
Phylogenetic and Molecular Variability Studies Reveal a New Genetic Clade of Citrus leprosis virus C

 ,
, 
 ,
, 
Abstract
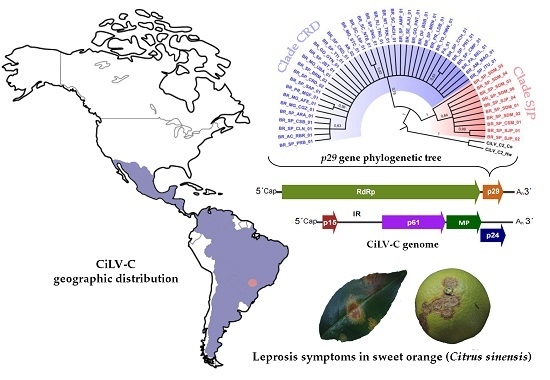
:
1. Introduction
2. Materials and Methods
2.1. Leprosis Surveys, Virus Detection and Isolates
2.2. Sequencing of the RNA2-Intergenic Region and p29, p15 and Partial MP Genes of CiLV-C Isolates
2.3. Phylogenetic Analysis and Estimation of Population Genetic Parameters
2.4. Estimation of Natural Selection Pressure
2.5. Recombination Analyses
2.6. Secondary Structure Prediction of P29 Proteins of Cileviruses
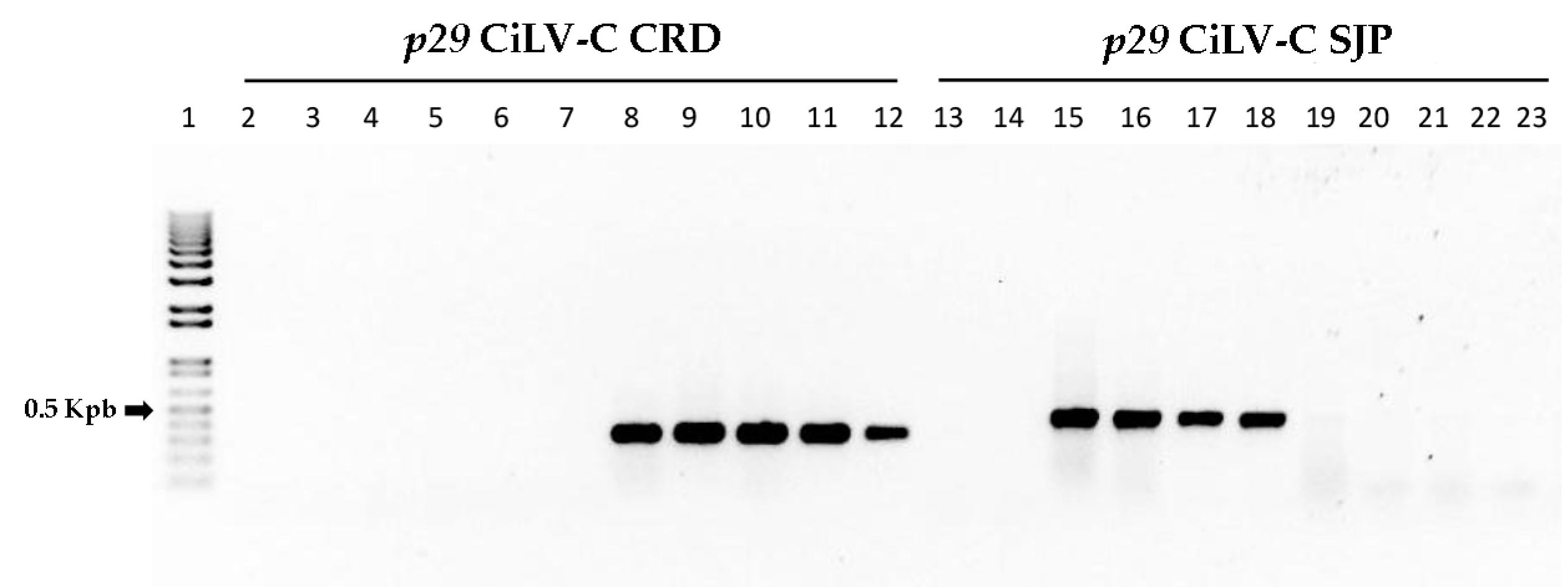
2.7. RT-PCR Test for Differential Detection of CiLV-C Clade-Specific Isolates
2.8. Characterization of the Isolate BR_SP_SJP_01
3. Results
3.1. CiLV-C Is the Prevalent Causal Agent of Citrus Leprosis in Brazil
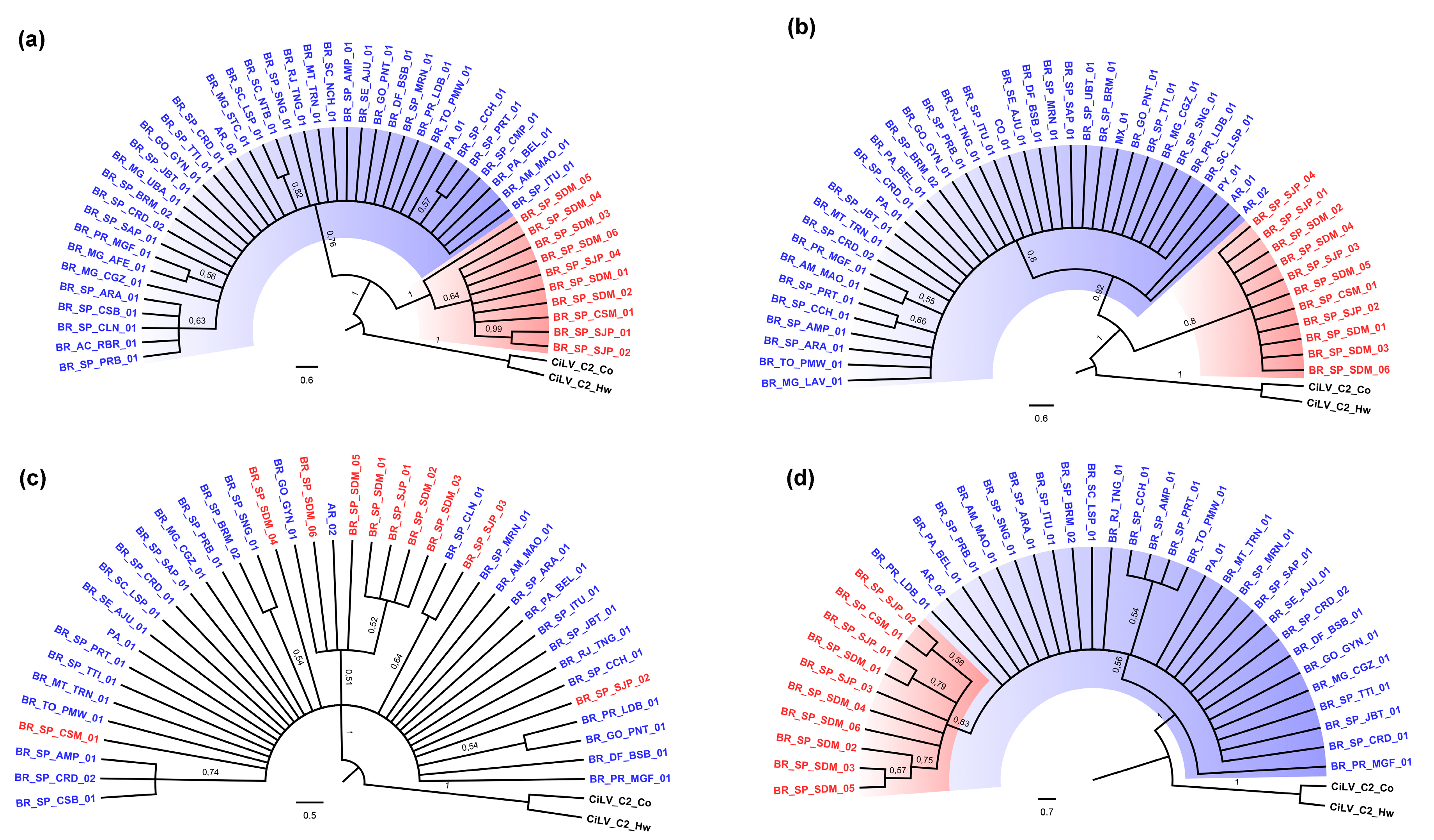
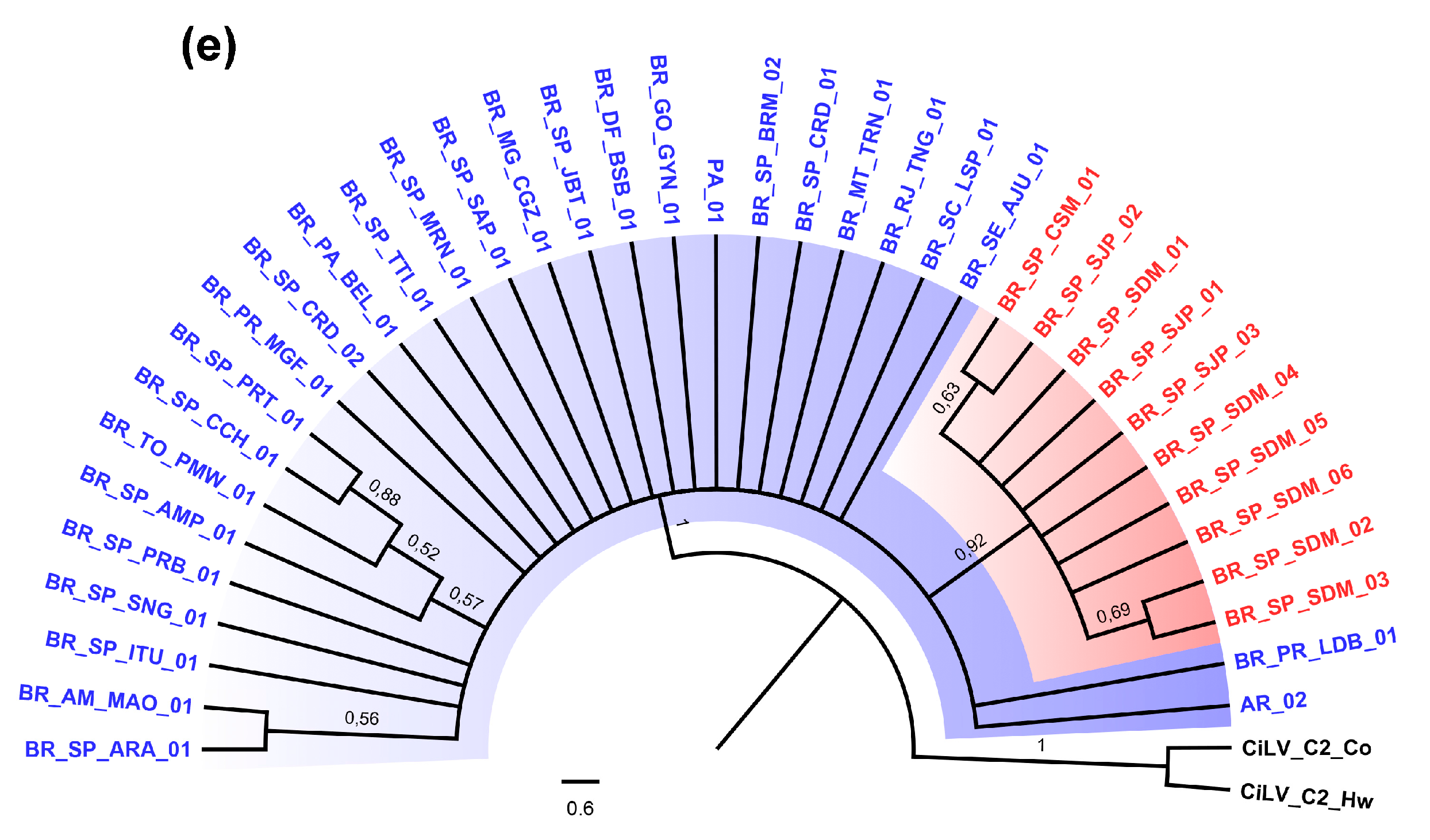
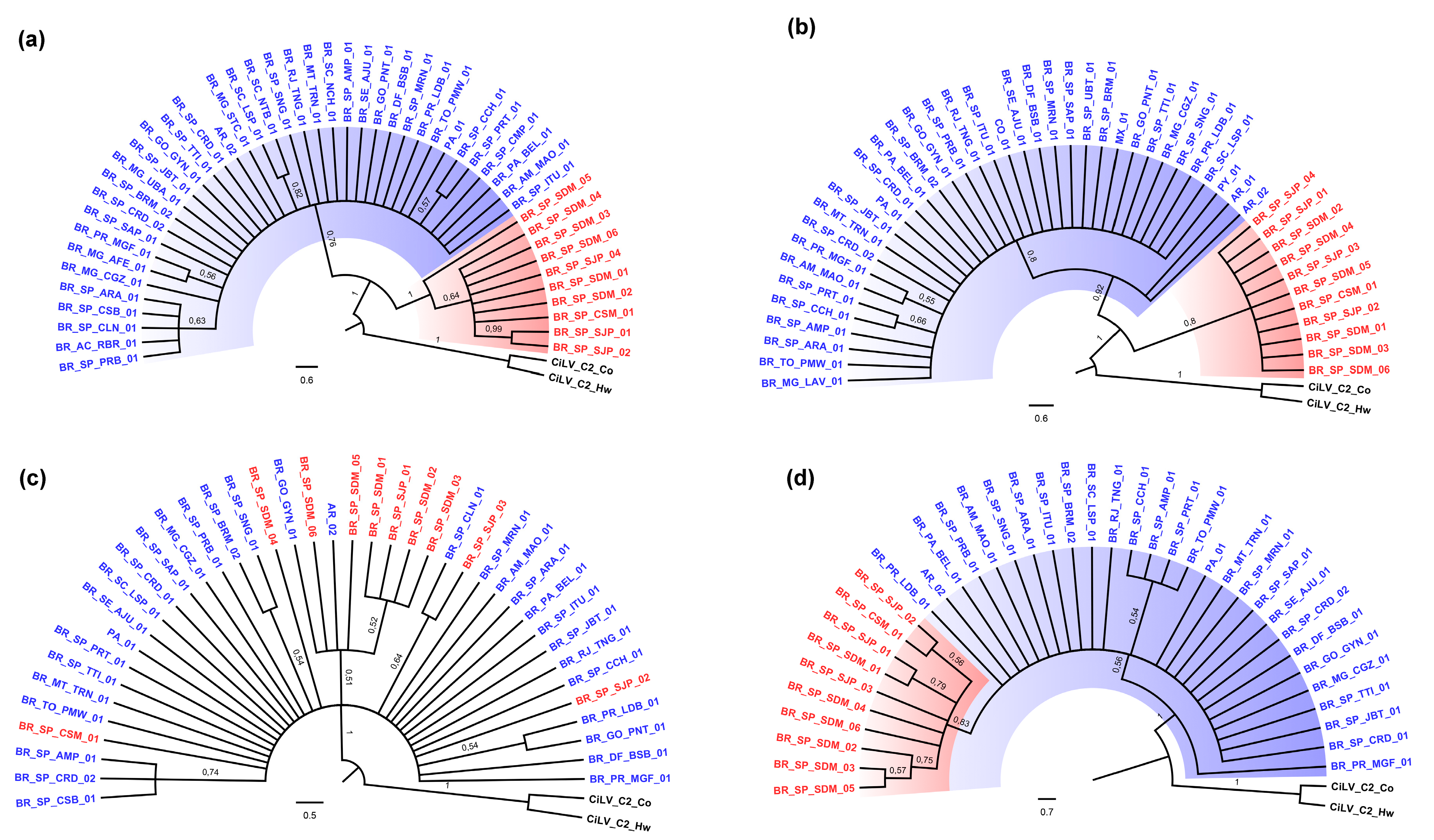
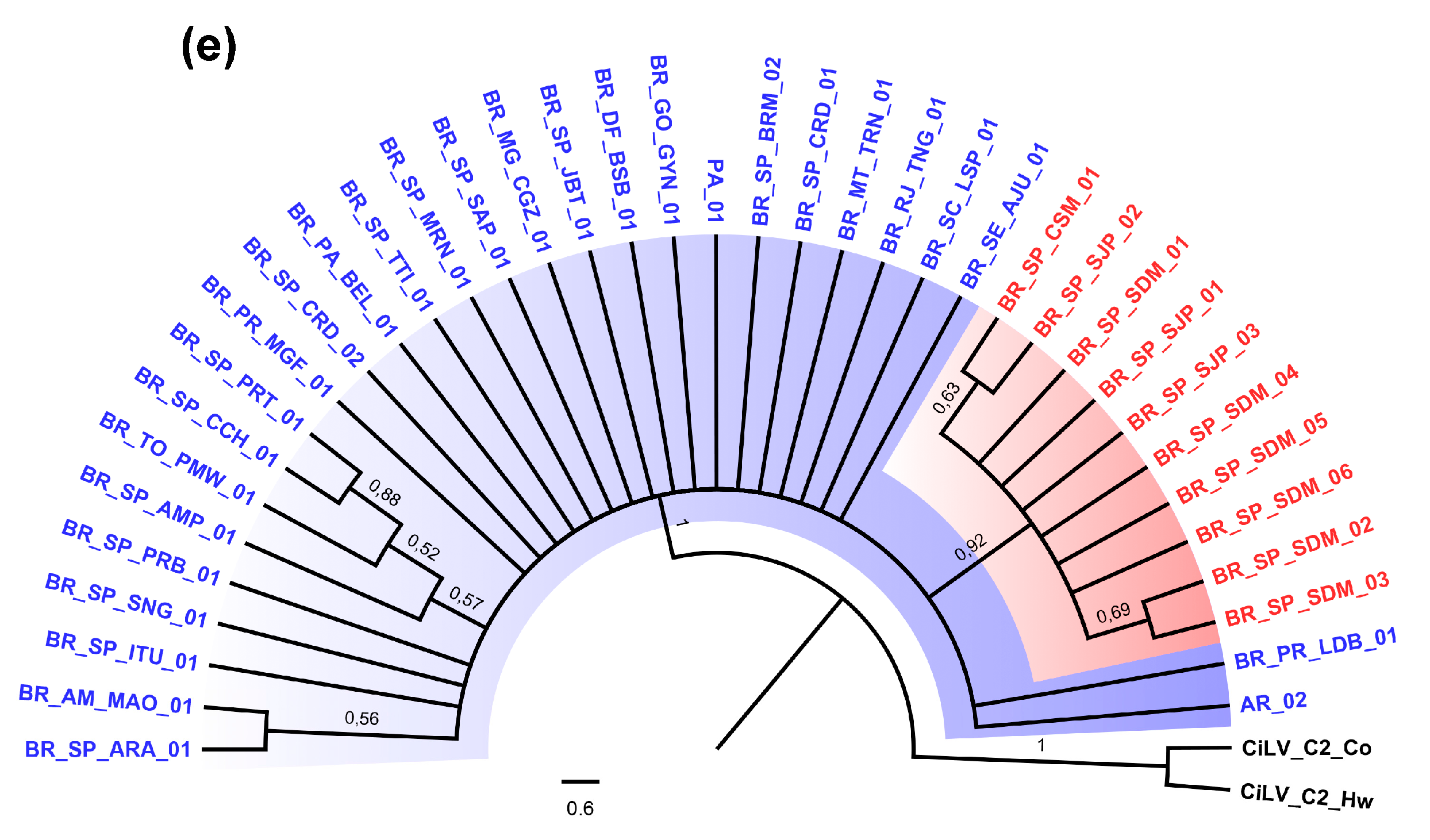
3.2. Phylogenetic Analysis of the p29 and MP Genes Indicate a Second Clade within the CiLV-C Population
3.3. p15 and IR-Based Trees Suggest Recombination
3.4. Genetic Data Support the Occurrence of Two CiLV-C Clades
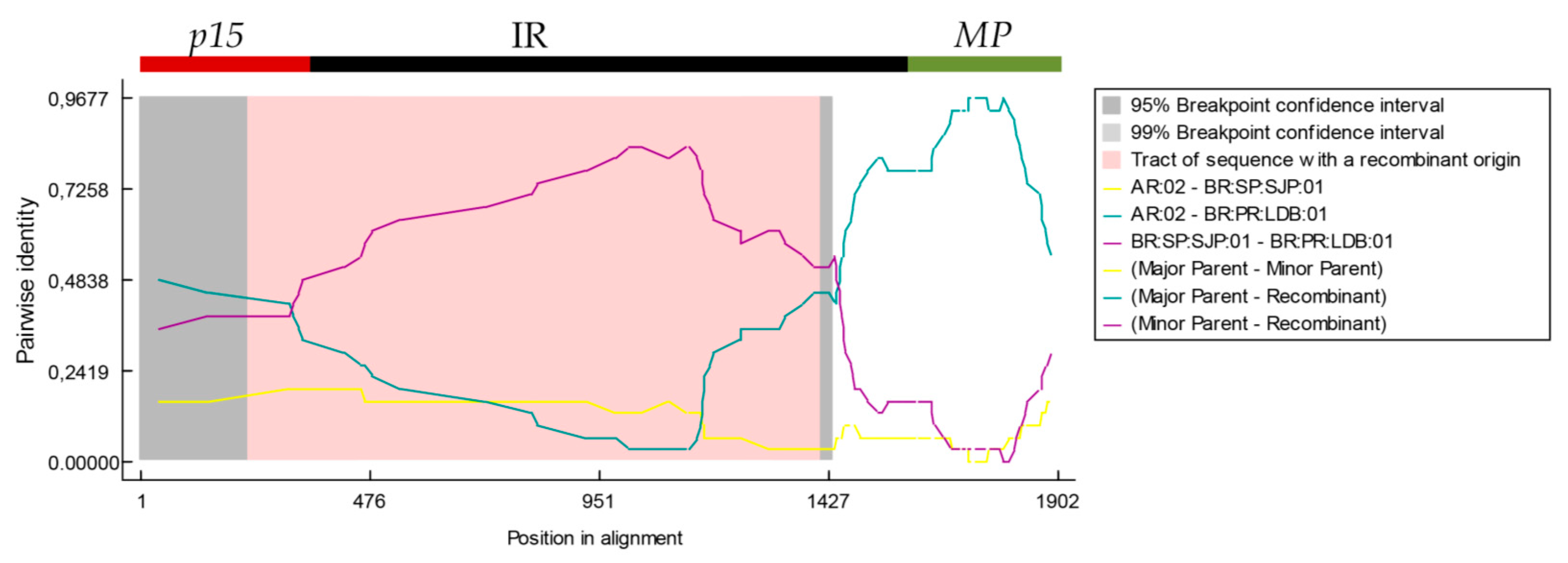
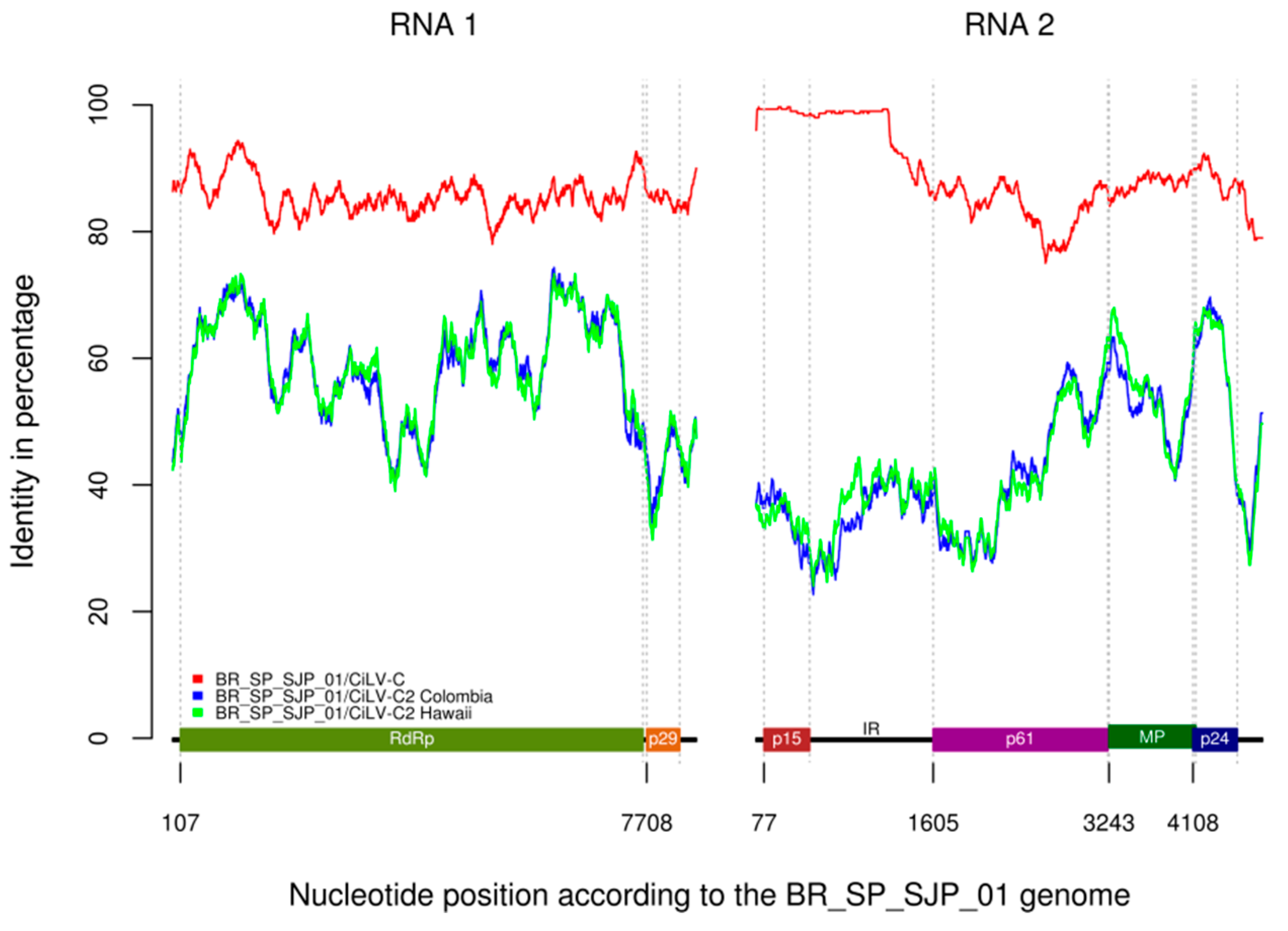
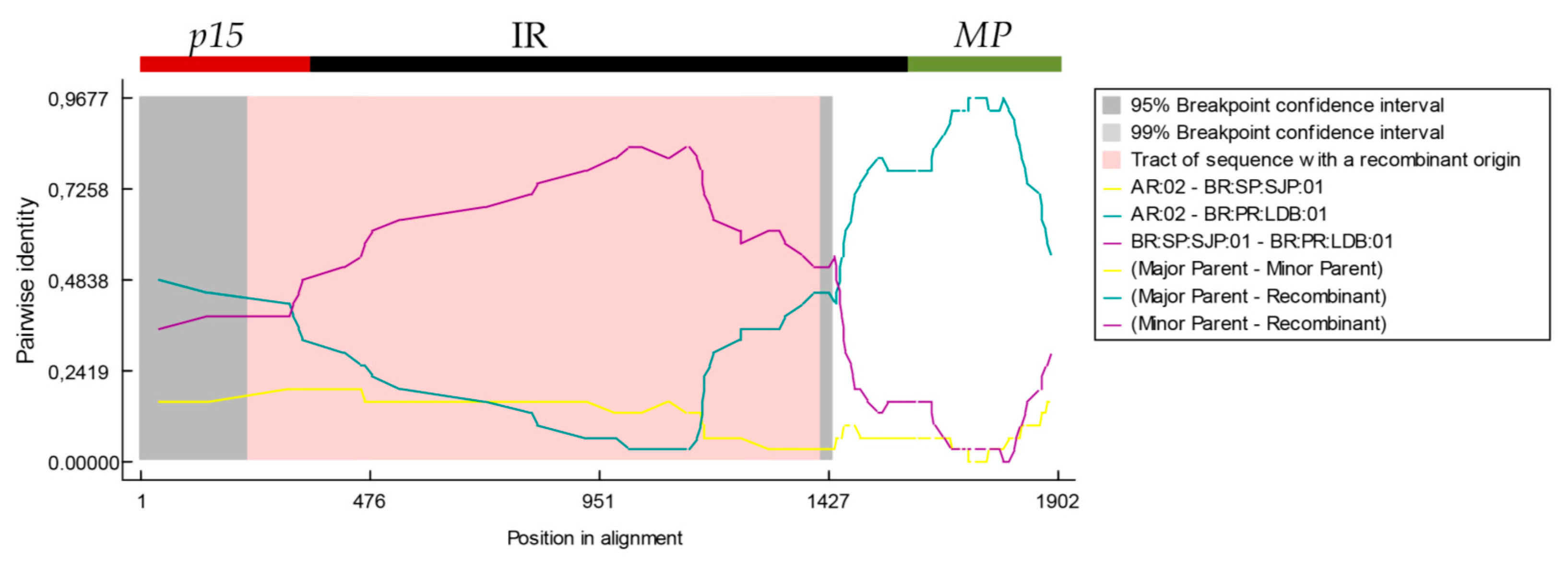
3.5. 5′-Proximal Region of CiLV-C RNA2 Harbors Recombination Signatures
3.6. Purifying Selection Is Acting on CiLV-C
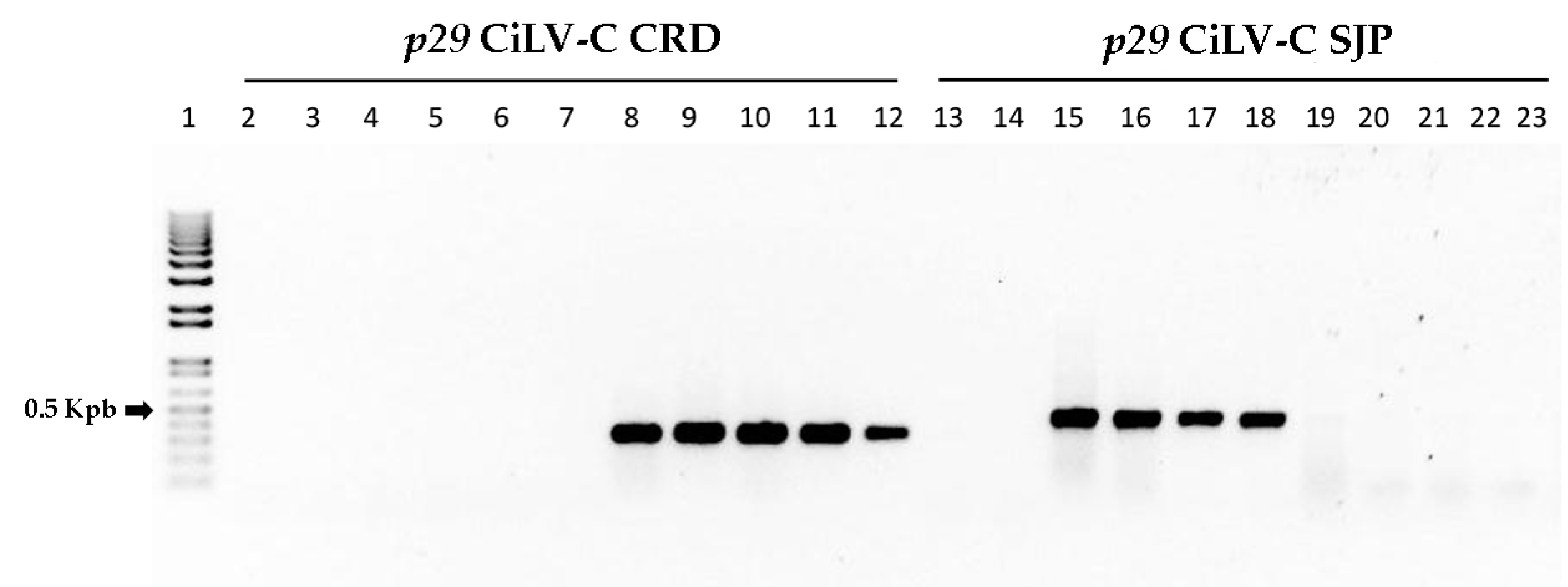
3.7. Clade-Specific CiLV-C Isolates Can Be Differentially Detected by RT-PCR
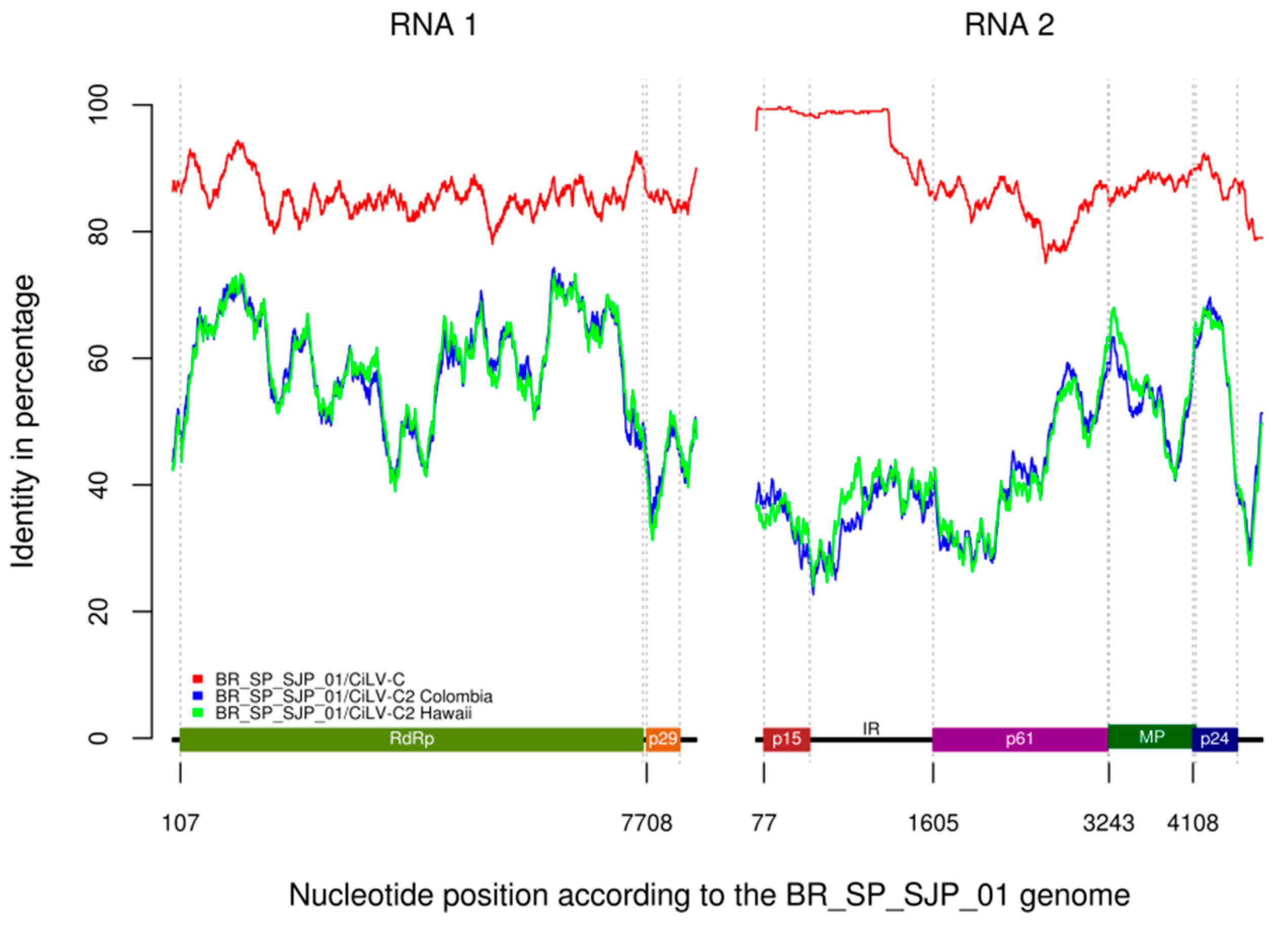
3.8. Genomic Characterization of CiLV-C Isolate BR_SP_SJP_01
3.9. Mite-Mediated Transmission of the Isolate BR_SP_SJP_01
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roy, A.; Hartung, J.S.; Schneider, W.L.; Shao, J.; Leon, M.G.; Melzer, M.J.; Beard, J.J.; Otero-Colina, G.; Bauchan, G.R.; Ochoa, R.; et al. Role bending: Complex relationships between viruses, hosts, and vectors related to citrus leprosis, an emerging disease. Phytopathology 2015, 105, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Bitancourt, A. Estudos sobre a leprose dos citros. Arq. Inst. Biológico 1955, 22, 161–231. [Google Scholar]
- Bitancourt, A. Relação das doenças e fungos parasitas observados na secção de fitopatologia durante os anos de 1931 e 1932. Arq. do Inst. Biológico 1934, 5, 185–196. [Google Scholar]
- Fawcett, H.S. Scarly bark of citrus. In Florida Agricultural Experimental Station Annual Report; Rolfs, P.H., Ed.; Pepper Pub and Ptg. Co.: Gainesville Fla, 1909; pp. 75–80. [Google Scholar]
- Spegazzini, C. Sobre algunas enfermedades y hongos que afectan las plantas de agrios en el Paraguay. An. Soc. Científica Argentina 1920, 90, 155–188. [Google Scholar]
- Childers, C.C.; Rodrigues, J.C.; Derrick, K.S.; Achor, D.S.; French, J.V.; Welbourn, W.C.; Ochoa, R.; Kitajima, E.W. Citrus leprosis and its status in Florida and Texas: past and present. Exp. Appl. Acarol. 2003, 30, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Bastianel, M.; Freitas-Astúa, J.; Kitajima, E.W.; Machado, M.A. The citrus leprosis pathosystem. Summa Phytopathol. 2006, 32, 211–220. [Google Scholar] [CrossRef]
- Cáceres, S.; Aguirre, A.; Costa, N.; de Coll, O.; Gonzáles Segnana, L.; Fariña, N.; Tassi, A.D.; Calegario, R.F.; Moraes, G.J.de; Freitas-Astúa, J.; et al. Present status of citrus leprosis in Argentina and Paraguay. Trop. Plant Pathol. 2013, 38, 282–294. [Google Scholar] [CrossRef]
- Castillo, I.I.; Diaz, L.F.; Mendez, W.; Otero-Colina, G.; Freitas-Astúa, J.; Locali-Fabris, E.C.; Moraes, G.J.; Calegario, R.F.; Tassi, A.D.; Kitajima, E.W. Confirmation of the presence of the citrus leprosis virus C (CiLV-C) in Southern Mexico. Trop. Plant Pathol. 2011, 36, 400–403. [Google Scholar]
- Leon, G.A.; Realpe, C.E.; Garzon, P.A.; Rodriguez, J.A.; Moreno, M.G.; Childers, C.C.; Achor, D.; Freitas-Astúa, J.; Antonioli-Luizon, R.; Salaroli, R.B.; et al. Occurrence of citrus leprosis virus in Llanos Orientales, Colombia. Plant Dis. 2006, 90, 682. [Google Scholar] [CrossRef]
- Dominguez, F.; Bernal, A.; Childers, C.C.; Kitajima, E.W.; de Dominguez, F.S.; Bernal, A.; Childers, C.C.; Kitajima, E.W. First report of citrus leprosis virus in Panama. Plant Dis. 2001, 85, 228. [Google Scholar] [CrossRef]
- Cruz-Jaramillo, J.L.; Ruiz-Medrano, R.; Rojas-Morales, L.; López-Buenfil, J.A.; Morales-Galván, O.; Chavarín-Palacio, C.; Ramírez-Pool, J.A.; Xoconostle-Cázares, B. Characterization of a proposed dichorhavirus associated with the citrus leprosis disease and analysis of the host response. Viruses 2014, 6, 2602–2622. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Stone, A.L.; Shao, J.; Otero-Colina, G.; Wei, G.; Choudhary, N.; Achor, D.; Levy, L.; Nakhla, M.K.; Hartung, J.S.; et al. Identification and molecular characterization of nuclear citrus leprosis virus, a member of the proposed Dichorhavirus genus infecting multiple citrus species in Mexico. Phytopathology 2015, 105, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Choudhary, N.; Guillermo, L.M.; Shao, J.; Govindarajulu, A.; Achor, D.; Wei, G.; Picton, D.D.; Levy, L.; Nakhla, M.K.; et al. A novel virus of the genus Cilevirus causing symptoms similar to citrus leprosis. Phytopathology 2013, 103, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.C.V.; Zuniga Reyes, J.A.; Achor, D.S.; Childers, C.C.; Kitajima, E.W. Occurrence and distribution of citrus leprosis virus in Honduras. Plant Pathol. 2007, 56, 344. [Google Scholar] [CrossRef]
- Gómez, E.C.; Vargas, M.R.; Rivadameira, C.; Locali, E.C.; Freitas-Astúa, J.; Astúa-Monge, G.; Rodrigues, J.C.V.; Mesa Cobo, N.C.; Kitajima, E.W. First report of citrus leprosis virus on citrus in Santa Cruz, Bolivia. Plant Dis. 2005, 89, 686. [Google Scholar] [CrossRef]
- León, M.G.; Becerra, C.H.; Freitas-Astúa, J.; Salaroli, R.B.; Kitajima, E.W. Natural infection of Swinglea glutinosa by the citrus leprosis virus cytoplasmic type (CiLV-C) in Colombia. Plant Dis. 2008, 92, 1364. [Google Scholar] [CrossRef]
- Araya-Gonzáles, J. Leprosis de los cítricos. Actual. Fitosanit. 2001, 1, 4. [Google Scholar]
- Mejia, L.; Paniagua, A.; Cruz, N.; Porras, M.; Palmieri, M. Citrus leprosis, disease that endangers plantations in Guatemala. Proc. Annu. Meet. Am. Phytopathol. Soc. Caribb. Div. 2002, 42, 17–19. [Google Scholar]
- Melzer, M.J.; Sether, D.M.; Borth, W.B.; Hu, J.S. Characterization of a virus infecting Citrus volkameriana with citrus leprosis-like symptoms. Phytopathology 2013, 102, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.J.; Simbajon, N.; Carillo, J.; Borth, W.B.; Freitas-Astúa, J.; Kitajima, E.W.; Neupane, K.R.; Hu, J.S. A cilevirus infects ornamental hibiscus in Hawaii. Arch. Virol. 2013, 158, 2421–2424. [Google Scholar] [CrossRef] [PubMed]
- Bastianel, M.; Novelli, V.M.; Kitajima, E.W.; Kubo, K.S.; Bassanezi, R.B.; Machado, M.A.; Freitas-Astúa, J. Citrus Leprosis: Centennial of an unusual mite–virus pathosystem. Plant Dis. 2010, 94, 284–292. [Google Scholar] [CrossRef]
- Rodrigues, J.C.V.; Childers, C.C. Brevipalpus mites (Acari: Tenuipalpidae): Vectors of invasive, non-systemic cytoplasmic and nuclear viruses in plants. Exp. Appl. Acarol. 2013, 59, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, E.W.; Chagas, C.M.; Rodrigues, J.C. Brevipalpus-transmitted plant virus and virus-like diseases: Cytopathology and some recent cases. Exp. Appl. Acarol. 2003, 30, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.J.; Ochoa, R.; Braswell, W.E.; Bauchan, G.R. Brevipalpus phoenicis (Geijskes) species complex (Acari: Tenuipalpidae)-a closer look. Zootaxa 2015, 3944, 1–67. [Google Scholar] [CrossRef] [PubMed]
- Garita, L.; Tassi, A.D.; Calegario, R.F.; Kitajima, E.W.; Carbonell, S.A.M.; Freitas-Astúa, J. Common bean (Phaseolus vulgaris L.): Experimental local lesion host for citrus leprosis virus C (CiLV-C) and some other cytoplasmic-type Brevipalpus-transmitted viruses. Plant Dis. 2013, 97, 1346–1351. [Google Scholar] [CrossRef]
- Garita, L.C.; Tassi, A.D.; Calegario, R.F.; Freitas-Astúa, J.; Salaroli, R.B.; Romão, G.O.; Kitajima, E.W. Experimental host range of citrus leprosis virus C (CiLV-C). Trop. Plant Pathol. 2014, 39, 43–55. [Google Scholar] [CrossRef]
- Afonso, C.L.; Amarasinghe, G.K.; Bányai, K.; Bào, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.-X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, (in press). [Google Scholar] [CrossRef] [PubMed]
- Locali-Fabris, E.C.; Freitas-Astúa, J.; Machado, M.A. Genus Cilevirus. In Virus Taxonomy - Ninth Report of the International Committee on Taxonomy of Viruses; King, A., Adams, M., Carstens, E., Lefkowitz, E., Eds.; Elsevier/Avademic Press: London, United Kingdom, 2011; pp. 1139–1142. [Google Scholar]
- Dietzgen, R.G.; Kuhn, J.H.; Clawson, A.N.; Freitas-Astúa, J.; Goodin, M.M.; Kitajima, E.W.; Kondo, H.; Wetzel, T.; Whitfield, A.E. Dichorhavirus: A proposed new genus for Brevipalpus mite-transmitted, nuclear, bacilliform, bipartite, negative-strand RNA plant viruses. Arch. Virol. 2014, 159, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Leon, M.G.; Stone, A.L.; Schneider, W.L.; Hartung, J.; Brlansky, R.H. First report of citrus leprosis virus nuclear type in sweet orange in Colombia. Plant Dis. 2014, 98, 1162. [Google Scholar] [CrossRef]
- Locali-Fabris, E.C.; Freitas-Astúa, J.; Souza, A.A.; Takita, M.A.; Astúa-Monge, G.; Antonioli-Luizon, R.; Rodrigues, V.; Targon, M.L.; Machado, M.A. Complete nucleotide sequence, genomic organization and phylogenetic analysis of citrus leprosis virus cytoplasmic type. J. Gen. Virol. 2006, 87, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhatla, D.B.; Sherman, W.A.; Chung, B.Y.W.; Cook, S.; Schneider, G.; Eisenhaber, B.; Karlin, D.G. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J. Virol. 2014, 88, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Pascon, R.C.; Kitajima, J.P.; Breton, M.C.; Assumpção, L.; Greggio, C.; Zanca, A.S.; Okura, V.K.; Alegria, M.C.; Camargo, M.E.; Silva, G.G.; et al. The complete nucleotide sequence and genomic organization of citrus leprosis associated virus, cytoplasmatic type (CiLV-C). Virus Genes 2006, 32, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Weeks, A.R.; Marec, F.; Breeuwer, J.A. A mite species that consists entirely of haploid females. Science 2001, 292, 2479–2482. [Google Scholar] [CrossRef] [PubMed]
- Childers, C.C.; French, J.V.; Rodrigues, J.C. Brevipalpus californicus, B. obovatus, B. phoenicis, and B. lewisi (Acari: Tenuipalpidae): A review of their biology, feeding injury and economic importance. Exp. Appl. Acarol. 2003, 30, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, E.W.; Alberti, G. Anatomy and fine structure of Bevipalpus mites (Tenuipalpidae)—Economically important plant virus vectors. Part 7. Ultrastructural detection of cytoplasmic and nuclear types of Brevipalpus transmitted viruses. Zoologica 2014, 160, 174–192. [Google Scholar]
- Domingo-Calap, P.; Sanjuán, R. Experimental evolution of RNA versus DNA viruses. Evolution 2011, 65, 2987–2994. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, A.; Calisher, C.; García-Arenal, F. Molecular basis of virus evolution, 1st ed.; Cambrige University Press: New York, NY, USA, 1995. [Google Scholar]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Bujarski, J.J. Genetic recombination in plant-infecting messenger-sense RNA viruses: Overview and research perspectives. Front. Plant Sci. 2013, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Mechanisms of plant virus evolution. Annu. Rev. Phytopathol. 1997, 35, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solińska, J.; Urbanowicz, A.; Figlerowicz, M.; Bujarski, J.J. RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 2011, 49, 415–443. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, B.D.; Li, O.T.; Poon, L.L.; Levine, A.J.; Rabadan, R. Viral reassortment as an information exchange between viral segments. Proc. Natl. Acad. Sci. USA 2012, 109, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, S.; Michalakis, Y.; Blanc, S. Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2012, 2, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Leal, R.; Duffy, S.; Xiong, Z.; Hammond, R.W.; Elena, S.F. Advances in plant virus evolution: translating evolutionary insights into better disease management. Phytopathology 2011, 101, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.F.; Trombin, V.G.; Lopes, F.F.; Kalaki, R.; Milan, P. The Orange Juice Business: A Brazilian Perspective; Wageningen Academic Publishers: Wageningen, The Netherlands, 2011. [Google Scholar]
- Nunes, M.A.; Bergamini, M.P.; Coerini, L.F.; Bastianel, M.; Novelli, V.M.; Kitajima, E.W.; Freitas-Astúa, J. Citrus leprosis virus C naturally infecting Commelina benghalensis, a prevalent monocot weed of citrus orchards in Brazil. Plant Dis. 2012, 96, 770. [Google Scholar] [CrossRef]
- Guerra, A.; Manjunath, K.; Rangel, E.; Brlansky, R.H.; Lee, R.F. Citrus leprosis symptoms can be associated with the presence of two different viruses, cytoplasmic and nuclear, the former having a multipartite RNA genome. (unpublished).
- Locali, E.C.; Freitas-Astúa, J.; de Souza, A.A.; Takita, M.A.; Astúa-Monge, G.; Antonioli, R.; Kitajima, E.W.; Machado, M.A. Development of a molecular tool for the diagnosis of leprosis, a major threat to citrus production in the Americas. Plant Dis. 2003, 87, 1317–1321. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 25 October 2015).
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Datamonkey: Rapid Detection of Positive Selection. Available online: http://www.datamonkey.org/dataupload.php (accessed on 4 June 2015).
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Pei, J.; Grishin, N.V. PROMALS: Towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 2007, 23, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- PlantGDB. Arabidopsis thaliana Genome. Available online: http://www.plantgdb.org/AtGDB/ (accessed on 4 June 2015).
- Citrus Genome Database. Available online: https://www.citrusgenomedb.org/ (accessed on 4 June 2015).
- Knudsen, T.; Knudsen, B. CLC genomics benchwork 6. Available online: http://www.clcbio.com (accessed on 25 October 2015).
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Rozanov, M.N.; Koonin, E.V.; Gorbalenya, A.E. Conservation of the putative methyltransferase domain: A hallmark of the “Sindbis-like” supergroup of positive-strand RNA viruses. J. Gen. Virol. 1992, 73, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Wierzchoslawski, R.; Dzianott, A.; Bujarski, J. Dissecting the requirement for subgenomic promoter sequences by RNA recombination of brome mosaic virus in vivo: Evidence for functional separation of transcription and recombination. J. Virol. 2004, 78, 8552–8564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wierzchoslawski, R.; Dzianott, A.; Kunimalayan, S.; Bujarski, J.J. A transcriptionally active subgenomic promoter supports homologous crossovers in a plus-strand RNA virus. J. Virol. 2003, 77, 6769–6776. [Google Scholar] [CrossRef] [PubMed]
- Martin, S. Genetic variation of populations of Citrus psorosis virus. J. Gen. Virol. 2006, 87, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Ayllón, M.A.; Kong, P.; Fernández, A.; Polek, M.; Guerri, J.; Moreno, P.; Falk, B.W. Genetic variation of Citrus tristeza virus isolates from California and Spain: Evidence for mixed infections and recombination. J. Virol. 2001, 75, 8054–8062. [Google Scholar] [CrossRef] [PubMed]
- Vives, M.C.; Rubio, L.; Galipienso, L.; Navarro, L.; Moreno, P.; Guerri, J. Low genetic variation between isolates of Citrus leaf blotch virus from different host species and of different geographical origins. J. Gen. Virol. 2002, 83, 2587–2591. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.S.; Roy, A.; Fu, S.; Shao, J.; Schneider, W.L.; Brlansky, R.H. History and diversity of citrus leprosis virus recorded in herbarium specimens. Phytopathology 2015, 105, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Bastianel, M.; Freitas-Astúa, J.; Nicolini, F.; Segatti, N.; Novelli, V.M.; Rodrigues, V.; Medina, C.L.; Machado, M.A. Response of mandarin cultivars and hybrids to citrus leprosis virus. J. Plant Pathol. 2008, 90, 305–301. [Google Scholar]
- Arena, G.D.; Bergamini, M.P.; Tassi, A.D.; Kitajima, E.W.; Kubo, K.S.; Freitas-Astúa, J. Citrus leprosis virus C infects Arabidopsis thaliana, the model for plant-pathogen interactions. J. Plant Pathol. 2013, 95, 448. [Google Scholar]
- Nunes, M.A.; Lameiro, P.; Calegario, R.F.; Bergamini, M.; Fender, L.; Kitajima, E.W.; Bastianel, M.; Novelli, V.M.; Freita-Astua, J. Tropical spiderwort (Commelina benghalensis L.) as source of inoculum for citrus leprosis virus C. Citrus Res. Technol. 2012, 33, 1–9. [Google Scholar] [CrossRef]
- Pu, Y.; Kikuchi, A.; Moriyasu, Y.; Tomaru, M.; Jin, Y.; Suga, H.; Hagiwara, K.; Akita, F.; Shimizu, T.; Netsu, O.; et al. Rice dwarf viruses with dysfunctional genomes generated in plants are filtered out in vector insects: Implications for the origin of the virus. J. Virol. 2011, 85, 2975–2979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sin, S.-H.; McNulty, B.C.; Kennedy, G.G.; Moyer, J.W. Viral genetic determinants for thrips transmission of Tomato spotted wilt virus. Proc. Natl. Acad. Sci. USA 2005, 102, 5168–5173. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.R.; Elena, S.F. Evolution of plant virus movement proteins from the 30K superfamily and of their homologs integrated in plant genomes. Virology 2015, 476C, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.H.; Bujarski, J.J. Multiple functions of capsid proteins in (+) stranded RNA viruses during plant-virus interactions. Virus Res. 2015, 196C, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Arrivabem, F.; Rodrigues, V.; Freitas-Astúa, J.; Bastianel, M.; Locali-Fabris, E.C.; Antonioli-Luizon, R.; Novelli, V.M.; Kitajima, E.W.; Machado, M.A. Transmissão diferencial do vírus da leprose dos citros por populações de Brevipalpus phoenicis. Summa Phytopathol. 2005, 31, 80. [Google Scholar]
- Sánchez-Velázquez, E.J.; Santillán-Galicia, M.T.; Novelli, V.M.; Nunes, M.A.; Mora-Aguilera, G.; Valdez-Carrasco, J.M.; Otero-Colina, G.; Freitas-Astúa, J. Diversity and genetic variation among Brevipalpus populations from Brazil and Mexico. PLoS ONE 2015, 10, e0133861. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Identification 1 and Host 2 of Collection | Collection Year | Viral Genomic Region | GenBank Accession No. | Reference |
|---|---|---|---|---|
| Brazilian isolates | ||||
| BR_MG_AFE_01 | 2012 | p29 | KR093040 | This work |
| BR_SP_AMP_01 | 2012 | p29 | KR093041 | This work |
| p15 | KR093078 | |||
| IR | KR093139 | |||
| MP | KR093106 | |||
| BR_SE_AJU_01 | 2012 | p29 | KR093042 | This work |
| p15 | KR093079 | |||
| IR | KR093140 | |||
| MP | KR093107 | |||
| BR_SP_ARA_01 | 2014 | p29 | KR093043 | This work |
| p15 | KR093080 | |||
| IR | KR093141 | |||
| MP | KR093108 | |||
| BR_PA_BEL_01 | 2012 | p29 | KR093045 | This work |
| p15 | KR136415 | |||
| IR | KR093143 | |||
| MP | KR093110 | |||
| BR_SP_BRM_01 (Commelina benghalensis) | 2012 | MP | JQ944802 | [49] |
| BR_SP_BRM_02 | 2012 | p29 | KR093046 | This work |
| p15 | KR093084 | |||
| IR | KR093144 | |||
| MP | KR093112 | |||
| BR_DF_BSB_01 | 2012 | p29 | KR093047 | This work |
| p15 | KR093085 | |||
| IR | KR093145 | |||
| MP | KR093113 | |||
| BR_SP_CMP_01 | 2012 | p29 | KR093049 | This work |
| BR_SP_CSB_01 | 2012 | p29 | KR093050 | This work |
| p15 | KR093086 | |||
| BR_SP_CLN_01 | 2012 | p29 | KR093051 | This work |
| p15 | KR093082 | |||
| BR_MG_CGZ_01 | 2012 | p29 | KR093048 | This work |
| p15 | KR093087 | |||
| IR | KR093146 | |||
| MP | KR093114 | |||
| BR_SP_CCH_01 | 2012 | p29 | KR093052 | This work |
| p15 | KR093088 | |||
| IR | KR093147 | |||
| MP | KR093115 | |||
| BR_SP_CRD_01 | 2006 | RNA1 | DQ352194 | [32] |
| RNA2 | DQ352195 | |||
| BR_SP_CRD_02 | 2012 | p29 | KR093053 | This work |
| p15 | KR093089 | |||
| IR | KR093148 | |||
| MP | KR093116 | |||
| BR_SP_CRD_03 | 2012 | MP | KR093117 | This work |
| BR_SP_CSM_01 | 2012 | p29 | KR093054 | This work |
| p15 | KR093083 | |||
| IR | KR093149 | |||
| MP | KR093111 | |||
| BR_GO_GYN_01 | 2012 | p29 | KR093055 | This work |
| p15 | KR093090 | |||
| IR | KR093150 | |||
| MP | KR093118 | |||
| BR_MG_LAV_01 | 2012 | MP | KR093120 | This work |
| BR_SC_LSP_01 | 2012 | p29 | KR093056 | This work |
| p15 | KR093091 | |||
| IR | KR093151 | |||
| MP | KR093119 | |||
| BR_PR_LDB_01 | 2012 | p29 | KR093057 | This work |
| p15 | KR093092 | |||
| IR | KR093152 | |||
| MP | KR093121 | |||
| BR_AM_MAO_01 | 2012 | p29 | KR093058 | This work |
| p15 | KR093093 | |||
| IR | KR093153 | |||
| MP | KR093122 | |||
| BR_PR_MGF_01 | 2012 | p29 | KR093059 | This work |
| p15 | KR093094 | |||
| IR | KR093154 | |||
| MP | KR093123 | |||
| BR_SP_MRN_01 | 2012 | p29 | KR093060 | This work |
| p15 | KR136416 | |||
| IR | KR093155 | |||
| MP | KR093124 | |||
| BR_SC_NCH_01 | 2012 | p29 | KR093061 | This work |
| BR_SC_NTB_01 | 2012 | p29 | KR093062 | This work |
| BR_TO_PMW_01 | 2012 | p29 | KR093063 | This work |
| p15 | KR093105 | |||
| IR | KR093166 | |||
| MP | KR093134 | |||
| BR_SP_PRB_01 | 2014 | p29 | KR093064 | This work |
| p15 | KR093095 | |||
| IR | KR093156 | |||
| MP | KR093125 | |||
| BR_GO_PNT_01 | 2012 | p29 | KR093065 | This work |
| p15 | KR093096 | |||
| MP | KR093126 | |||
| BR_SP_PRT_01 | 2012 | p29 | KR093066 | This work |
| p15 | KR093097 | |||
| IR | KR093157 | |||
| MP | KR093127 | |||
| BR_AC_RBR_01 | 2012 | p29 | KR093067 | This work |
| BR_SP_ITU_01 | 2012 | p29 | KR093070 | This work |
| p15 | KR093099 | |||
| IR | KR093159 | |||
| MP | KR093129 | |||
| BR_MG_STC_01 | 2012 | p29 | KR093069 | This work |
| BR_SP_SAP_01 | 2012 | p29 | KR093068 | This work |
| p15 | KR093098 | |||
| IR | KR093158 | |||
| MP | KR093128 | |||
| BR_SP_JBT_01 | 2006 | RNA1 | DQ157466 | [34] |
| RNA2 | DQ157465 | |||
| BR_SP_SNG_01 | 2012 | p29 | KR093071 | This work |
| p15 | KR093100 | |||
| IR | KR093160 | |||
| MP | KR093130 | |||
| BR_SP_SJP_01 | 2012 | RNA1 | KP336746 | This work |
| RNA2 | KP336747 | |||
| BR_SP_SJP_02 | 2012 | p29 | KR093072 | This work |
| p15 | KR093101 | |||
| IR | KR093161 | |||
| MP | KR093135 | |||
| BR_SP_SJP_03 | 2012 | p15 | KR093167 | This work |
| IR | KR093162 | |||
| MP | KR093136 | |||
| BR_SP_SJP_04 (Commelina benghalensis) | 2013 | p29 | KR093077 | This work |
| MP | KR093137 | |||
| BR_SP_SDM_01 | 2015 | p29 | KT253463 | This work |
| p15 | KT253469 | |||
| IR | KT253475 | |||
| MP | KT253481 | |||
| BR_SP_SDM_02 | 2015 | p29 | KT253464 | This work |
| p15 | KT253470 | |||
| IR | KT253476 | |||
| MP | KT253482 | |||
| BR_SP_SDM_03 | 2015 | p29 | KT253465 | This work |
| p15 | KT253471 | |||
| IR | KT253477 | |||
| MP | KT253483 | |||
| BR_SP_SDM_04 | 2015 | p29 | KT253466 | This work |
| p15 | KT253472 | |||
| IR | KT253478 | |||
| MP | KT253484 | |||
| BR_SP_SDM_05 | 2015 | p29 | KT253467 | This work |
| p15 | KT253473 | |||
| IR | KT253479 | |||
| MP | KT253485 | |||
| BR_SP_SDM_06 | 2015 | p29 | KT253468 | This work |
| p15 | KT253474 | |||
| IR | KT253480 | |||
| MP | KT253486 | |||
| BR_RJ_TNG_01 | 2012 | p29 | KR093073 | This work |
| p15 | KR093102 | |||
| IR | KR093163 | |||
| MP | KR093131 | |||
| BR_SP_TTI_01 | 2012 | p29 | KR093074 | This work |
| p15 | KR093103 | |||
| IR | KR093164 | |||
| MP | KR093132 | |||
| BR_MT_TRN_01 | 2012 | p29 | KR093075 | This work |
| p15 | KR093104 | |||
| IR | KR093165 | |||
| MP | KR093133 | |||
| BR_MG_UBA_01 | 2012 | p29 | KR093076 | This work |
| MP | KR093138 | |||
| Isolates from other countries | ||||
| AR_01 | 2012 | MP | JX163907 | [8] |
| AR_02 | 2012 | p29 | KR093044 | This work |
| p15 | KR093081 | |||
| IR | KR093142 | |||
| MP | KR093109 | |||
| CO_01 | 2005 | MP | DQ272491 | [10] |
| MX_01 | 2010 | MP | HQ292778 | [9] |
| PA_01 | 2006 | RNA1 | DQ388512 | [50] |
| RNA2 | DQ388513 | |||
| PY_01 | 2012 | MP | JX163908 | [8] |
| Genomic Region | Region Length (nt) | Dataset 1/# of Isolates | π | h | Hd | dS | dN | ω (dN/dS) |
|---|---|---|---|---|---|---|---|---|
| p29 | 792 | All/47 | 0.053 ± 0.009 | 40 | 0.990 | 0.252 ± 0.037 2 | 0.018 ± 0.003 2 | 0.07 |
| CRD/38 | 0.009 ± 0.001 | 32 | 0.986 | 0.022 ± 0.004 | 0.004 ± 0.001 | 0.18 | ||
| p15 | 393 | All/41 | 0.010 ± 0.001 | 30 | 0.961 | 0.014 ± 0.004 | 0.007 ± 0.002 | 0.50 |
| CRD/31 | 0.010 ± 0.001 | 24 | 0.953 | 0.014 ± 0.004 | 0.008 ± 0.002 | 0.57 | ||
| MP | 300 | All/46 | 0.056 ± 0.009 | 22 | 0.854 | 0.202 ± 0.043 | 0.021 ± 0.007 | 0.10 |
| CRD/36 | 0.007 ± 0.001 | 17 | 0.778 | 0.019 ± 0.006 | 0.005 ± 0.002 | 0.26 | ||
| IR | 940 | All/38 | 0.021 ± 0.003 | 38 | 1 | - | - | - |
| CRD/28 | 0.016 ± 0.003 | 28 | 1 | - | - | - |
| ORF | Nucleotide Substitution Model | Methods | Selection Pressure | Amino Acid Position in the Sequence of Isolate BR_SP_SJP_01 |
|---|---|---|---|---|
| p29 | TrN | SLAC-FEL-REL | purification | 27, 39, 113, 156, 157, 173, 207, 211, 227, 259 |
| FEL-REL | purification | 2, 7, 23, 30, 47, 49, 57, 58, 76, 87, 99, 100, 104, 116, 118, 133, 137, 140, 151, 154, 165, 171, 172, 180, 181, 190, 196, 202, 213, 214, 217, 219, 245, 254 | ||
| p15 | HKY85 | SLAC-FEL-REL | - | - |
| FEL-REL | positive | 91 | ||
| MP 1 | GTR | SLAC-FEL-REL | purification | 71, 112, 148 |
| FEL-REL | purification | 59, 74, 80, 84, 85, 92, 93, 96, 97, 98, 99, 102, 103, 111, 115, 118, 121, 136, 146 |
| BR_SP_SJP_01 | CiLV-C | CiLV-C2 Isolate Colombia | CiLV-C2 Isolate Hawaii | |||
|---|---|---|---|---|---|---|
| nt | aa | nt | aa | nt | aa | |
| RNA1 | 85.6 | - | 60.1 | - | 60.1 | - |
| ORF RdRp | 85.4 | 93.1 | 61.4 | 59.3 | 61.4 | 59.1 |
| ORF p29 | 85.0 | 90.5 | 49.2 | 36.0 | 49.0 | 35.0 |
| RNA2 | 88.4 | - | 52.6 | - | 52.0 | - |
| ORF p15 | 99.5 | 100.0 | 44.7 | 20.8 | 45.2 | 24.6 |
| ORF p61 | 81.8 | 84.0 | 52.0 | 32.0 | 51.2 | 33.1 |
| ORF MP | 86.8 | 91.9 | 56.6 | 51.7 | 54.9 | 51.4 |
| ORF p24 | 87.4 | 93.9 | 63.9 | 63.1 | 62.0 | 61.6 |
| IR | 96.7 | - | 46.0 | - | 46.0 | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-González, P.L.; Chabi-Jesus, C.; Guerra-Peraza, O.; Breton, M.C.; Arena, G.D.; Nunes, M.A.; Kitajima, E.W.; Machado, M.A.; Freitas-Astúa, J. Phylogenetic and Molecular Variability Studies Reveal a New Genetic Clade of Citrus leprosis virus C. Viruses 2016, 8, 153. https://doi.org/10.3390/v8060153
Ramos-González PL, Chabi-Jesus C, Guerra-Peraza O, Breton MC, Arena GD, Nunes MA, Kitajima EW, Machado MA, Freitas-Astúa J. Phylogenetic and Molecular Variability Studies Reveal a New Genetic Clade of Citrus leprosis virus C. Viruses. 2016; 8(6):153. https://doi.org/10.3390/v8060153
Chicago/Turabian StyleRamos-González, Pedro Luis, Camila Chabi-Jesus, Orlene Guerra-Peraza, Michèle Claire Breton, Gabriella Dias Arena, Maria Andreia Nunes, Elliot Watanabe Kitajima, Marcos Antonio Machado, and Juliana Freitas-Astúa. 2016. "Phylogenetic and Molecular Variability Studies Reveal a New Genetic Clade of Citrus leprosis virus C" Viruses 8, no. 6: 153. https://doi.org/10.3390/v8060153
APA StyleRamos-González, P. L., Chabi-Jesus, C., Guerra-Peraza, O., Breton, M. C., Arena, G. D., Nunes, M. A., Kitajima, E. W., Machado, M. A., & Freitas-Astúa, J. (2016). Phylogenetic and Molecular Variability Studies Reveal a New Genetic Clade of Citrus leprosis virus C. Viruses, 8(6), 153. https://doi.org/10.3390/v8060153