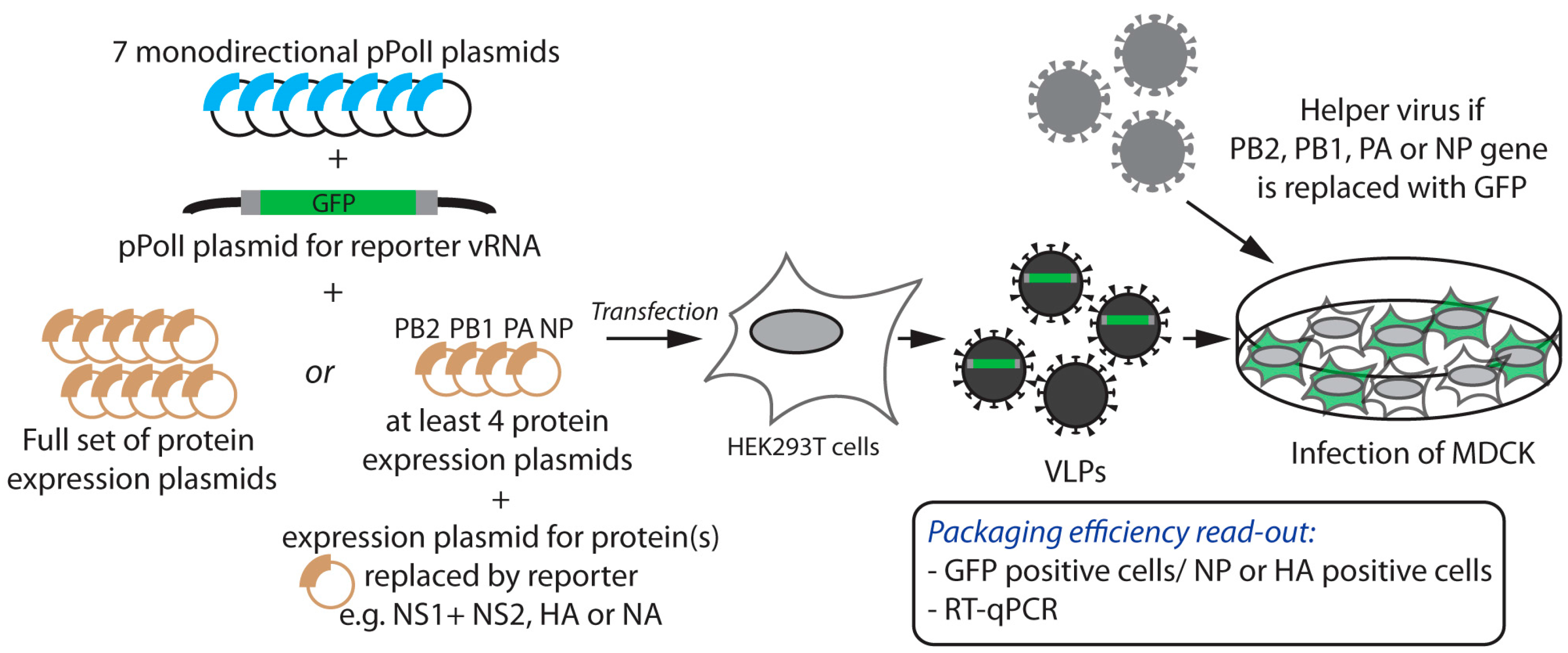
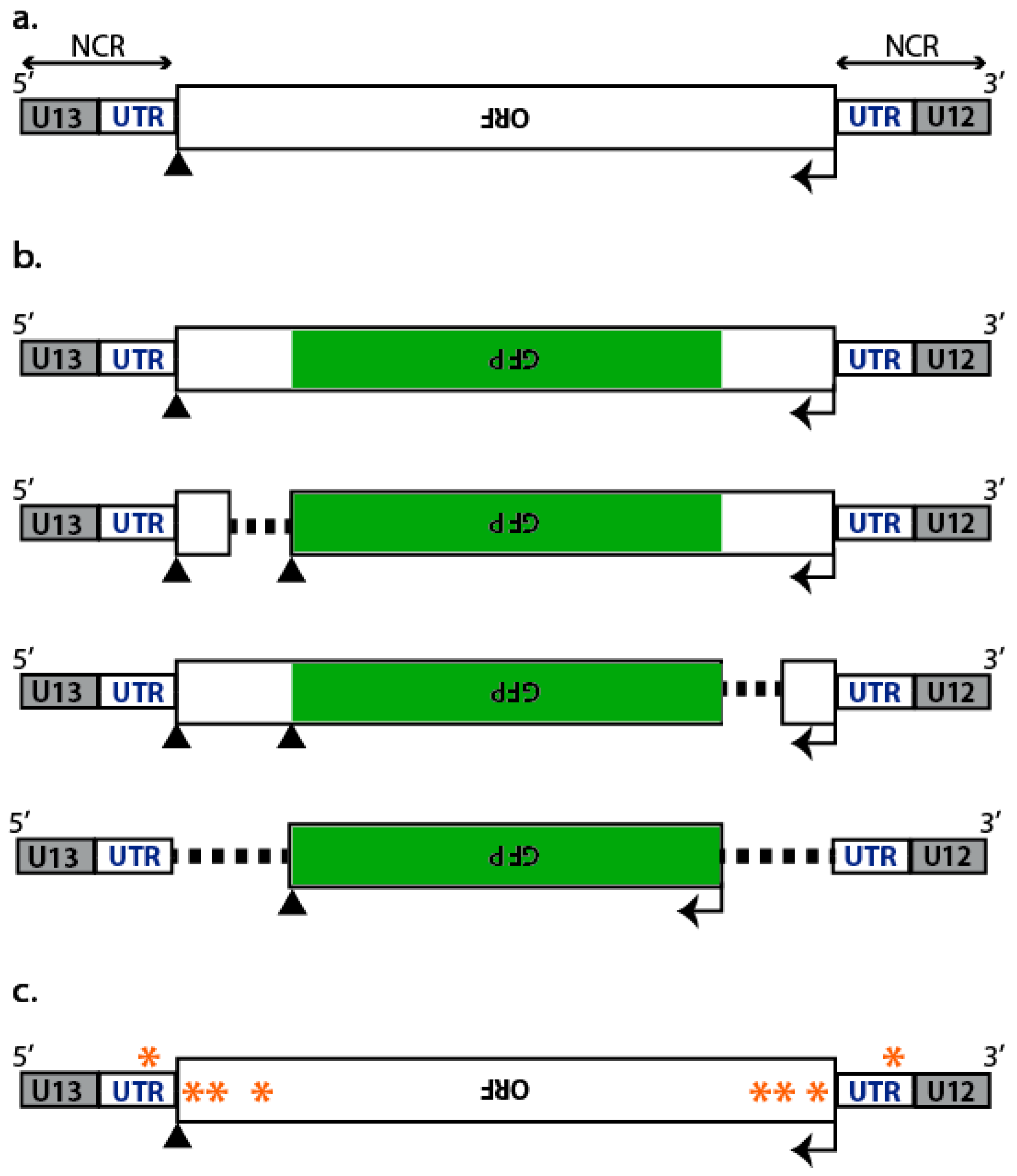
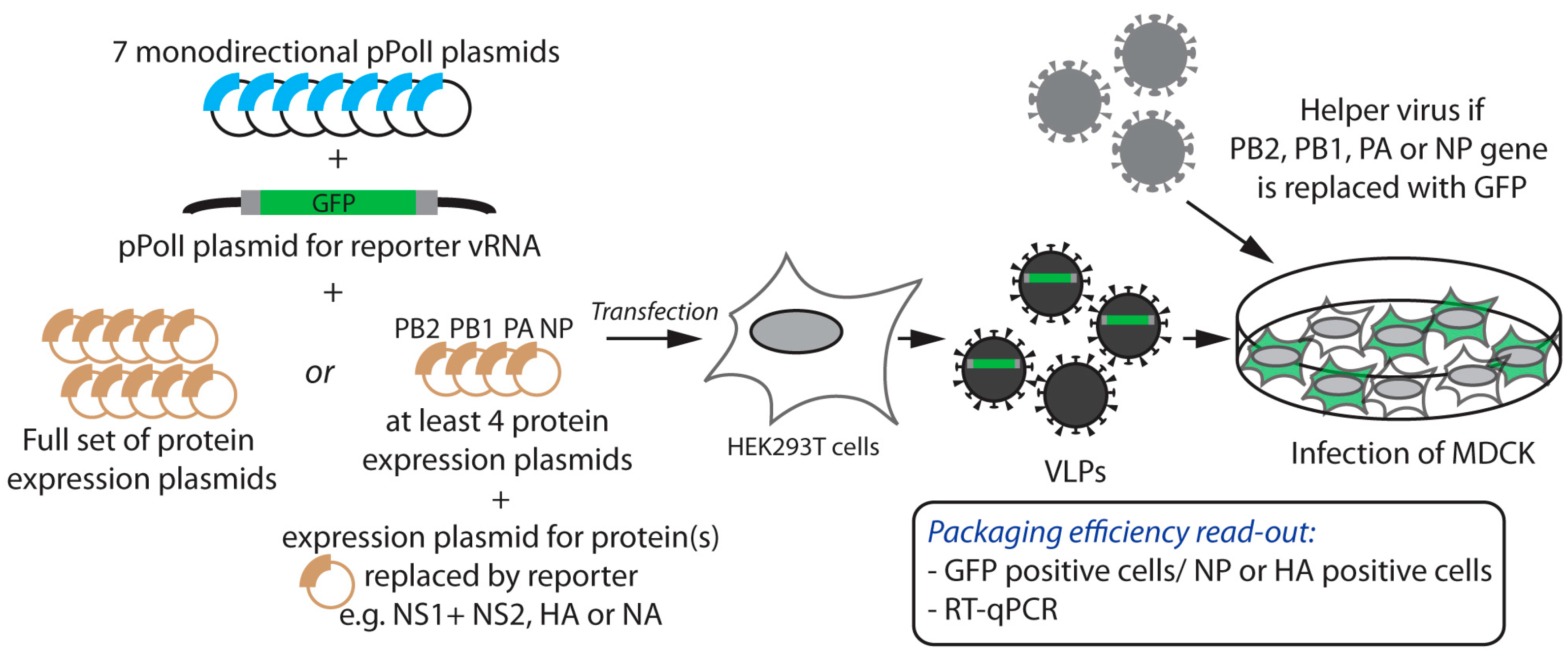
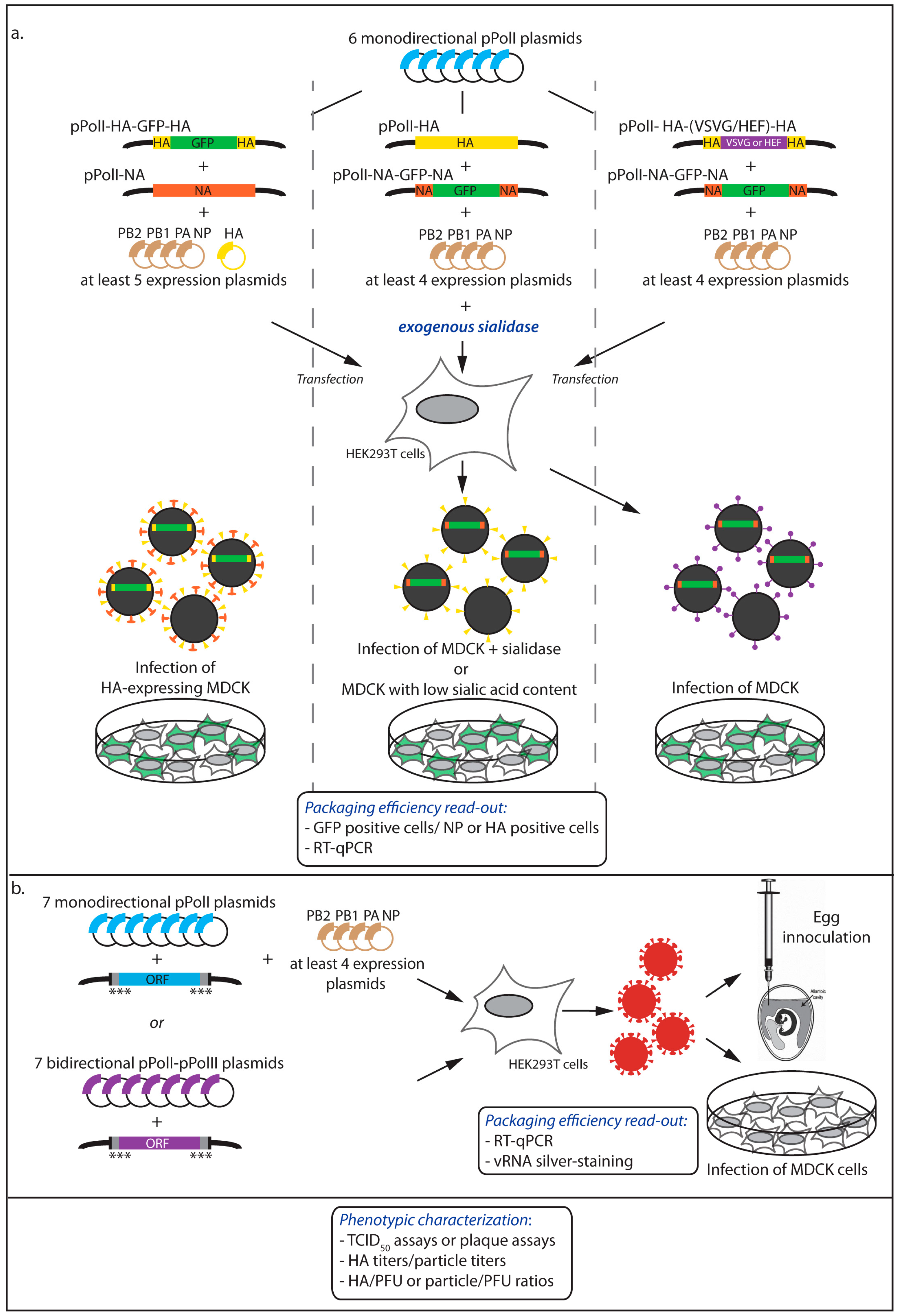
2.1. Incorporation of Reporter vRNAs into VLPs
2.2. Incorporation of Engineered vRNAs into Replication-Competent Viruses
2.2.1. Phenotypic Characterization of the Viruses
2.2.2. Quantitative Measurement of the Viral RNA Content
2.3. Relevant Experimental Controls
3. Unraveling the Mechanism for Co-Packaging of Eight Distinct vRNAs
3.1. Co-Packaging Assays
3.1.1. The Rewiring Approach
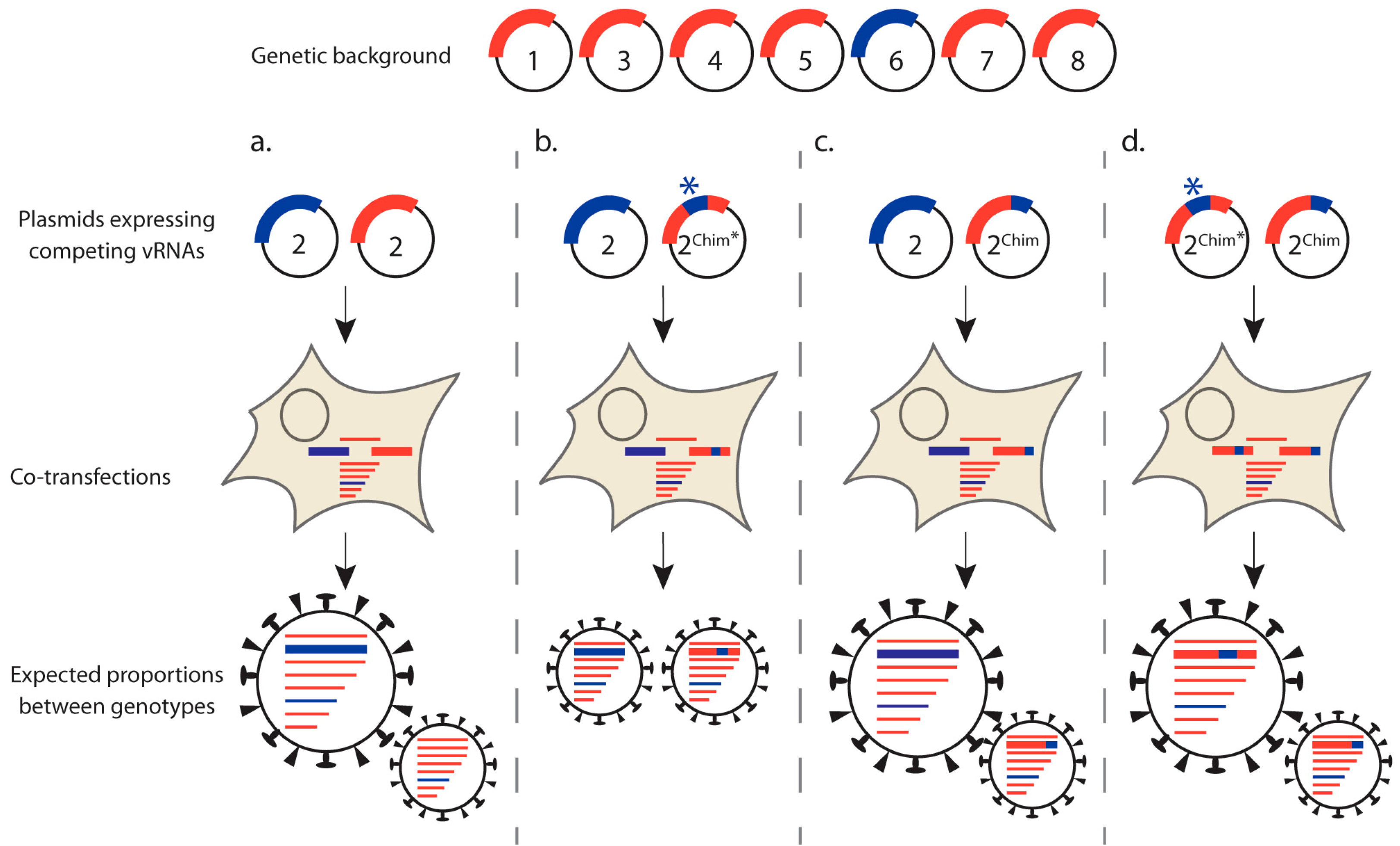
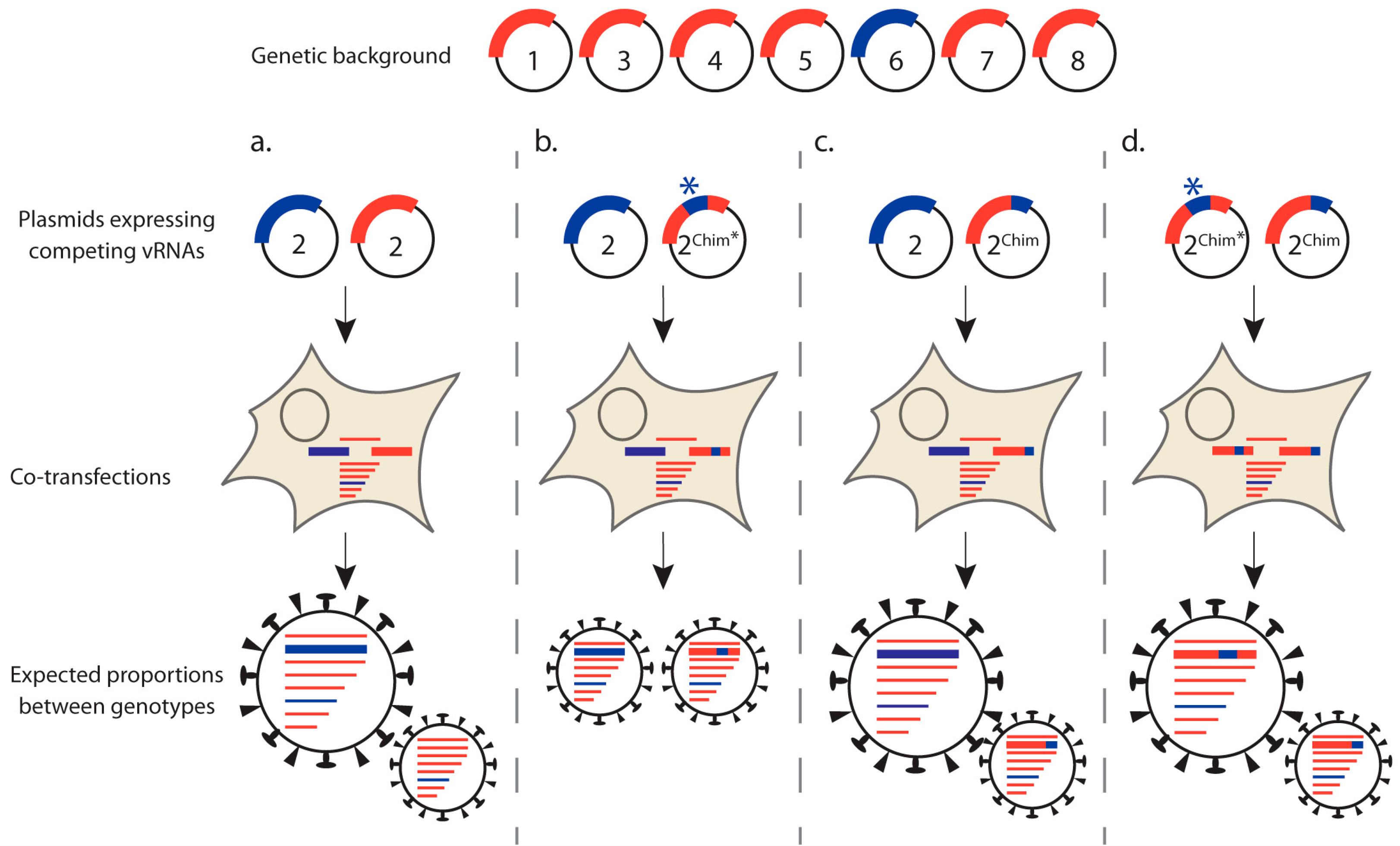
3.1.2. Competitive Reverse Genetics
3.2. Visualization of vRNP Transport and Bundling
3.2.1. Single-Molecule Fluorescence In Situ Hybridization (smFISH)
3.2.2. Electron Microscopy and Tomography
3.3. In Vitro vRNA-vRNA Interactions Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tomescu, A.I.; Robb, N.C.; Hengrung, N.; Fodor, E.; Kapanidis, A.N. Single-molecule FRET reveals a corkscrew RNA structure for the polymerase-bound influenza virus promoter. Proc. Natl. Acad. Sci. USA 2014, 111, E3335–E3342. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Giese, S.; Bolte, H.; Schwemmle, M. The feat of packaging eight unique genome segments. Viruses 2016, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Laver, W.G.; Downie, J.C. Influenza virus recombination. I. Matrix protein markers and segregation during mixed infections. Virology 1976, 70, 105–117. [Google Scholar] [CrossRef]
- Nakajima, K.; Sugiura, A. Three-factor cross of influenza virus. Virology 1977, 81, 486–489. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.G. Many ways to make an influenza virus—Review of influenza virus reverse genetics methods. Influenza Other Respir. Viruses 2013, 7, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Devenish, L.; Engelhardt, O.G.; Palese, P.; Brownlee, G.G.; Garcia-Sastre, A. Rescue of influenza A virus from recombinant DNA. J. Virol. 1999, 73, 9679–9682. [Google Scholar] [PubMed]
- Neumann, G.; Watanabe, T.; Ito, H.; Watanabe, S.; Goto, H.; Gao, P.; Hughes, M.; Perez, D.R.; Donis, R.; Hoffmann, E.; et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 1999, 96, 9345–9350. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Neumann, G.; Kawaoka, Y.; Hobom, G.; Webster, R.G. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 2000, 97, 6108–6113. [Google Scholar] [CrossRef] [PubMed]
- Gultyaev, A.P.; Fouchier, R.A.; Olsthoorn, R.C. Influenza virus RNA structure: Unique and common features. Int. Rev. Immunol. 2010, 29, 533–556. [Google Scholar] [CrossRef] [PubMed]
- Gultyaev, A.P.; Tsyganov-Bodounov, A.; Spronken, M.I.; van der Kooij, S.; Fouchier, R.A.; Olsthoorn, R.C. RNA structural constraints in the evolution of the influenza A virus genome NP segment. RNA Biol. 2014, 11, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, E.; Kesy, J.; Ruszkowska, A.; Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Turner, D.H.; Kierzek, R.; Kierzek, E. Self-folding of naked segment 8 genomic RNA of influenza A virus. PLoS ONE 2016, 11, e0148281. [Google Scholar] [CrossRef] [PubMed]
- Baudin, F.; Bach, C.; Cusack, S.; Ruigrok, R.W. Structure of influenza virus RNP. I. Influenza virus nucleoprotein melts secondary structure in panhandle rna and exposes the bases to the solvent. EMBO J. 1994, 13, 3158–3165. [Google Scholar] [PubMed]
- Gog, J.R.; Afonso Edos, S.; Dalton, R.M.; Leclercq, I.; Tiley, L.; Elton, D.; von Kirchbach, J.C.; Naffakh, N.; Escriou, N.; Digard, P. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res. 2007, 35, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; Rabadan, R.; Levine, A.J.; Palese, P. Highly conserved regions of influenza A virus polymerase gene segments are critical for efficient viral RNA packaging. J. Virol. 2008, 82, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Watanabe, S.; Noda, T.; Fujii, Y.; Kawaoka, Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 2003, 77, 10575–10583. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Fujii, Y.; Noda, T.; Muramoto, Y.; Watanabe, T.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005, 79, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Muramoto, Y.; Takada, A.; Fujii, K.; Noda, T.; Iwatsuki-Horimoto, K.; Watanabe, S.; Horimoto, T.; Kida, H.; Kawaoka, Y. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J. Virol. 2006, 80, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Fujii, K.; Muramoto, Y.; Yamada, S.; Yamayoshi, S.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007, 81, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hong, Y.; Parslow, T.G. Cis-acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; Hatami, R.; Palese, P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 2007, 81, 9727–9736. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.T.; McGregor, M.; Suzuki, T.; Suzuki, Y.; Kawaoka, Y. Adaptation of influenza A viruses to cells expressing low levels of sialic acid leads to loss of neuraminidase activity. J. Virol. 2001, 75, 3766–3770. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Brydon, E.W.; Palese, P. A seven-segmented influenza A virus expressing the influenza C virus glycoprotein HEF. J. Virol. 2008, 82, 6419–6426. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Curran, M.D.; Read, E.K.; Gog, J.R.; Digard, P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J. Virol. 2008, 82, 11869–11879. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Wise, H.M.; Kudryavtseva, K.; Curran, M.D.; Digard, P. Characterisation of influenza A viruses with mutations in segment 5 packaging signals. Vaccine 2009, 27, 6270–6275. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Huang, T.; Ly, H.; Parslow, T.G.; Liang, Y. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J. Virol. 2008, 82, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Isel, C.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res. 2012, 40, 2197–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Sugita, Y.; Aoyama, K.; Hirase, A.; Kawakami, E.; Miyazawa, A.; Sagara, H.; Kawaoka, Y. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat. Commun. 2012, 3, 639. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Chou, Y.Y.; Doganay, S.; Vafabakhsh, R.; Ha, T.; Palese, P. The influenza A virus PB2, PA, NP, and M segments play a pivotal role during genome packaging. J. Virol. 2012, 86, 7043–7051. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Muramoto, Y.; Noda, T.; Kawaoka, Y. The genome-packaging signal of the influenza A virus genome comprises a genome incorporation signal and a genome-bundling signal. J. Virol. 2013, 87, 11316–11322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Peng, Y.; Zhou, K.; Cao, M.; Wang, J.; Wang, X.; Jiang, T.; Deng, T. New insights into the nonconserved noncoding region of the subtype-determinant hemagglutinin and neuraminidase segments of influenza A viruses. J. Virol. 2014, 88, 11493–11503. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Wu, Y.; Wawrzusin, P.; Kabat, J.; Broadbent, A.J.; Lamirande, E.W.; Fodor, E.; Altan-Bonnet, N.; Shroff, H.; Subbarao, K. Influenza A virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 2014, 10, e1003971. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hatta, M.; Watanabe, S.; Neumann, G.; Kawaoka, Y. Compatibility among polymerase subunit proteins is a restricting factor in reassortment between equine H7N7 and human H3N2 influenza viruses. J. Virol. 2008, 82, 11880–11888. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Van Hoeven, N.; Chen, L.M.; Maines, T.R.; Cox, N.J.; Katz, J.M.; Donis, R.O. Reassortment between avian H5N1 and human H3N2 influenza viruses in ferrets: A public health risk assessment. J. Virol. 2009, 83, 8131–8140. [Google Scholar] [CrossRef] [PubMed]
- Octaviani, C.P.; Ozawa, M.; Yamada, S.; Goto, H.; Kawaoka, Y. High level of genetic compatibility between swine-origin H1N1 and highly pathogenic avian H5N1 influenza viruses. J. Virol. 2010, 84, 10918–10922. [Google Scholar] [CrossRef] [PubMed]
- Essere, B.; Yver, M.; Gavazzi, C.; Terrier, O.; Isel, C.; Fournier, E.; Giroux, F.; Textoris, J.; Julien, T.; Socratous, C.; et al. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E3840–E3848. [Google Scholar] [CrossRef] [PubMed]
- Cobbin, J.C.; Ong, C.; Verity, E.; Gilbertson, B.P.; Rockman, S.P.; Brown, L.E. Influenza virus PB1 and neuraminidase gene segments can cosegregate during vaccine reassortment driven by interactions in the PB1 coding region. J. Virol. 2014, 88, 8971–8980. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, C.; Isel, C.; Fournier, E.; Moules, V.; Cavalier, A.; Thomas, D.; Lina, B.; Marquet, R. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: Comparison with a human H3N2 virus. Nucleic Acids Res. 2013, 41, 1241–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ince, W.L.; Gueye-Mbaye, A.; Bennink, J.R.; Yewdell, J.W. Reassortment complements spontaneous mutation in influenza A virus NP and M1 genes to accelerate adaptation to a new host. J. Virol. 2013, 87, 4330–4338. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef] [PubMed]
- Zeldovich, K.B.; Liu, P.; Renzette, N.; Foll, M.; Pham, S.T.; Venev, S.V.; Gallagher, G.R.; Bolon, D.N.; Kurt-Jones, E.A.; Jensen, J.D.; et al. Positive selection drives preferred segment combinations during influenza virus reassortment. Mol. Biol. Evol. 2015, 32, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Shcherbik, S.V.; Pearce, N.C.; Levine, M.L.; Klimov, A.I.; Villanueva, J.M.; Bousse, T.L. Rapid strategy for screening by pyrosequencing of influenza virus reassortants—Candidates for live attenuated vaccines. PLoS ONE 2014, 9, e92580. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Vafabakhsh, R.; Doganay, S.; Gao, Q.; Ha, T.; Palese, P. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc. Natl. Acad. Sci. USA 2012, 109, 9101–9106. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Heaton, N.S.; Gao, Q.; Palese, P.; Singer, R.H.; Lionnet, T. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Lubeck, E.; Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 2012, 9, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef] [PubMed]
- Crosetto, N.; Bienko, M.; van Oudenaarden, A. Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 2015, 16, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, C.; Yver, M.; Isel, C.; Smyth, R.P.; Rosa-Calatrava, M.; Lina, B.; Moules, V.; Marquet, R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc. Natl. Acad. Sci. USA 2013, 110, 16604–16609. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Sagara, H.; Noda, T.; Kawaoka, Y. Configuration of viral ribonucleoprotein complexes within the influenza A virion. J. Virol. 2013, 87, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- Arranz, R.; Coloma, R.; Chichon, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortin, J.; Martin-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Kirchdoerfer, R.N.; Potter, C.S.; Carragher, B.; Wilson, I.A. Organization of the influenza virus replication machinery. Science 2012, 338, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Juozapaitis, M.; Aguiar Moreira, E.; Mena, I.; Giese, S.; Riegger, D.; Pohlmann, A.; Hoper, D.; Zimmer, G.; Beer, M.; Garcia-Sastre, A.; et al. An infectious bat-derived chimeric influenza virus harbouring the entry machinery of an influenza A virus. Nat. Commun. 2014, 5, 4448. [Google Scholar] [CrossRef] [PubMed]
- Sherry, L.; Punovuori, K.; Wallace, L.E.; Prangley, E.; DeFries, S.; Jackson, D. Identification of cis-acting packaging signals in the coding regions of the influenza B virus HA gene segment. J. Gen. Virol. 2016, 97, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, T., Jr.; Sung, P.Y.; Roy, P. Disruption of specific RNA-RNA interactions in a double-stranded RNA virus inhibits genome packaging and virus infectivity. PLoS Pathog. 2015, 11, e1005321. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; McCrae, M.A.; Boyce, P.; Kim, J.T. Inter-segment complementarity in orbiviruses: A driver for co-ordinated genome packaging in the Reoviridae? J. Gen. Virol. 2016, 97, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Elderfield, R.A.; Hartgroves, L.C.S.; Barclay, W.S. Using reverse genetics to improve influenza vaccines. In Reverse Genetics of RNA Viruses; Bridgen, A., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 224–249. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isel, C.; Munier, S.; Naffakh, N. Experimental Approaches to Study Genome Packaging of Influenza A Viruses. Viruses 2016, 8, 218. https://doi.org/10.3390/v8080218
Isel C, Munier S, Naffakh N. Experimental Approaches to Study Genome Packaging of Influenza A Viruses. Viruses. 2016; 8(8):218. https://doi.org/10.3390/v8080218
Chicago/Turabian StyleIsel, Catherine, Sandie Munier, and Nadia Naffakh. 2016. "Experimental Approaches to Study Genome Packaging of Influenza A Viruses" Viruses 8, no. 8: 218. https://doi.org/10.3390/v8080218
APA StyleIsel, C., Munier, S., & Naffakh, N. (2016). Experimental Approaches to Study Genome Packaging of Influenza A Viruses. Viruses, 8(8), 218. https://doi.org/10.3390/v8080218