
ESCRT Requirements for Murine Leukemia Virus Release



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
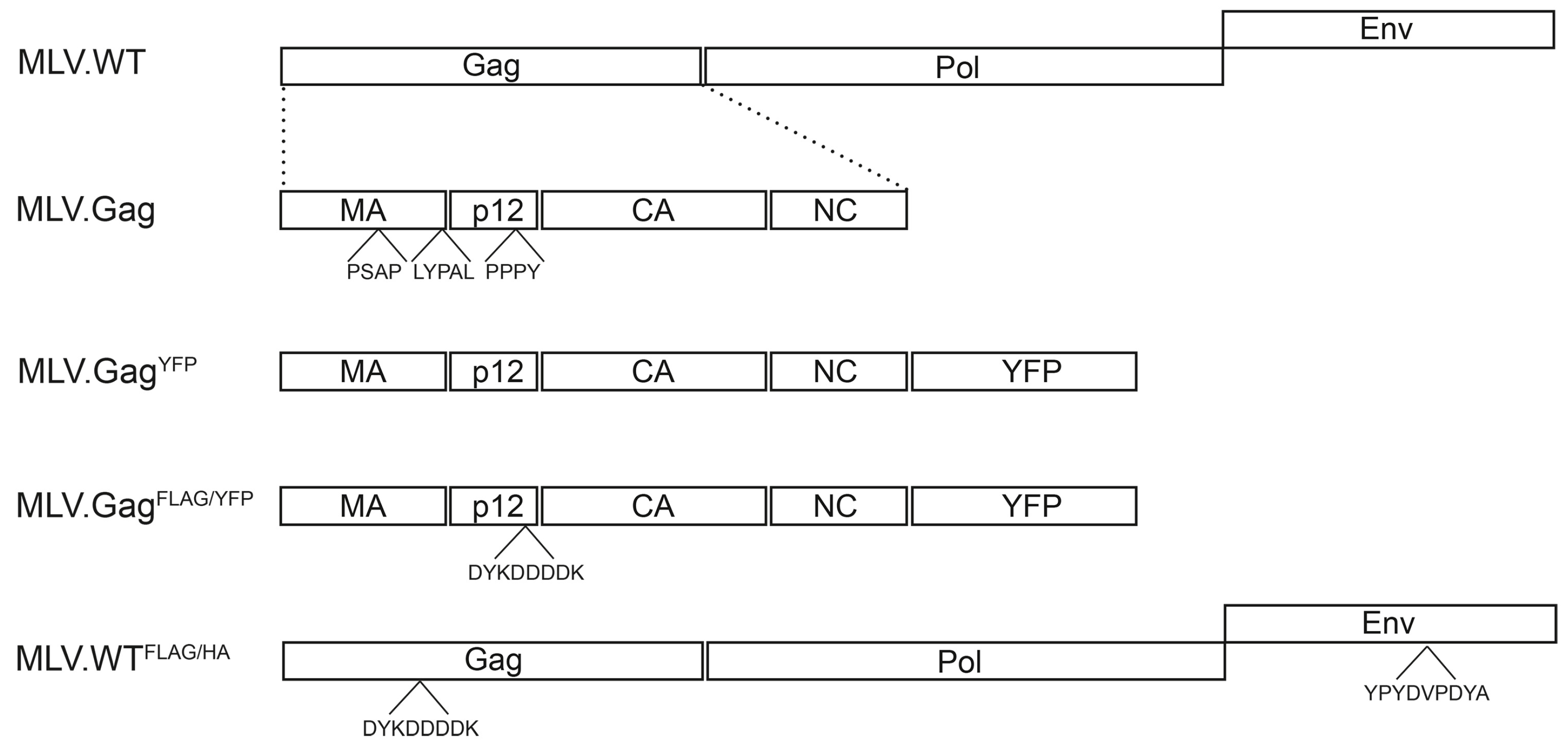
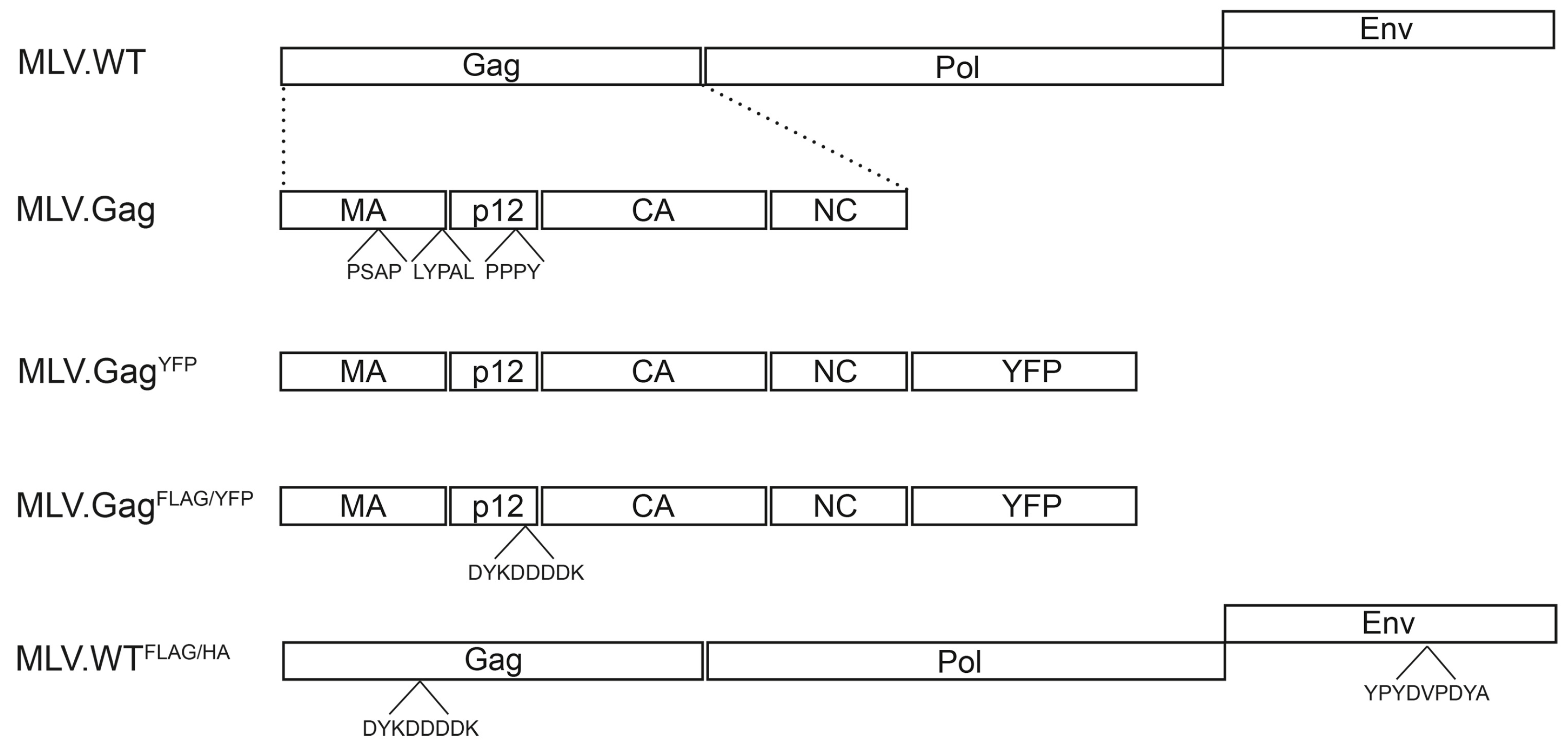
2.1. Expression Constructs
2.2. Antibodies
2.3. Cells and Transfection
2.4. Cell Lysis and Viral Particle Analysis
2.5. Quantitative Reverse Transcriptase (qRT)-PCR Analysis
2.6. MLV Infection Assay
2.7. Fluorescence Microscopy
3. Results
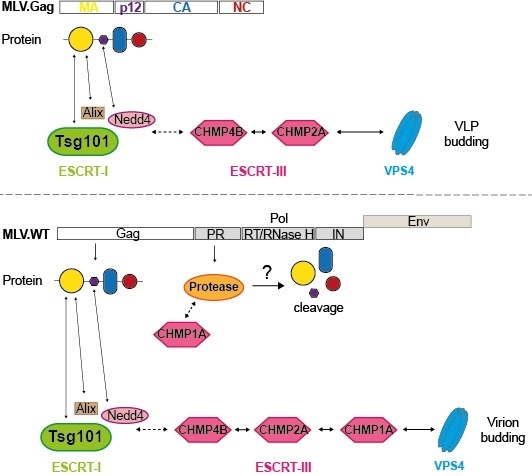
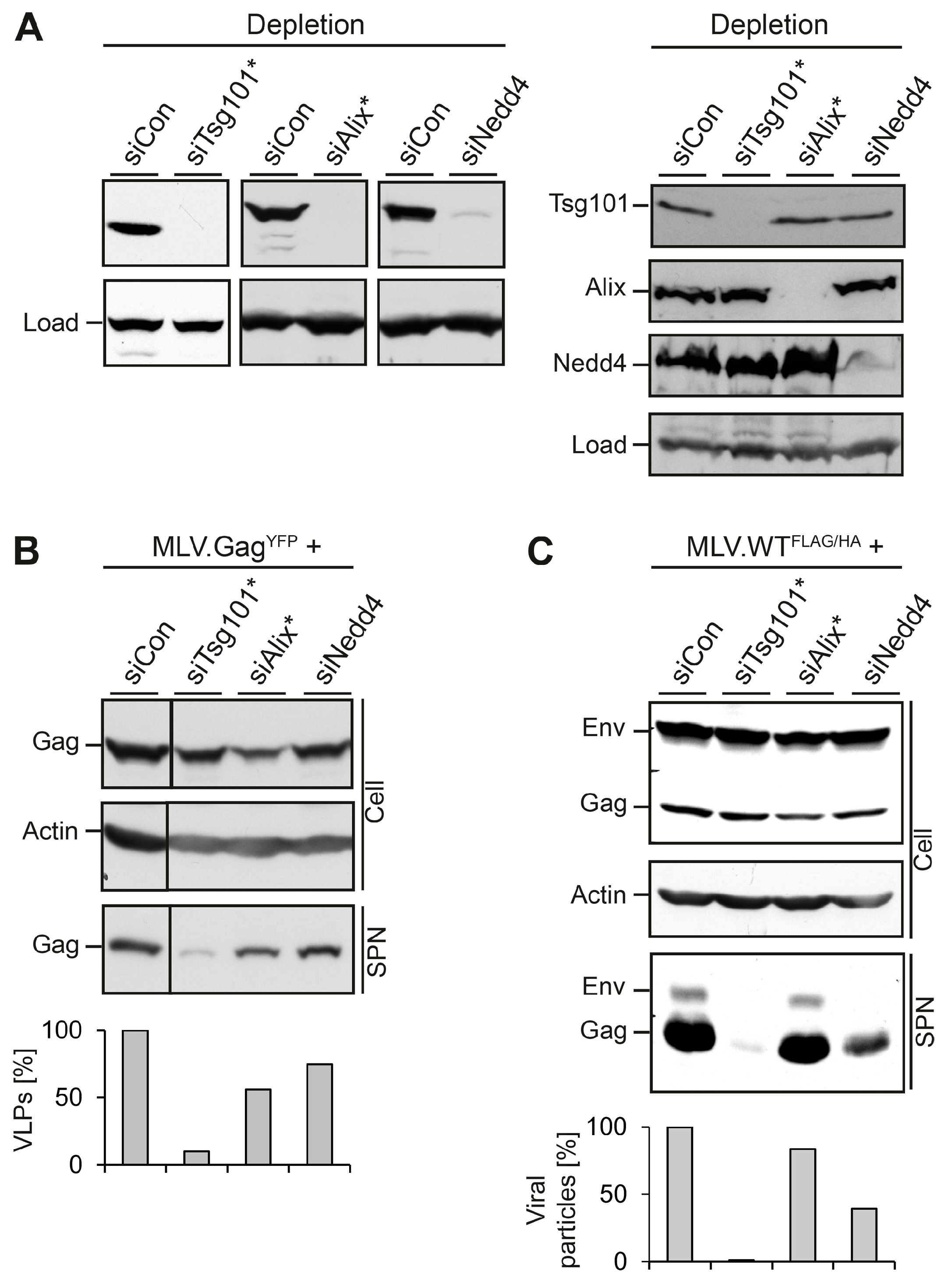
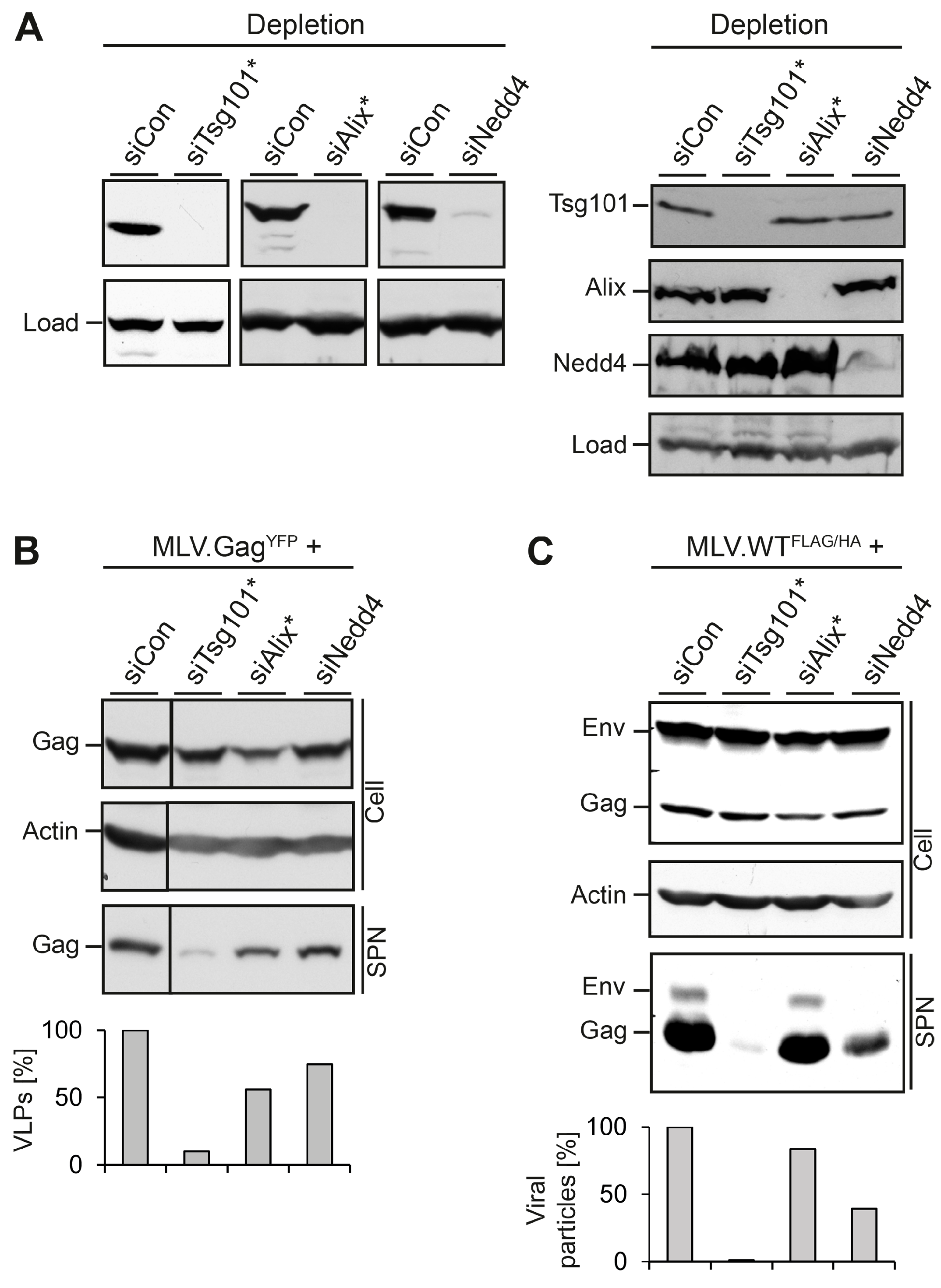
3.1. Role of MVB Adaptors and ESCRT-I in MLV VLP and Virion Release
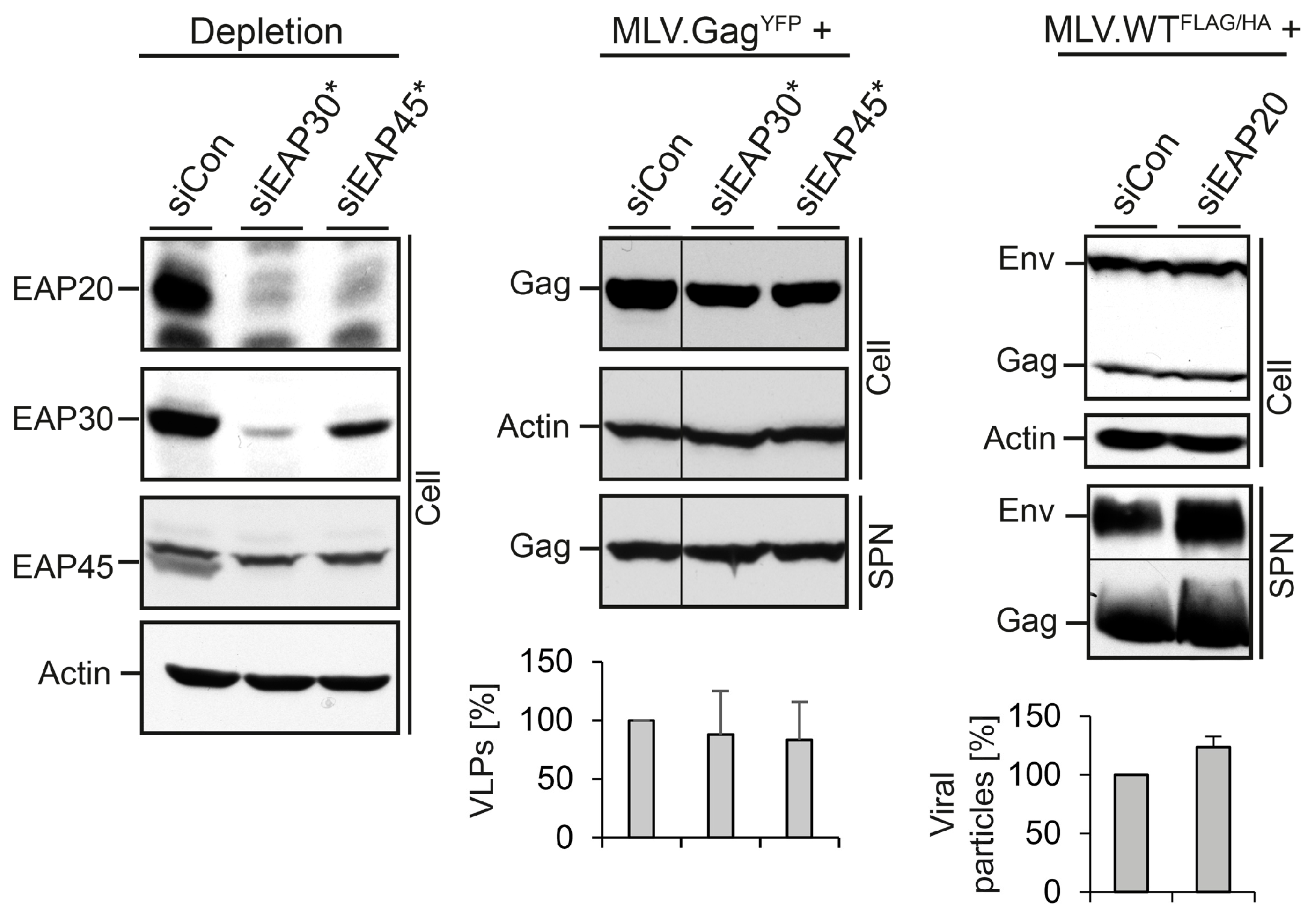
3.2. Role of ESCRT-II in MLV VLP and Virion Release
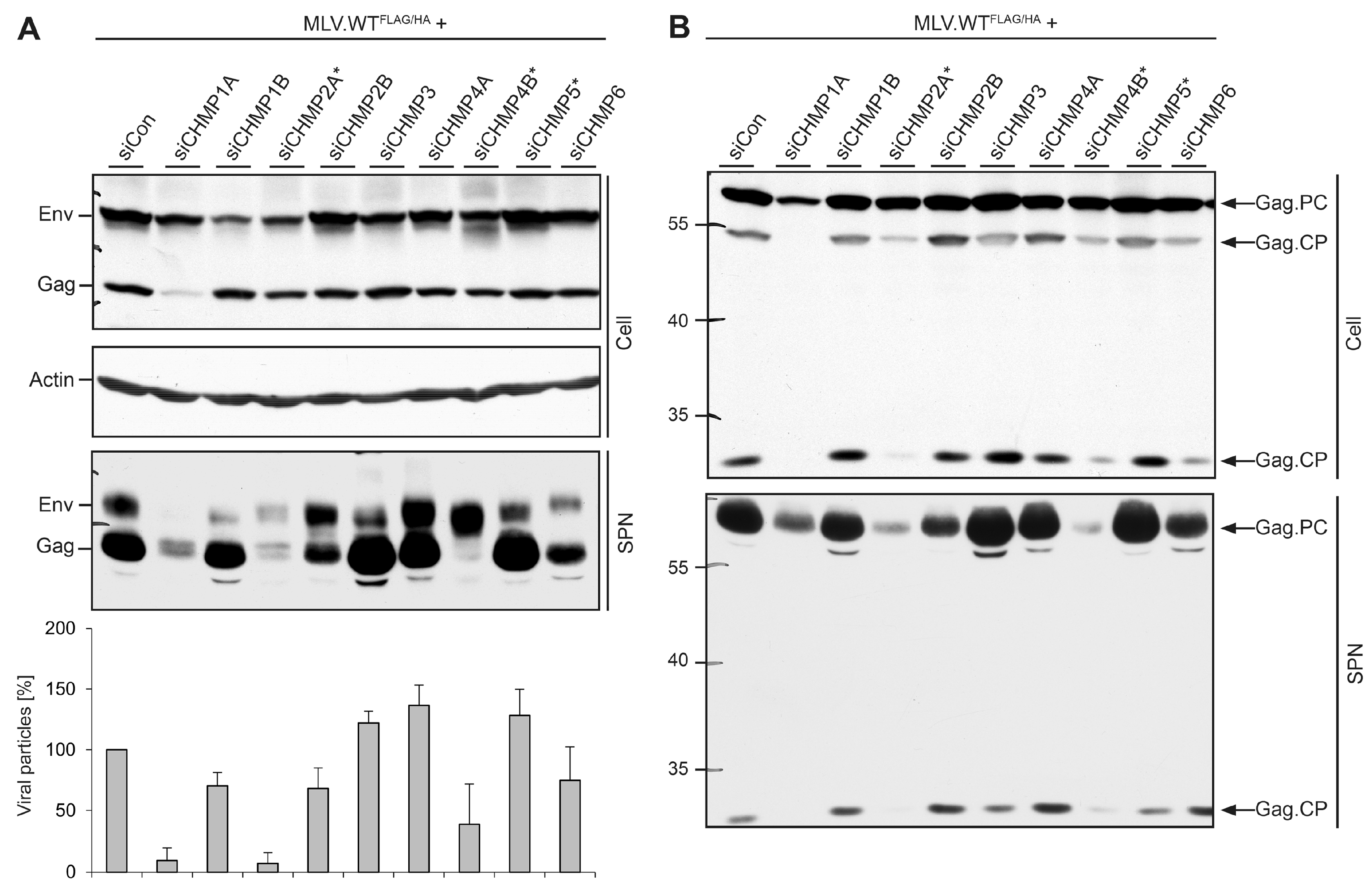
3.3. Role of ESCRT-III in MLV VLP Release
3.4. Role of ESCRT-III in MLV Virion Release
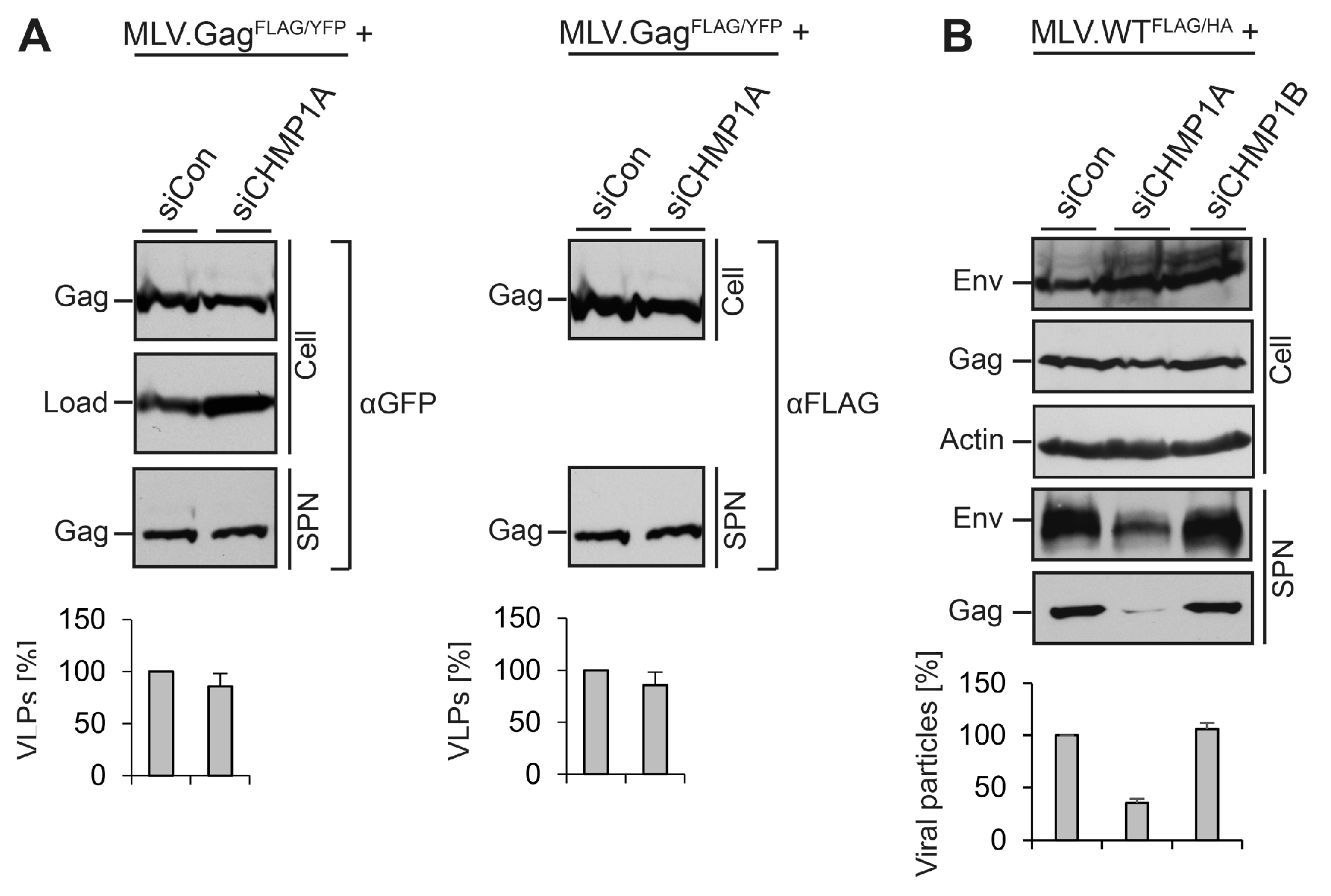
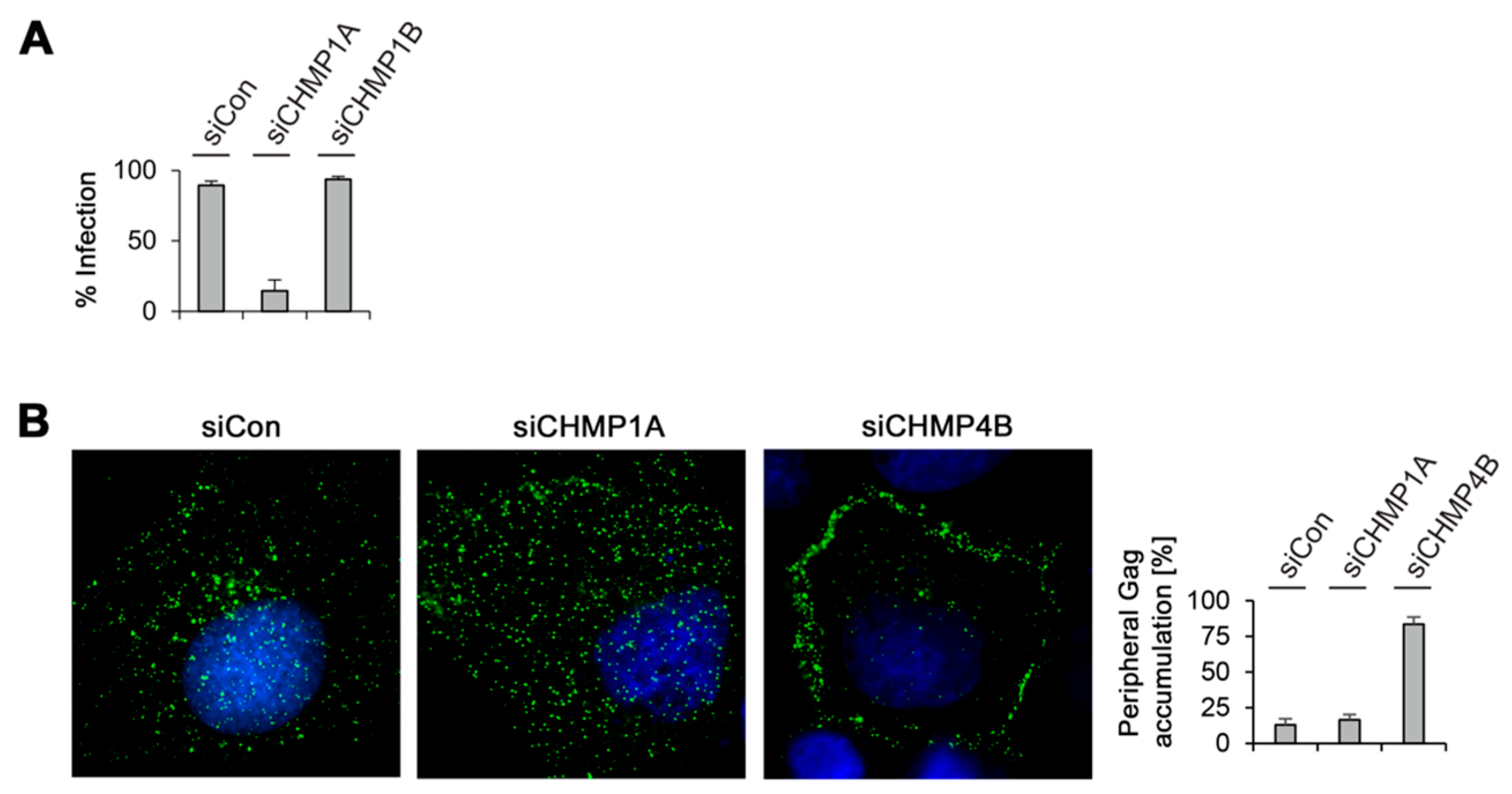
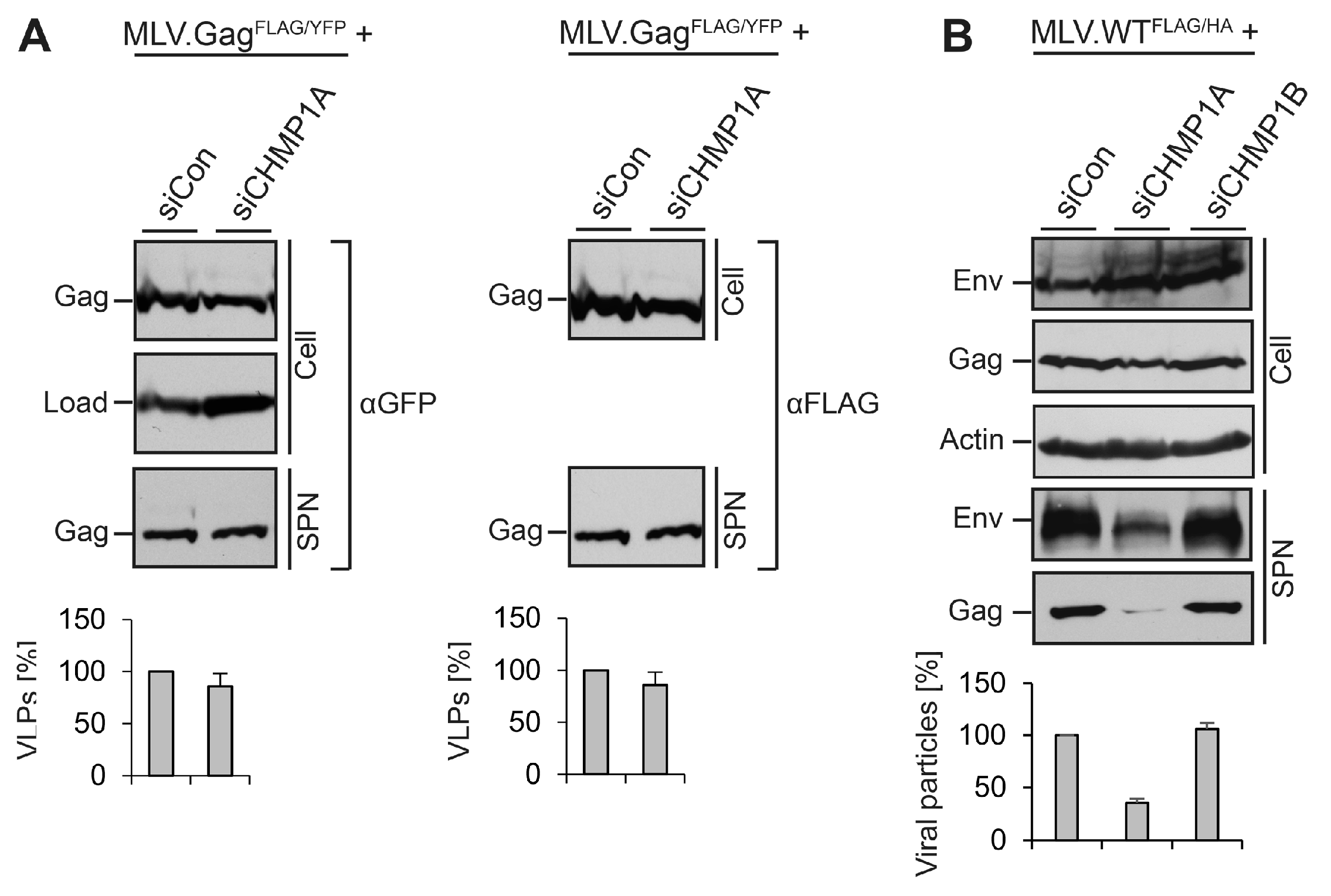
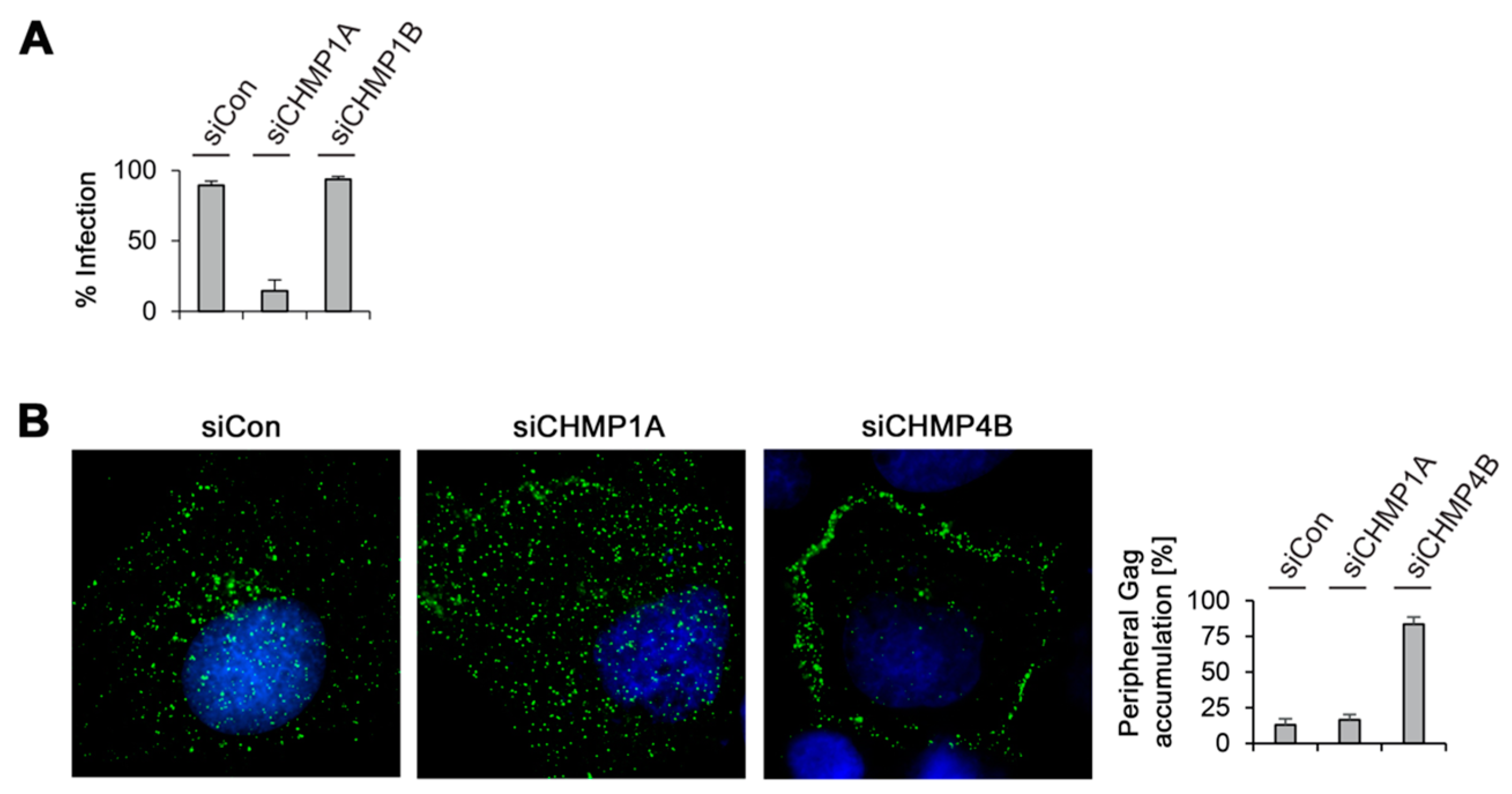
3.5. Divergent Role of CHMP1A in MLV VLP and Virion Release
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MLV | Murine leukemia virus |
| ESCRT | endosomal sorting complex required for transport |
| VLP | viral-like particles |
| MVB | multivesicular body |
| HIV-1 | human immunodeficiency virus type 1 |
| RSV | Rous sarcoma virus |
| HBV | Hepatitis B virus |
| ASV | Avian sarcoma virus |
| EIAV | equine infectious anemia virus |
| ART | Arrestin-related trafficking |
| YFP | yellow fluorescent protein |
| Pol | Polymerase |
| Env | Envelope |
| ORFs | open reading frames |
| aa | Amino acid |
| WB | Western blotting |
| qRT | quantitative reverse transcriptase |
| MA | Matrix |
| CA | Capsid |
| NC | Nucleocapsid |
| MIT | Microtubule-interacting and transport |
| MIM | MIT-interacting motifs |
References
- McCullough, J.; Colf, L.A.; Sundquist, W.I. Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 2013, 82, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Neil, S.J. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 2011, 9, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Morita, E. Differential requirements of mammalian ESCRTs in multivesicular body formation, virus budding and cell division. FEBS J. 2012, 279, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Peel, S.; Macheboeuf, P.; Martinelli, N.; Weissenhorn, W. Divergent pathways lead to ESCRT-III-catalyzed membrane fission. Trends Biochem. Sci. 2011, 36, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Langelier, C.; von Schwedler, U.K.; Fisher, R.D.; de Domenico, I.; White, P.L.; Hill, C.P.; Kaplan, J.; Ward, D.; Sundquist, W.I. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 2006, 80, 9465–9480. [Google Scholar] [CrossRef] [PubMed]
- Yorikawa, C.; Shibata, H.; Waguri, S.; Hatta, K.; Horii, M.; Katoh, K.; Kobayashi, T.; Uchiyama, Y.; Maki, M. Human CHMP6, a myristoylated ESCRT-III protein, interacts directly with an ESCRT-II component EAP20 and regulates endosomal cargo sorting. Biochem. J. 2005, 387, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. Late budding domains and host proteins in enveloped virus release. Virology 2006, 344, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Freed, E.O. Retrovirus budding. Virus Res. 2004, 106, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Gottlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Wills, J.W.; Cameron, C.E.; Wilson, C.B.; Xiang, Y.; Bennett, R.P.; Leis, J. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol. 1994, 68, 6605–6618. [Google Scholar] [PubMed]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55Gag. Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; Hurley, J.H. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc. Natl. Acad. Sci. USA 2012, 109, 16928–16933. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Ip, N.C.; Prestwood, L.J.; Abbink, T.E.; Lever, A.M. Evidence that the endosomal sorting complex required for transport-II (ESCRT-II) is required for efficient human immunodeficiency virus-1 (HIV-1) production. Retrovirology 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Stieler, J.T.; Prange, R. Involvement of ESCRT-II in hepatitis B virus morphogenesis. PLoS ONE 2014, 9, e91279. [Google Scholar] [CrossRef] [PubMed]
- Pincetic, A.; Medina, G.; Carter, C.; Leis, J. Avian sarcoma virus and human immunodeficiency virus, type 1 use different subsets of ESCRT proteins to facilitate the budding process. J. Biol. Chem. 2008, 283, 29822–29830. [Google Scholar] [CrossRef] [PubMed]
- Segura-Morales, C.; Pescia, C.; Chatellard-Causse, C.; Sadoul, R.; Bertrand, E.; Basyuk, E. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J. Biol. Chem. 2005, 280, 27004–27012. [Google Scholar] [CrossRef] [PubMed]
- Jadwin, J.A.; Rudd, V.; Sette, P.; Challa, S.; Bouamr, F. Late domain-independent rescue of a release-deficient Moloney murine leukemia virus by the ubiquitin ligase itch. J. Virol. 2010, 84, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Martin-Serrano, J. Multiple interactions between the ESCRT machinery and arrestin-related proteins: Implications for PPXY-dependent budding. J. Virol. 2011, 85, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Kikonyogo, A.; Bouamr, F.; Vana, M.L.; Xiang, Y.; Aiyar, A.; Carter, C.; Leis, J. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 2001, 98, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.R.; Popova, E.; Yamanaka, H.; Kim, H.C.; Huibregtse, J.M.; Gottlinger, H. Rescue of HIV-1 release by targeting widely divergent NEDD4-type ubiquitin ligases and isolated catalytic HECT domains to Gag. PLoS Pathog. 2010, 6, e1001107. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Morita, E.; von Schwedler, U.; Muller, B.; Krausslich, H.G.; Sundquist, W.I. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Li, X.; Goff, S.P. Mutations altering the moloney murine leukemia virus p12 Gag protein affect virion production and early events of the virus life cycle. EMBO J. 1999, 18, 4700–4710. [Google Scholar] [CrossRef] [PubMed]
- Rein, A. Murine leukemia viruses: Objects and organisms. Adv. Virol. 2011, 2011, 403419. [Google Scholar] [CrossRef] [PubMed]
- Andrawiss, M.; Takeuchi, Y.; Hewlett, L.; Collins, M. Murine leukemia virus particle assembly quantitated by fluorescence microscopy: Role of Gag-Gag interactions and membrane association. J. Virol. 2003, 77, 11651–11660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Ingmundson, A.; Horner, S.M.; Cicchetti, G.; Allen, P.G.; Pypaert, M.; Cunningham, J.M.; Mothes, W. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 2003, 4, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Costanzi, E.; Haas, M.; Verma, I.M. The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996, 70, 5701–5705. [Google Scholar] [PubMed]
- Yueh, A.; Goff, S.P. Phosphorylated serine residues and an arginine-rich domain of the moloney murine leukemia virus p12 protein are required for early events of viral infection. J. Virol. 2003, 77, 1820–1829. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Erlwein, O.; Buchholz, C.J.; Schnierle, B.S. The proline-rich region of the ecotropic Moloney murine leukaemia virus envelope protein tolerates the insertion of the green fluorescent protein and allows the generation of replication-competent virus. J. Gen. Virol. 2003, 84, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Seed, B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003, 31, 1–8. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Eastman, S.W.; Chung, W.; Bieniasz, P.D. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 2005, 168, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Goliand, I.; Nachmias, D.; Gershony, O.; Elia, N. Inhibition of ESCRT-II-CHMP6 interactions impedes cytokinetic abscission and leads to cell death. Mol. Biol. Cell 2014, 25, 3740–3748. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Doring, T.; Prange, R. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J. Virol. 2007, 81, 9050–9060. [Google Scholar] [CrossRef] [PubMed]
- Agromayor, M.; Martin-Serrano, J. Interaction of AMSH with ESCRT-III and deubiquitination of endosomal cargo. J. Biol. Chem. 2006, 281, 23083–23091. [Google Scholar] [CrossRef] [PubMed]
- Houzet, L.; Gay, B.; Morichaud, Z.; Briant, L.; Mougel, M. Intracellular assembly and budding of the Murine Leukemia Virus in infected cells. Retrovirology 2006, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zang, T.; Wilson, S.J.; Johnson, M.C.; Bieniasz, P.D. Clathrin facilitates the morphogenesis of retrovirus particles. PLoS Pathog. 2011, 7, e1002119. [Google Scholar] [CrossRef] [PubMed]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Shimazu, Y.; Yoshida, T.; Sakaguchi, T. The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J. Virol. 2007, 81, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Gosselin-Grenet, A.S.; Marq, J.B.; Abrami, L.; Garcin, D.; Roux, L. Sendai virus budding in the course of an infection does not require Alix and VPS4A host factors. Virology 2007, 365, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.F.; Tsai, M.L.; Huang, J.Y.; Chang, Y.S.; Shih, C. The dual role of an ESCRT-0 component HGS in HBV transcription and naked capsid secretion. PLoS Pathog. 2015, 11, e1005123. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Sandrin, V.; McCullough, J.; Katsuyama, A.; Baci Hamilton, I.; Sundquist, W.I. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 2011, 9, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M.; Vaughn, M.B.; Shiflett, S.L.; White, P.L.; Pollock, A.L.; Hill, J.; Schnegelberger, R.; Sundquist, W.I.; Kaplan, J. The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J. Biol. Chem. 2005, 280, 10548–10555. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Shibata, H.; Hatta, K.; Maki, M. CHMP4b is a major binding partner of the ALG-2-interacting protein Alix among the three CHMP4 isoforms. Arch. Biochem. Biophys. 2004, 421, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Bodon, G.; Chassefeyre, R.; Pernet-Gallay, K.; Martinelli, N.; Effantin, G.; Hulsik, D.L.; Belly, A.; Goldberg, Y.; Chatellard-Causse, C.; Blot, B.; et al. Charged multivesicular body protein 2B (CHMP2B) of the endosomal sorting complex required for transport-III (ESCRT-III) polymerizes into helical structures deforming the plasma membrane. J. Biol. Chem. 2011, 286, 40276–40286. [Google Scholar] [CrossRef] [PubMed]
- Effantin, G.; Dordor, A.; Sandrin, V.; Martinelli, N.; Sundquist, W.I.; Schoehn, G.; Weissenhorn, W. ESCRT-III CHMP2A and CHMP3 form variable helical polymers in vitro and act synergistically during HIV-1 budding. Cell Microbiol. 2013, 15, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Stuchell-Brereton, M.D.; Skalicky, J.J.; Kieffer, C.; Karren, M.A.; Ghaffarian, S.; Sundquist, W.I. ESCRT-III recognition by VPS4 ATPases. Nature 2007, 449, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Wunder, C.; Lippincott-Schwartz, J.; Hurley, J.H. Membrane scission by the ESCRT-III complex. Nature 2009, 458, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.; Merrill, S.A.; Hanson, P.I. Novel interactions of ESCRT-III with LIP5 and VPS4 and their implications for ESCRT-III disassembly. Mol. Biol. Cell 2008, 19, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Vild, C.J.; Li, Y.; Guo, E.Z.; Liu, Y.; Xu, Z. A novel mechanism of regulating the ATPase VPS4 by its cofactor LIP5 and the endosomal sorting complex required for transport (ESCRT)-III protein CHMP5. J. Biol. Chem. 2015, 290, 7291–7303. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, V.; Sundquist, W.I. ESCRT requirements for EIAV budding. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.D.; Piper, S.C.; Bright, N.A.; Evans, J.L.; Boname, J.M.; Bowers, K.; Lehner, P.J.; Luzio, J.P. A non-canonical ESCRT pathway, including histidine domain phosphotyrosine phosphatase (HD-PTP), is used for down-regulation of virally ubiquitinated MHC class I. Biochem. J. 2015, 471, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Maemoto, Y.; Shibata, H.; Maki, M. Identification of phosphorylation sites in the C-terminal region of charged multivesicular body protein 1A (CHMP1A). Biosci. Biotechnol. Biochem. 2013, 77, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Tsang, H.T.; Connell, J.W.; Brown, S.E.; Thompson, A.; Reid, E.; Sanderson, C.M. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics 2006, 88, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Park, M. Posttranslational modifications of CHMP1A and their effects on human pancreatic cancer cells. In Proceedings of the AACR Special Conference on Pancreatic Cancer: Innovations in Research and Treatment, New Orleans, LA, USA, 18–21 May 2014; AACR: Philadelphia, PA, USA, 2015; Volume 75. [Google Scholar]
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science 2012, 336, 220–225. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartusch, C.; Prange, R. ESCRT Requirements for Murine Leukemia Virus Release. Viruses 2016, 8, 103. https://doi.org/10.3390/v8040103
Bartusch C, Prange R. ESCRT Requirements for Murine Leukemia Virus Release. Viruses. 2016; 8(4):103. https://doi.org/10.3390/v8040103
Chicago/Turabian StyleBartusch, Christina, and Reinhild Prange. 2016. "ESCRT Requirements for Murine Leukemia Virus Release" Viruses 8, no. 4: 103. https://doi.org/10.3390/v8040103
APA StyleBartusch, C., & Prange, R. (2016). ESCRT Requirements for Murine Leukemia Virus Release. Viruses, 8(4), 103. https://doi.org/10.3390/v8040103