1. Introduction
A lambda (λ) prophage is maintained within the chromosome of
Escherichia coli (
E. coli) by an action of the CI repressor encoded by gene
cI. The inhibition of replication initiation from the prophage
oriλ site positioned midway within gene
O [
1,
2,
3] (
Figure 1A) is blocked by CI protein binding to operator sites overlapping promoters directing transcription leftward from
pL, e.g., copying genes
N-int, or rightward from
pR for expressing genes
cro-cII-O-P-Q. The product of gene
P (gp
P, P) participates in loading the
E. coli DnaB helicase [
4] onto DNA during formation of a DnaB:P:O:
oriλ preprimosomal complex [
5,
6,
7], each factor being required for replication initiation from
oriλ. DnaB unwinds double-stranded (ds) DNA at the replication fork, by encircling and translocating along the 5′ lagging strand using energy provided by ATP hydrolysis [
4]. The interaction between DnaG primase and the N-terminal end of DnaB [
8] increases both the NTPase and helicase activities of DnaB and the synthesis of RNA primers by DnaG [
4,
8,
9,
10,
11,
12,
13,
14,
15]. DnaB can promote the progression of Holliday junctions, believed important in the repair of DNA damage occurring near advancing replication forks [
16,
17]. DnaB functions as a hexamer, with about 20 hexamers per cell [
18,
19], with each binding up to six ATP [
20,
21]. It forms a complex with the host replication initiation protein DnaC to which the majority of DnaB in a cell is bound [
22]. This interferes with the intrinsic single-stranded (ss) DNA binding activity of DnaB [
20,
22,
23,
24,
25,
26]. The DnaC-DnaB complex acquires cryptic ssDNA binding activity specific to the bacterial origin of replication,
oriC, where it begins unwinding dsDNA after a number of complex changes [
27,
28,
29,
30,
31,
32,
33,
34].
The λ P protein has evolved to compete for and dissociate DnaC-DnaB complexes [
5,
38] and two to six P monomers can bind to every DnaB hexamer [
5,
39,
40], forming an enzymatically inactive “dead-end” complex [
22]. High levels of P were found deleterious to host cells [
41,
42] and it was suggested that P sequestered,
i.e., bound-up, DnaB and thus interfered with the initiation of host DNA synthesis. ColE1 replication establishment or propagation [
43] is extremely sensitive to P [
35]. The effect is suppressed by alleles of
dnaB or
P, which suggests that P can impact a cellular replication event by acting outside of the DnaA and
oriC-dependent replication initiation step. This same study showed that P-dependent cellular filamentation arose in cells defective for SOS induction, suggesting that P was influencing replication propagation or restart mechanisms. For example, the sequestration of DnaB by P could perturb or impede origin-independent stable DNA replication [
44,
45,
46,
47,
48], where both cellular replication restart and ColE1 replication depend upon PriA helicase activity [
49,
50,
51,
52] at R-loops.
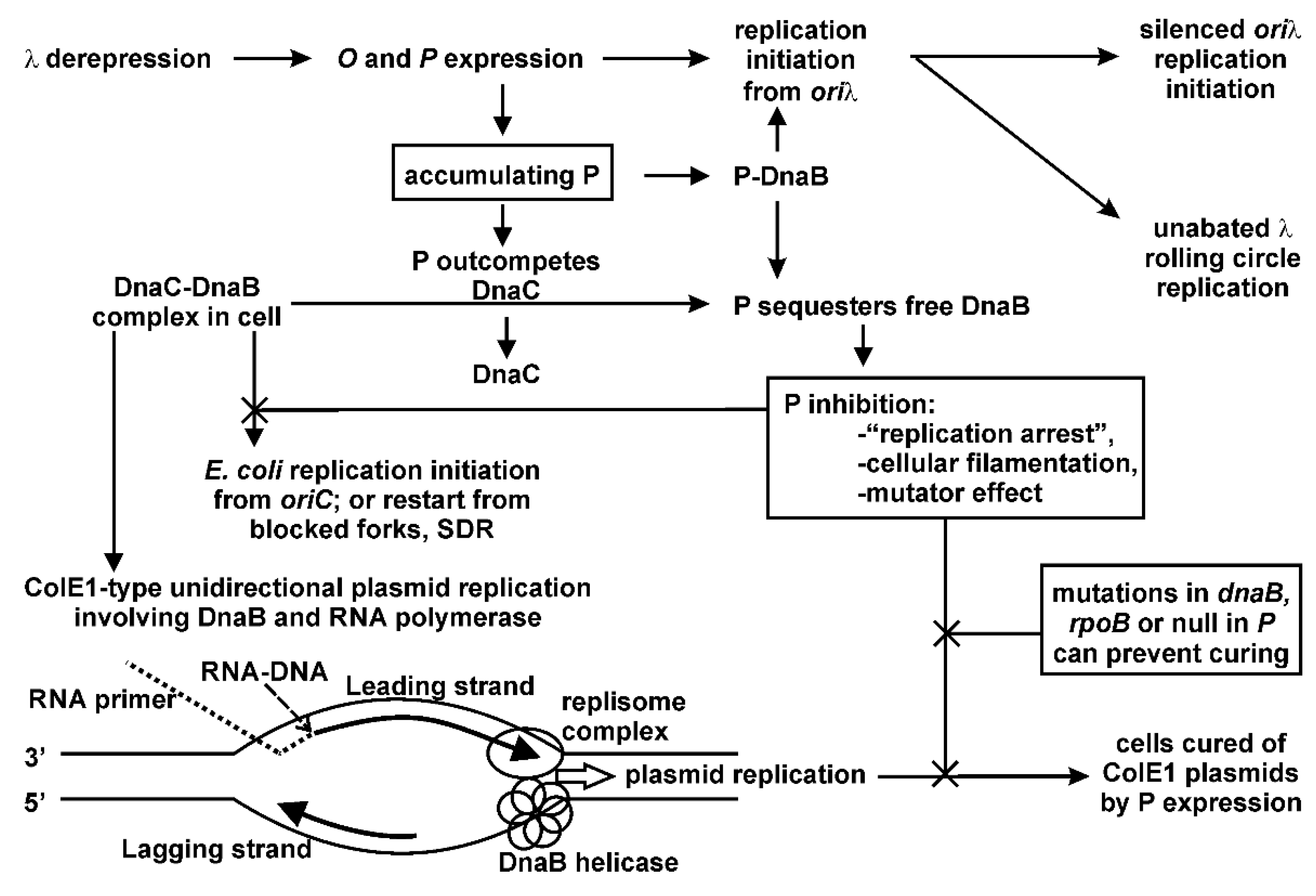
Multiple possibilities present for the P-lethality/inhibition observations, and are summarized in
Figure 2. The 233-amino acid P protein is very stable, with a half-life of an hour [
53,
54]. In addition to binding Oλ [
55], P interacts with the host proteins DnaA [
56,
57,
58], DnaB [
59,
60,
61], GrpE [
62], DnaJ and DnaK [
63], and possibly RNA polymerase (RNAP) [
64]. As is understood from
in vitro studies, the Hsp70 chaperone complex removes P from DnaB when the P-DnaB complex is bound to
oriλ:O [
40] in a two-step reaction involving DnaJ binding to complexed P-DnaB, which enhances P-DnaK binding, ATP hydrolysis, and the release of a P-DnaK-ADP complex. There is a requirement for a high concentration of DnaK unless GrpE is present, and a proposal that DnaK changes the conformation of P from a native to a folded state that is no longer able to bind DnaB [
65]. An existing hypothesis for P-lethality/inhibition is that by binding up DnaB (or DnaA), P will inhibit chromosomal replication initiation from
oriC. Greater understanding of the extent of intracellular binding between P and DnaB, which occurs beyond that involved in the formation of an
oriλ preprimosomal complex is required to help explain DnaB sequestration. We showed that: (a) very low levels of P are necessary for the limited
oriλ initiation required for λ replication [
35]; and (b) that λ replication resulting in high phage burst from an induced λ
cI[Ts]857
Sam7 prophage blocked for cell lysis occurred in cells that were simultaneously fully derepressed for
P expression from pcIpR-
P-timm—conditions where DnaB would be sequestered.
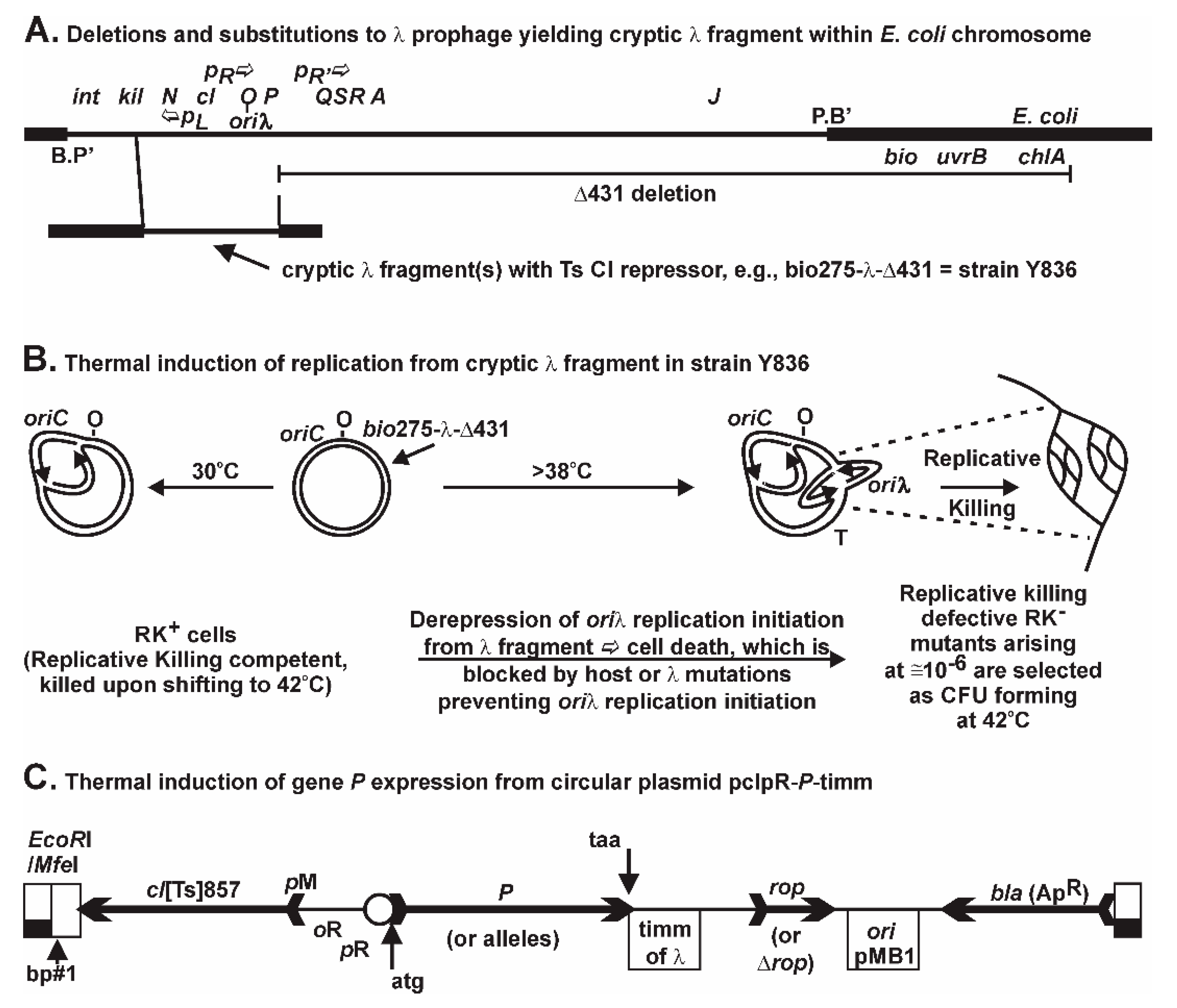
The initiation of
oriλ replication from a chromosomally integrated, but non-excisable λ gene fragment (
Figure 1A) is repressed by the
cI [Ts] repressor. When CI[Ts] is inactivated upon shifting cells growing at 30 °C to 42 °C,
O-P are transcribed, replication initiation arises at
oriλ, and the Replicative Killing, RK
+, phenotype is triggered, resulting in massive cell death [
66]. Rare mutations nullifying the RK
+ phenotype permit the selection for colony forming units (CFU) at 42 °C, each representing RK
− mutants in host or prophage genes (not merely in
P) that block some aspect of λ replication initiation [
66,
67,
68,
69]. For example, we characterized within RK
− mutants small deletions in
O that shift the reading frame and yield close-by stop codons polar for downstream gene expression. These RK
− mutants form CFU at 42 °C, likely surviving by limiting downstream
P expression. Thus, it appears these induced mutants can tolerate some level of λ
P expression (or that of any other toxic λ fragment protein, such as the unstable
cII gene product). Since P expression had been shown to be toxic, confusion existed relative to its contribution to the RK
+ phenotype. We suggested the terms
cis Replicative Killing and
trans P-lethality to distinguish mechanistically these two ideas. We found that the RK
+ phenotype was uniquely dependent upon multiple, non-repairable λ replication forks arising from
oriλ within a trapped prophage fragment, resulting in very rapid, nonreversible cell death, whereas there are likely multiple possibilities for P-lethality, which can be reversed even after hours of
P expression [
35]. In short, limited levels of P can be metabolized. The
dnaBgrpD55 allele [
70], which encodes missense mutations V256I and E426K [
35] was shown to fully prevent
oriλ replication initiation and to suppress cell killing by 10
6-fold from the lambda prophage fragment in strain Y836 (
Figure 2C in reference [
71]) when the cells were shifted from 30 °C to 42 °C, even though
P expression remained constitutive from the induced defective prophage. This allele was shown Ts for λ replication but not for
E. coli cell growth [
70]. When removing the complication of the defective prophage and simply expressing
P from a plasmid, we found that the same allele of
dnaB suppressed the various P-lethality phenotypic manifestations [
35]. This suggested that the P-lethality phenotype was dependent upon a P-DnaB interaction, possibly DnaB sequestration, and required further study.
In order to test whether P-DnaB interactions influence cellular replication events around the chromosome, rather than just at oriC, we explored a hypothesis that P buildup within a cell can perturb host replication fidelity. Since blocks to replication restart can arise around the chromosome, we first surveyed for an increase in the appearance of auxotrophic mutations of any type, and then for a targeted increase in rifampicin resistant (RifR) mutations arising in rpoB. The assumption made was that perturbing replication restart can be error prone. Herein, we observed a λ-dependent mutator effect that was linked to P. We observed that P expression creates the potential for sequestration of DnaB and results in a dramatic increase in cellular mutagenesis, which is nullified by inactivated or altered alleles of P and by an allele of dnaB.
3. Discussion
The initiation of replication from a trapped λ fragment (
Figure 1B) results in backward, or wrong-orientation replication forks that can be problematic for cell viability [
44,
48,
80,
81]. The leftward fork from
oriλ can undergo head-on collisions with the rightward replication fork arising from
oriC, or with rightward-directed transcription arising, e.g., from five of the seven
rrn operons:
rrnC,
A,
B,
E, and
H, positioned between
oriC and
oriλ. Such head-on collisions are likely responsible for the powerful RK
+ phenotype. We previously compared the kinetics of cell death resulting from
P expression from pcIpR-
P-timm or Replicative Killing by initiation from a trapped
oriλ [
35]. The
cis-RK
+ phenotype became irreversible by 20 min while
trans-P lethality/inhibition was slower and reversible for several hours. The RK
+ phenotype (
Figure 1B) seems uniquely dependent upon unresolved collisions between replication arising from
oriλ and the rightward fork from
oriC or with cellular transcripts. Of course, when the trapped λ fragment is induced, both
cis-RK
+ and
trans-P-lethality/inhibition phenotypes are jointly produced. Normal λ induction would likely escape the
cis-RK
+ phenotype because the prophage excises from the chromosome, which is not possible for the defective prophage. The combined lethal effects may amplify the selective pressure for survivors and be responsible for the 20-fold increase in mutants arising compared to situations where only
P is expressed. This extreme selective pressure may account for the very high frequency of co-selected auxotrophs among the selected RK
− mutants, especially for the RK
− CFU acquiring a mutation conferring Ts auxotrophy. For example, any co-selected Ts auxotrophic mutation arising in an amino acid synthesis or utilization pathway would inhibit protein synthesis at the limiting temperature and reduce λ fragment gene expression, in turn lowering the buildup of P, and help such RK
− mutants be selected for colony formation during 42 °C selection period. As previously noted, RK
− mutants arising on RM at 42 °C are defective for
oriλ replication and can form a CFU at 42 °C. Any host mutation that facilitates colony formation during cell growth at 42 °C could be co-selected. While never previously explored in the literature, co-selected Ts auxotrophs might be particularly supportive in situations where the RK
− mutation does not inactivate
P, for example mutations nullifying
O activity. However, none of the original characterized
O-defective
P+ RK
− starting strains employed herein had acquired an auxotrophic mutation when originally isolated (and all were then maintained at non-inducing temperatures to prevent further selective pressure). Nevertheless, a high proportion of auxotrophs appeared from these strains when re-isolated CFU were obtained from 42 °C plates when
P+ was expressed. Given the subsequent results suggesting that P conferred a mutator effect, we explain the high proportion of auxotrophs (refer to
Table 3 section “Induced defective λ lysogens”) as representing a combination of P mutagenesis combined with a powerful unrealized selection for cellular mutations that limited
P expression toxicity during cell growth at 42 °C. It is also entirely possible that some of the Rif
R rpoB CFU selected at 37 °C (cells with plasmid) or 42 °C served to reduce cellular toxicity to
P expression. Further investigation is required to explore potential manifestations of P toxicity on alternative mutation selection; however, this may simply be an inherent limitation in all studies where a mutagenic agent is also toxic, and where toxicity can influence selection. One possibility is that the selected mutants are resistant to the toxic agent. We explored this difficulty in FA#2 by accounting for both toxicity and recovered Rif
R mutants for each selection temperature. We could demonstrate that many Rif
R mutants were sensitive to P-toxicity, and, for selections at 36 °C, which were less toxic than those at 37 °C, there was a significant increase in the appearance of Rif
R mutant CFU (see below). A separate manuscript is in preparation on the complementation and examined toxicity of cloned
vs. defective prophage (single copy) expressed λ
imm-
rep gene products.
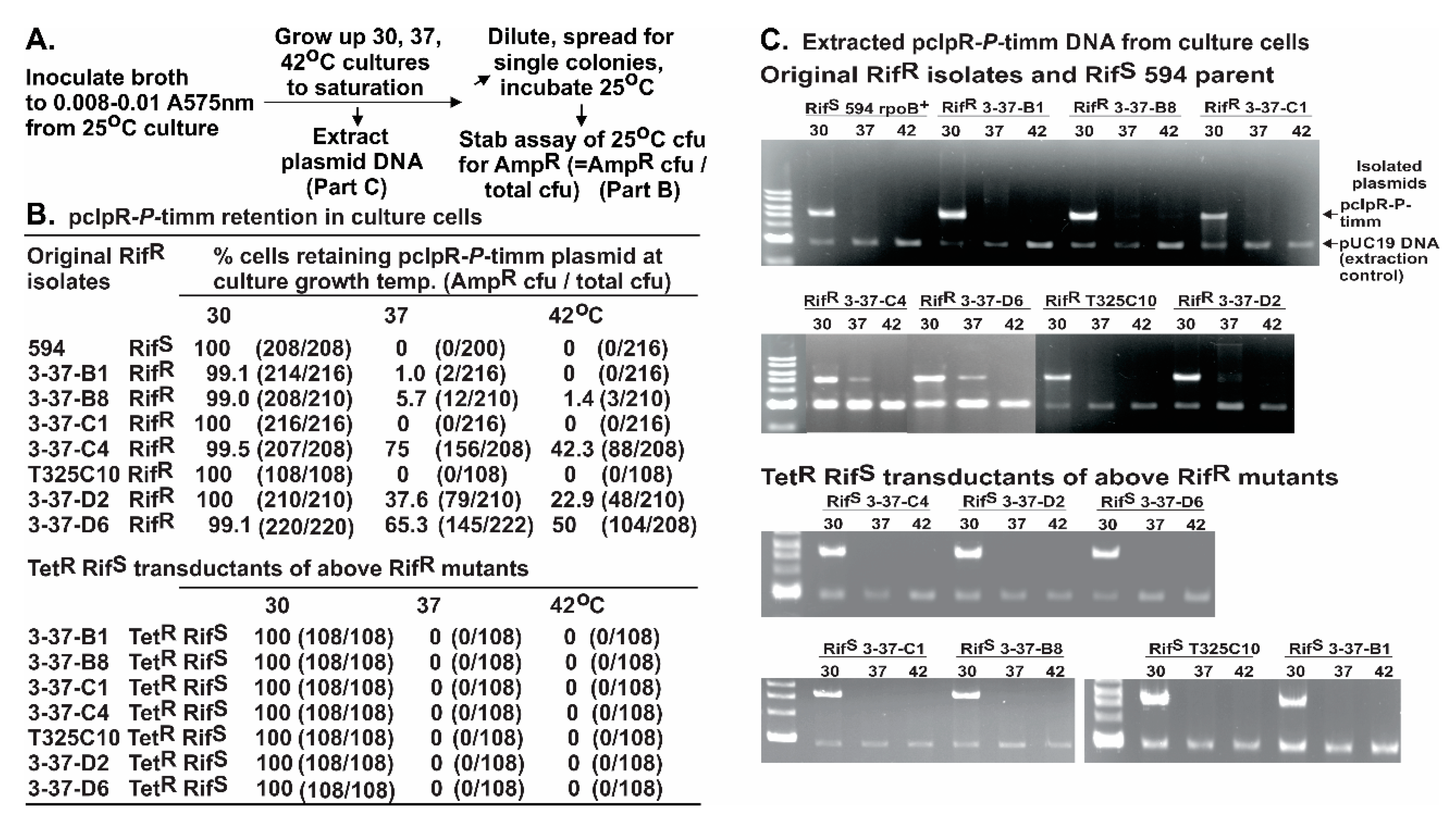
Fluctuation assays were used to determine if the RifR mutations arising in rpoB preexisted the expression of P. As noted, this was complicated by observations that 80-fold cellular toxicity occurred in cells with pcIpR-P-timm incubated at 37 °C, and that most of the RifR CFU were PS. On lowering the incubation temperature to 36 °C, P-lethality was reduced by half but the recovered RifR mutants increased by nine-fold compared to 30 °C. With incubation lowered yet another degree to 35 °C, the toxicity of P was minimal (1.3-fold), ColE1 curing of cells was slight (5%), yet the RifR mutants recovered were 3.9-fold higher than at 30 °C. These observations suggest that the increase in RifR mutants was dependent upon P expression, rather than selection conditions favoring preexisting PR RifR mutants.
Seven of the Rif
R mutants acquired a P
R phenotype. Two of these mutations, G537C and L571Q map close to the Rif binding pocket in RpoB [
79], but the remaining mutations Q148L, Q148P, ∆GEV440-442V, R451S, and S509R fell outside of the pocket. These mutations may help define a contact point(s) between RpoB and P that has long been suggested [
64]. Our P
R mutation Q148P in
rpoB is identical to rpo*148 [
82]. A small collection of rpo* mutations, which possess some level of Rif
R [
82], were isolated based on the discovery [
83] that mutations in RNAP alleviate the UV-sensitive phenotype of
ruv strains, e.g., rpo*148 suppresses the extreme sensitivity of UV treatment to a ∆
relA ∆
spoT ruv strain. This property was linked to the rpo* mutation destabilizing RNAP open complexes and stalled elongation complexes, thereby reducing the occurrence of stalled RNAP(s) at lesions in the DNA template [
82,
83]. Co-directional collisions between the replisome and RNAP in an arrested (backtracked) elongation complex can lead to DNA double strand breaks [
84]. Transcription pausing and stalling regulators include ppGpp which destabilizes open complexes, and the RNAP modulators DskA, GreA, GreB, and Mfd [
81,
84,
85]. The failure of rpo* RNAP to pause and backtrack, and their ability to reduce the accumulation of RNAP arrays [
81], helps explain why rpo* mutations can suppress the formation of DSB [
84]. Assuming that our P
R Q148P Rif
R mutation will reduce the accumulation of RNAP arrays, can this explain a P
R phenotype and the suppression of P-dependent ColE1 plasmid curing? Future studies require determining if the other P
R mutants share an rpo* phenotype.
Several unanswered problems arise in relation to the observed mutator effect caused by
P expression. Based on the low λ requirement for P, coupled with P sequestration of the low cellular amount of DnaB [
18,
19], either
P expression from λ requires a high degree of negative regulation to prevent P-lethality/inhibition (probably not fully appreciated), or there is an unknown mechanism for limiting the P
S phenotype. The apparent complete escape of λ replication from P is contrasted with the exquisite sensitivity of ColE1 plasmid replication to P, and its mutagenic effect on
E. coli. One possible explanation is that late λ rolling circle replication may be independent of replication restart. Whereas, the mechanism for initiation of ColE1 [
43] is similar to restart (see [
35]), and likely requires loading DnaB for each initiation event, which breaks down with P-DnaB sequestration. Just how P
R Rif
R mutants suppress ColE1 curing by P, and seemingly overcome P-DnaB sequestration remain open questions.
The NH
2-terminal portion of P was suggested to bind O and its C-terminal domain to interact with host proteins [
86,
87]. The NH
2-terminal domain of DnaC is involved in binding to DnaB. Both P and DnaC share a stretch of amino acids with high homology at their NH
2 termini [
88]. The in-frame ∆76 deletion within codons 9–86 at the NH
2-terminal end of P suppressed: (a) the curing of ColE1 plasmids; (b) P-lethality at 37–39 °C [
35]; and (c) prevented a P-dependent increase in Rif
R mutations or mutagenic effect. These observations suggest that the NH
2 end of P can compete for the DnaC binding site on DnaB, and that P binding at this site can account for P
S phenotype: its cellular toxicity, ColE1 curing, and mutator activity.
The cell construct,
Figure 1A, possessed a mutagenic activity that was linked to the expression of
P from the λ fragment within the chromosome. Independently, cells that included a plasmid where
P was the only inducible gene product exhibited a mutator phenotype when
P was expressed. The mutator activity was assessed by screening for auxotrophic mutations arising during Replicative Killing selection for RK
− clones, or the selection of Rif
R mutations within
rpoB. Since alleles of the host
dnaB replicative helicase, or of
P, can nullify the mutator phenotype, and since P interacts with DnaB to form an enzymatically inactive “dead-end” P-DnaB complex [
22],
i.e., the so-called sequestered DnaB, reducing or eliminating cellular DnaB activity is linked to the mutator phenotype.
Under conditions of DnaB sequestration, any replication fork collapse could be problematic for replication re-initiation and generate gaps in DNA synthesis needing repair. Studies reported with yeast suggested that the stalling of transcription at abasic (AP) sites is highly mutagenic [
89] and mechanisms, such as mutations arising via translesion synthesis through AP sites were proposed. Most of the auxotrophs we characterized acquired temperature sensitive mutations, characteristic of a missense arising from a point mutation. We did not find that blocking SOS induction, deleting
recA, or inactivating individual
E. coli mutator polymerases Pol IV or Pol V eliminated P-dependent mutagenesis. Since three
E. coli DNA polymerases, Pol II, Pol IV and Pol V, are involved in induced mutagenesis [
90], this was not evaluated rigorously as the lack of any one could be substituted for by another. Further work is needed to determine if the sequestration of DnaB by P stimulates the appearance of gaps and AP sites around the chromosome, linking them to the mutagenic state associated with elevated levels of P. This would agree with the observation that P can stimulate SOS-independent filamentation [
35]. We propose a model that P sequestration of DnaB has several cellular outcomes: (a) it prevents the loading of DnaB helicase needed for the initiation of ColE1 replication (
Figure 2) resulting in rapid, complete plasmid curing; (b) it prevents replication restart requiring the reloading of DnaB by DnaC; and (c) when a faster-moving Pol III replisome complex inevitably collides with RNAP, the displaced replisome components along with RNAP from the leading strand are uncoupled from DNA polymerase copying the lagging strand, leading to ssDNA regions that become a target for DNA damage which increase the probability for spontaneous mutation. We speculate that P may exacerbate this situation by gaining access to DnaB remaining bound to the lagging strand, and so interfere with rebinding of the β-clamp and Pol III, resulting in replication stuttering and the formation of leading strand gap(s) as illustrated for the co-directional collisions drawn in
Figure 5e of reference [
91]. We imagine that mutations in RNAP that limit P-interaction, or limit arrested RNAP complex formation on DNA, hence reducing collisions between the replisome and arrested RNAP, will lessen the influence of P-lethality/inhibition.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}