
A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae


Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Sample, Multiplication and Purification
2.2. Analysis of Permissiveness of BrMV in Different Amoebae
2.3. Genome Sequencing, Assembly and Annotation
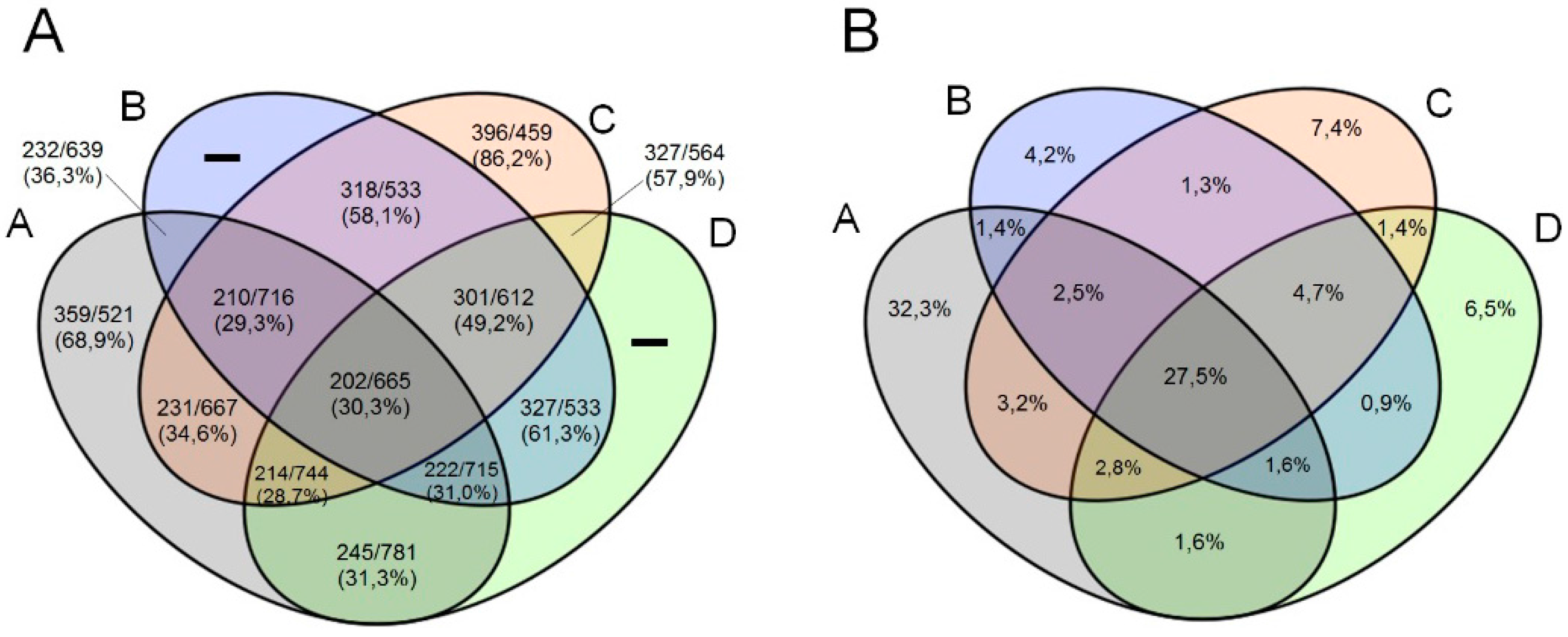
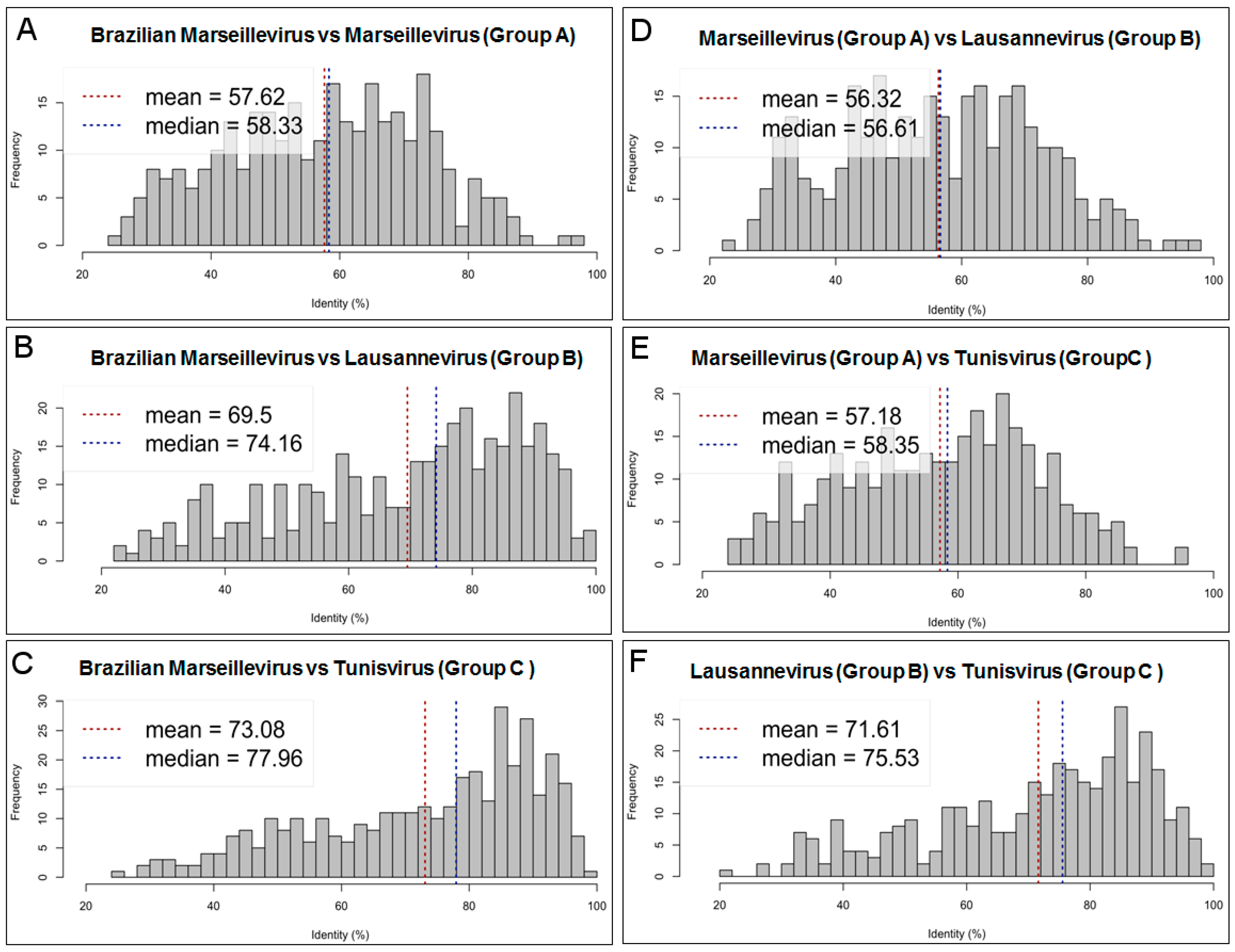
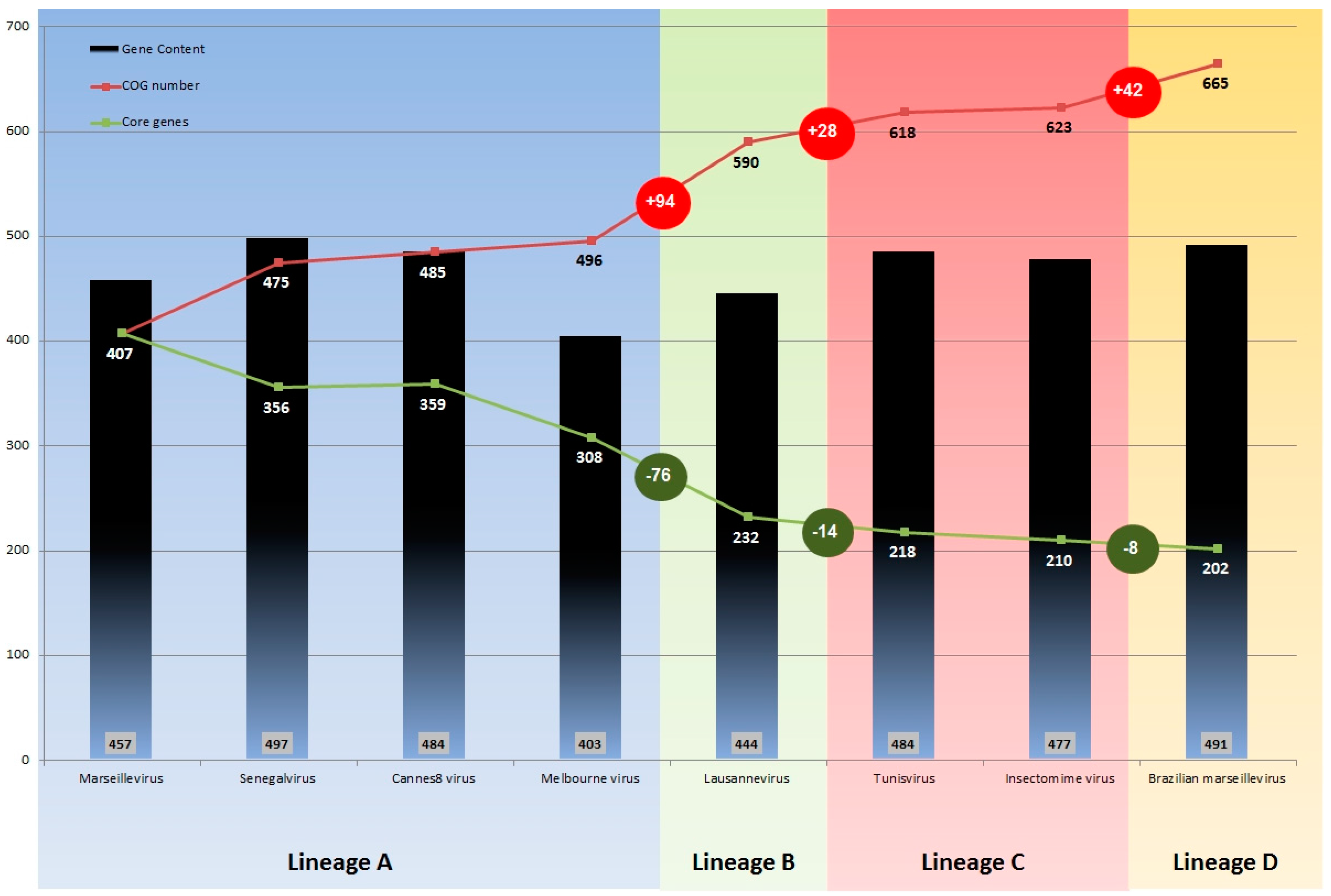
2.4. Comparative Genomic and Pan-Genome Analysis
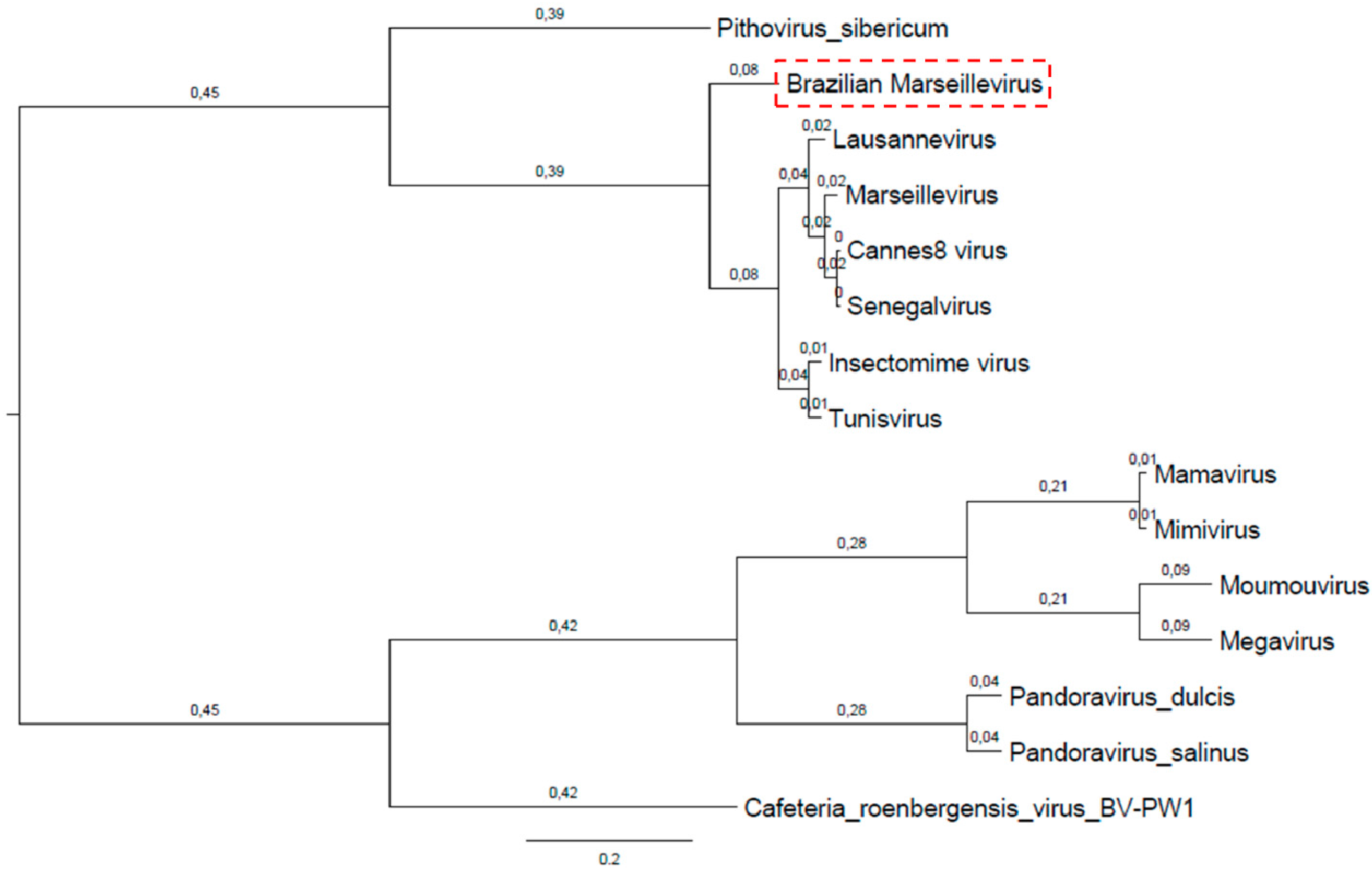
2.5. Phylogeny
3. Results
3.1. Brazilian Marseillevirus
3.2. Brazilian Marseillevirus Genome and Annotation
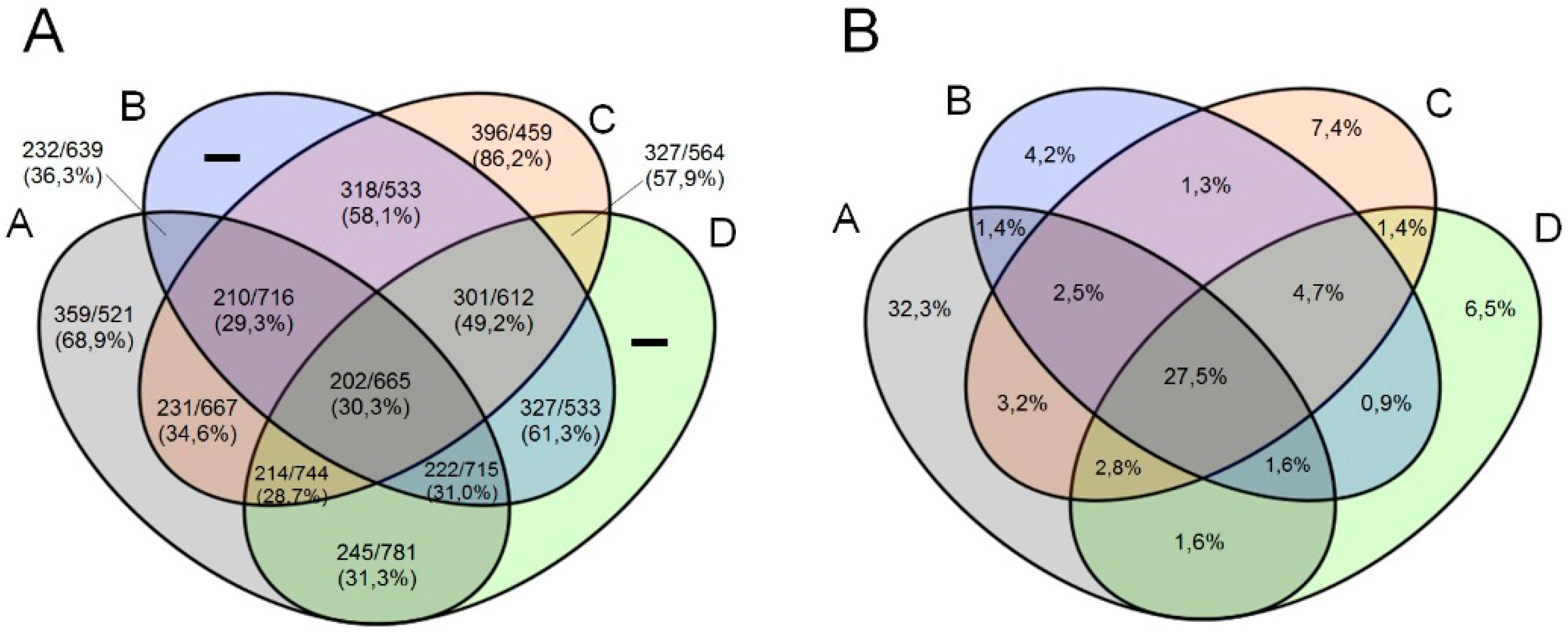
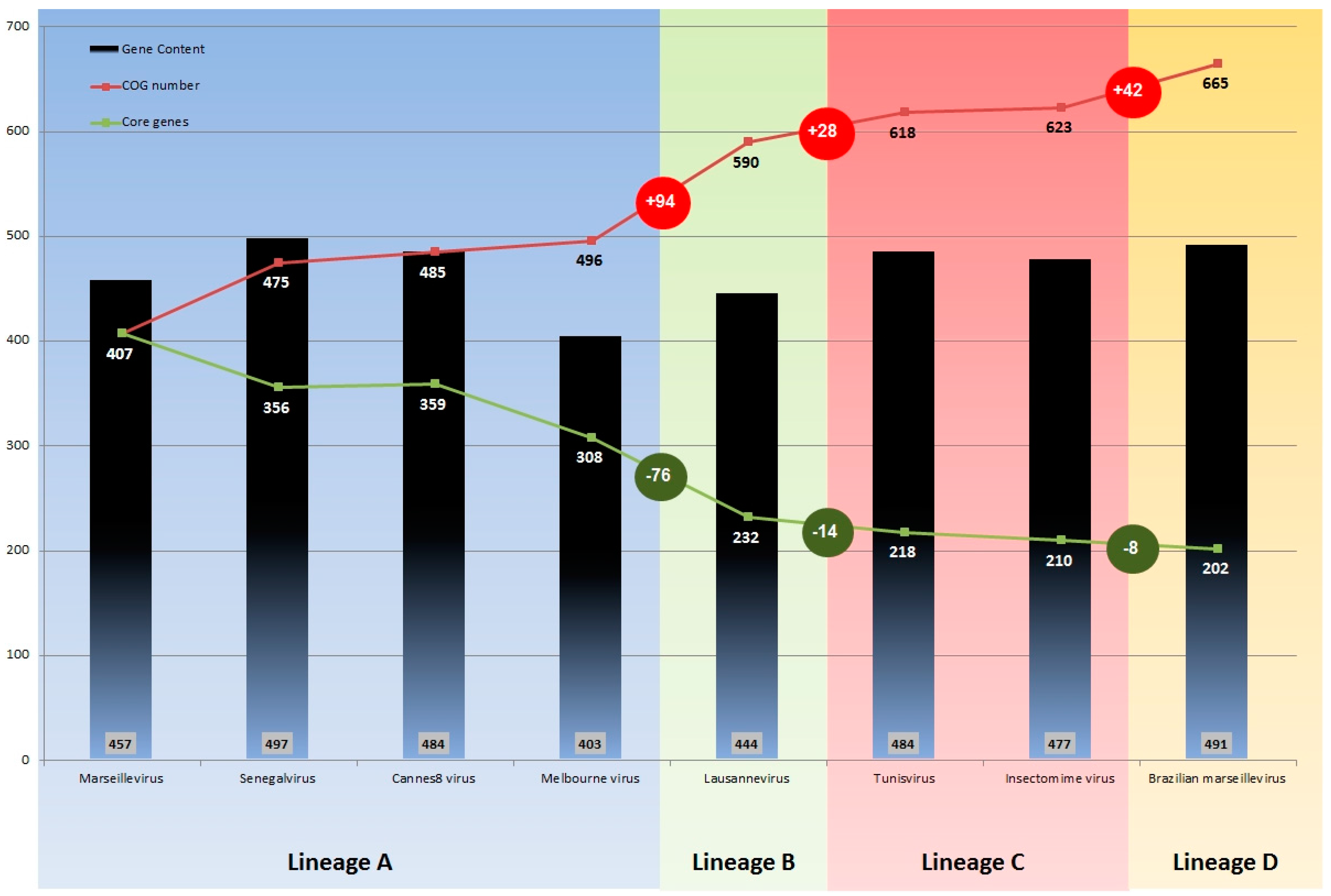
3.3. Comparative Genome and Pan-Genome Analysis
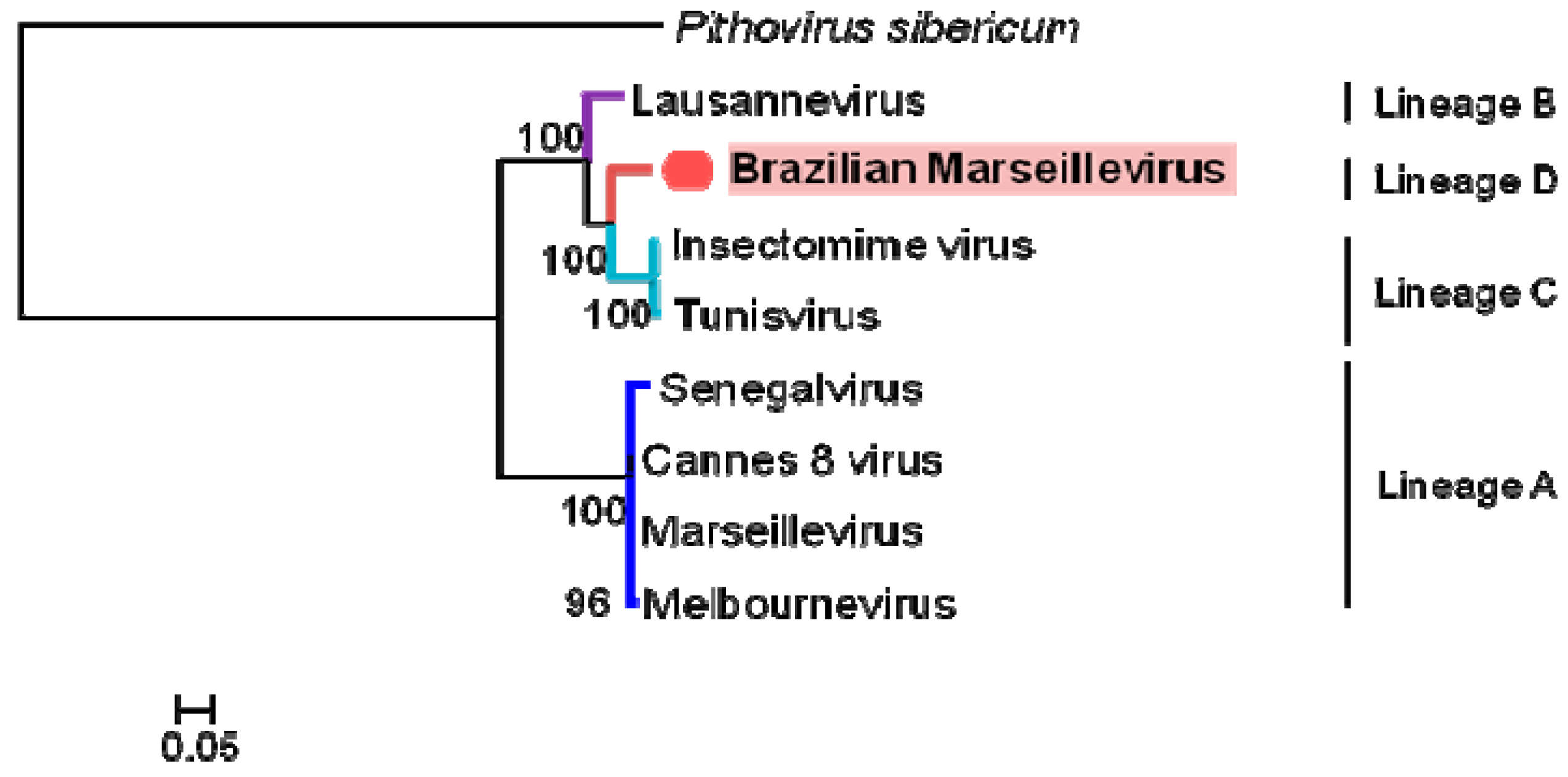
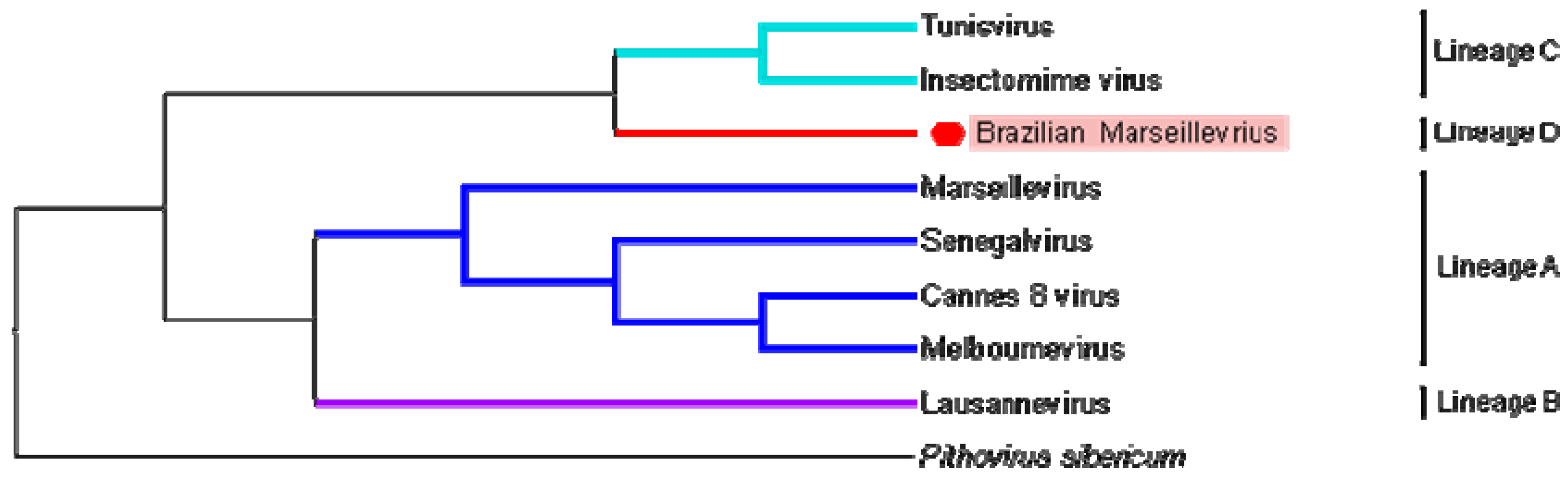
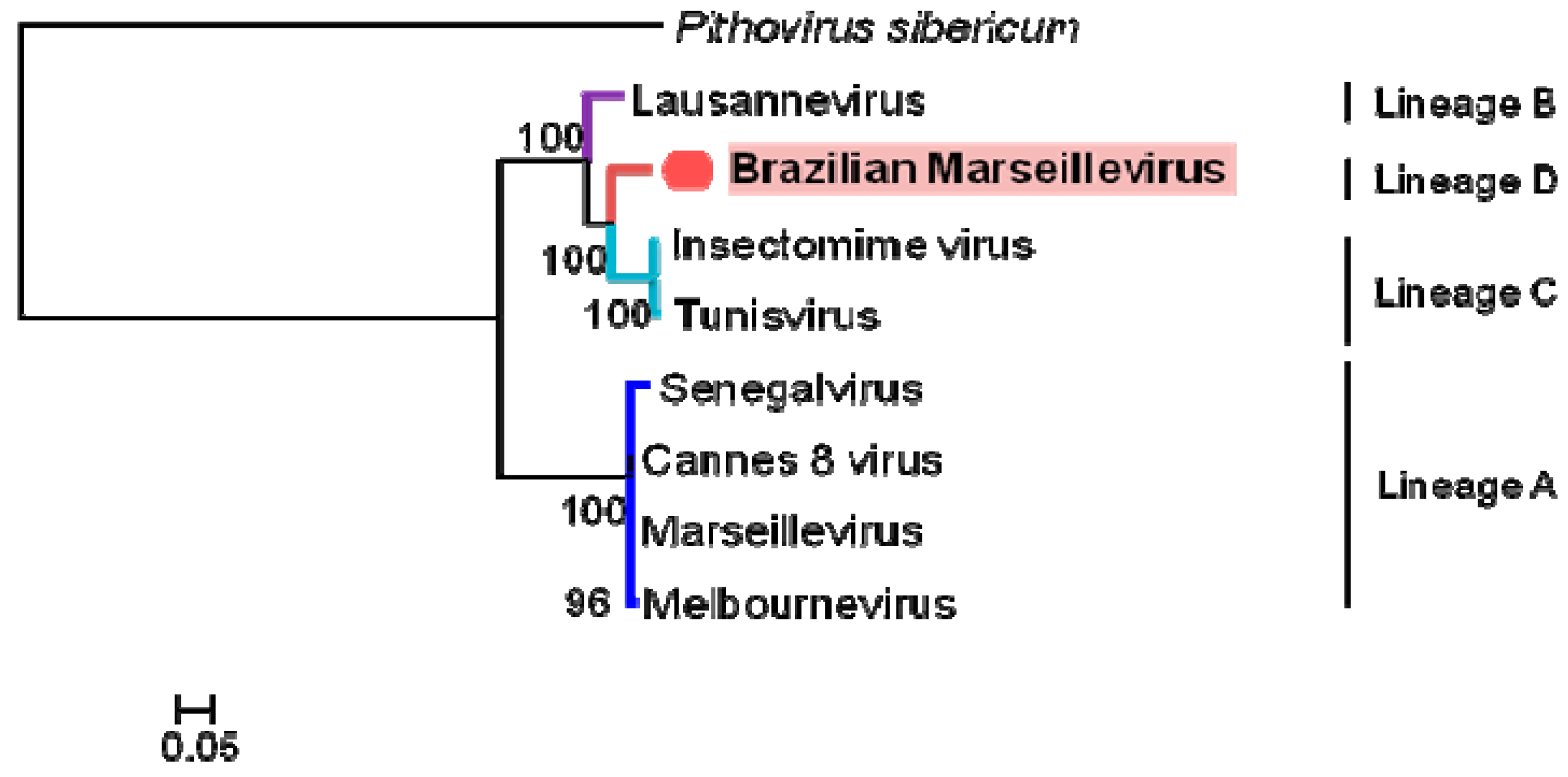
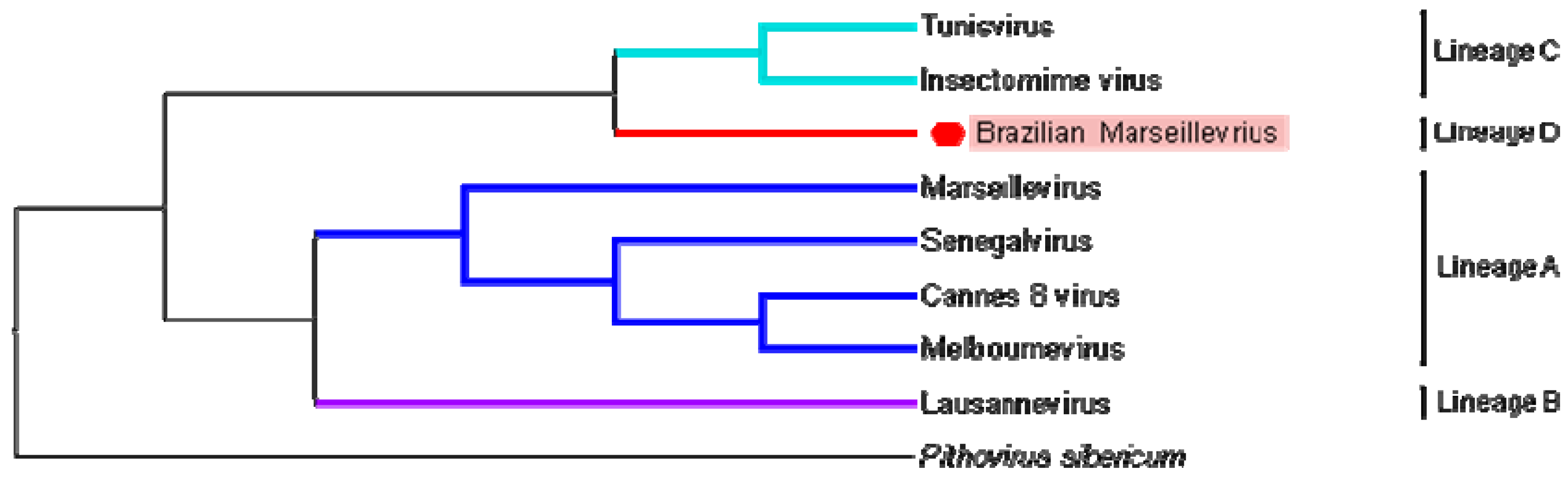
3.4. Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Etten, J.L.; Meints, R.H. Giant viruses infecting algae. Annu. Rev. Microbiol. 1999, 53, 447–494. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; Raoult, D. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [PubMed]
- Yoosuf, N.; Yutin, N.; Colson, P.; Shabalinan, S.A.; Pagnier, I.; Robert, C.; Azza, S.; Klose, T.; Wong, J.; Rossmann, M.G.; et al. Related giant viruses in distant locations and different habitats: Acanthamoeba polyphaga moumouvirus represents a third lineage of the Mimiviridae that is close to the megavirus lineage. Genome Biol. 2012, 4, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Pagnier, I.; Reteno, D.G.I.; Saadi, H.; Boughalmi, M.; Gaia, M.; Slimani, M.; Ngounga, T.; Bekliz, M.; Colson, P.; Raoult, D.; et al. A Decade of Improvements in Mimiviridae and Marseilleviridae Isolation from Amoeba. Intervirology 2013, 56, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Saadi, H.; Reteno, D.G.; Colson, P.; Aherfi, S.; Minodier, P.; Pagnier, I.; Raoult, D.; La Scola, B. Shan virus: A new mimivirus isolated from the stool of a Tunisian patient with pneumonia. Intervirology 2013, 56, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Saadi, H.; Pagnier, I.; Colson, P.; Cherif, J.K.; Beji, M.; Boughalmi, M.; Azza, S.; Armstrong, N.; Robert, C.; Fournous, G.; et al. First isolation of Mimivirus in a patient with pneumonia. Clin. Infect. Dis. 2013, 57, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba Viruses with Genomes Up to 2.5 Mb Reaching That of Parasitic Eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Scheid, P.; Balczun, C.; Schaub, G.A. Some secrets are revealed: Parasitic keratitis amoebae as vectors of the scarcely described pandoraviruses to humans. Parasitol. Res. 2014, 113, 3759–3764. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA. 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Reteno, D.G.; Benamar, S.; Khalil, J.B.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; La Scola, B. Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 2015, 89, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/taxonomyHistory.asp?taxnode_id=20133581&taxa_name=Marseilleviri-dae (accessed on 1 November 2015).
- Colson, P.; Pagnier, I.; Yoosuf, N.; Fournous, G.; La Scola, B.; Raoult, D. “Marseilleviridae”, a new family of giant viruses infecting amoebae. Arch. Virol. 2013, 158, 915–920. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Campocasso, A.; N’Dong, R.; Fournous, G.; Barrassi, L.; Flaudrops, C.; Raoult, D. Tentative characterization of new environmental giant viruses by MALDI-TOF mass spectrometry. Intervirology 2010, 53, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.; Bertelli, C.; Collyn, F.; Casson, N.; Telenti, A.; Goesmann, A.; Croxatto, A.; Greub, G. Lausannevirus, a giant amoebal virus encoding histone doublets. Environ. Microbiol. 2011, 13, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1893. [Google Scholar] [CrossRef] [PubMed]
- Aherfi, S.; Pagnier, I.; Fournous, G.; Raoult, D.; La Scola, B.; Colson, P. Complete genome sequence of Cannes 8 virus, a new member of the proposed family “Marseilleviridae”. Virus Genes 2013, 47, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Boughalmi, M.; Saadi, H.; Pagnier, I.; Colson, P.; Fournous, G.; Raoult, D.; La Scola, B. 2013 High-throughput isolation of giant viruses of the Mimiviridae and Marseilleviridae families in the Tunisian environment. Environ. Microbiol. 2013, 15, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Boughalmi, M.; Pagnier, I.; Aherfi, S.; Colson, P.; Raoult, D.; La Scola, B. First Isolation of a Marseillevirus in the Diptera Syrphidae Eristalis tenax. Intervirology 2013, 56, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Popgeorgiev, N.; Boyer, M.; Fancello, L.; Monteil, S.; Robert, C.; Rivet, R.; Nappez, C.; Azza, S.; Chiaroni, J.; Raoult, D.; et al. Marseillevirus-like virus recovered from blood donated by asymptomatic humans. J. Infect. Dis. 2013, 208, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Colson, P.; Raoult, D.; Koonin, E.V. Mimiviridae: Clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 2013, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Doutre, G.; Philippe, N.; Abergel, C.; Claverie, J.M. Genome analysis of the first Marseilleviridae representative from Australia indicates that most of its genes contribute to virus fitness. J. Virol. 2014, 88, 14340–14349. [Google Scholar] [CrossRef] [PubMed]
- Aherfi, S.; Boughalmi, M.; Pagnier, I.; Fournous, G.; La Scola, B.; Raoult, D.; Colson, P. Complete genome sequence of Tunisvirus, a new member of the proposed family Marseilleviridae. Arch. Virol. 2014, 159, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Dornas, F.P.; Khalil, J.Y.; Pagnier, I.; Raoult, D.; Abrahão, J.; La Scola, B. Isolation of new Brazilian giant viruses from environmental samples using a panel of protozoa. Front. Microbiol. 2015, 6, 1086. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Fgenes, V. Available online: http://linux1.softberry.com/berry.phtml?topic=virus&group=programs&subgroup=gfindv (accessed on 1 November 2015).
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadeze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Raoult, D.; Koonin, E.V. Eukaryotic large nucleo-cytoplasmic DNA viruses: Clusters of orthologous genes and reconstruction of viral genome evolution. Virol. J. 2009, 17, 223. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomics sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 28. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Mackey, A.J.; Stoeckert, C.J., Jr.; Ross, D.S. OrthoMCL-DB: Querying a comprehensive multi-species collection of orthologs groups. Nucleic Acids Res. 2006, 34, D363–D368. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of orthologs groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. By passing Cultivation to identify bacterial species. Microbe 2014, 9, 111–118. [Google Scholar]
- BLASTclust website. Available online: http://www.ncbi.nlm.nih.gov/Web/Newsltr/Spring04/blastlab.html (accessed on 4 March 2016).
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [PubMed]
- FigTree website. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 4 March 2016).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 7, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Whidden, C.; Zeh, N.; Beiko, R.G. Supertress based on the subtree prune-and-regraft distance. Syst. Biol. 2014, 63, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Baroudy, B.M.; Venkatesan, S.; Moss, B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell 1982, 28, 315–324. [Google Scholar] [CrossRef]
- Suzan-Monti, M.; La Scola, B.; Raoult, D. Genomic and evolutionary aspects of Mimivirus. Virus Res. 2005, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORFan ID | Protein Identification | Organism (1st and 2nd Best Hits) | Interation | Max Score | Total Score | Query Cover | e-Value | Ident | Accession Number |
|---|---|---|---|---|---|---|---|---|---|
| ORF_L46 | Methyltransferase | Rhizobium leguminosarum | 3 | 166 | 166 | 93% | 4 × 10−45 | 12% | WP_025395836.1 |
| Methyltransferase | Sinorhizobium meliloti | 165 | 165 | 93% | 7 × 10−45 | 12% | WP_015242269.1 | ||
| ORF_R48 | Transglycosylase | Streptomyces fulvoviolaceus | 2 | 256 | 256 | 93% | 4 × 10−78 | 24% | WP_030603268.1 |
| Transglycosylase | Streptomyces sp. WM6386 | 229 | 229 | 93% | 9 × 10−68 | 22% | WP_046261419.1 | ||
| ORF_R84 | hypothetical protein | Aquimarina megaterium | 3 | 58,1 | 58,1 | 20% | 8 × 10−5 | 18% | WP_025666489.1 |
| hypothetical protein | Novosphingobium tardaugens | 83,5 | 83,5 | 20% | 2 × 10−15 | 34% | WP_021691485.1 | ||
| ORF_R86 | cysteine protease ATG4B | Apaloderma vittatum | 2 | 145 | 145 | 83% | 10−38 | 23% | KFP75383.1 |
| cysteine protease ATG4B | Tyto alba | 135 | 135 | 83% | 10−34 | 23% | KFV59860.1 | ||
| ORF_L94 | cytidine and deoxycytidylate deaminase | Acanthocystis turfacea Chlorella virus | 3 | 102 | 102 | 77% | 2 × 10−25 | 24% | AGE49630.1 |
| cytidine and deoxycytidylate deaminase | Acanthocystis turfacea Chlorella virus | 101 | 101 | 69% | 7 × 10-25 | 23% | AGE55798.1 | ||
| ORF_R115 | DNA mismatch repair protein MutL | Deinococcus deserti | 3 | 170 | 170 | 95% | 2 × 10−46 | 18% | WP_012693648.1 |
| DNA mismatch repair protein MutL | Deinococcus deserti | 145 | 145 | 80% | 2 × 10−39 | 22% | WP_034401941.1 | ||
| ORF_R123 | protein-L-isoaspartate O-methyltransferase | Frankia sp. CcI3 | 2 | 124 | 124 | 64% | 8 × 10−31 | 29% | WP_049761110.1 |
| protein-L-isoaspartate O-methyltransferase | Frankia sp. BMG5.23 | 124 | 124 | 64% | 10−30 | 29% | WP_043591788.1 | ||
| ORF_R124 | ABC transporter substrate-binding protein | Rheinheimera texasensis | 2 | 170 | 170 | 83% | 10−48 | 29% | WP_031569037.1 |
| ABC transporter substrate-binding protein | Vibrio gazogenes | 127 | 127 | 87% | 2 × 10-32 | 23% | WP_027693958.1 | ||
| ORF_L133 | conserved signaling intermediate in Toll pathway | Nannospalax galili | 2 | 142 | 142 | 87% | 4 × 10−37 | 27% | XP_008831822.1 |
| conserved signaling intermediate in Toll pathway | Nannospalax galili | 141 | 141 | 87% | 7 × 10−37 | 27% | XP_008831824.1 | ||
| ORF_R218 | diguanylate phosphodiesterase | Vibrionales bacterium SWAT-3 | 3 | 147 | 147 | 70% | 3 × 10−38 | 18% | WP_008217346.1 |
| diguanylate phosphodiesterase | Vibrio crassostreae | 145 | 145 | 70% | 8 × 10−38 | 19% | WP_048663292.1 | ||
| ORF_R239 | rho GTPase-activating protein 1 | Equus caballus | 2 | 107 | 107 | 41% | 2 × 10−24 | 32% | XP_001490021.2 |
| rho GTPase-activating protein 1 isoform X2 | Equus caballus | 107 | 107 | 41% | 2 × 10−24 | 32% | XP_005598135.1 | ||
| ORF_L254 | leucine-rich repeat-containing protein 9-like | Lepisosteus oculatus | 2 | 130 | 130 | 64% | 2 × 10−31 | 28% | XP_006632383.1 |
| Peroxidase | Actinomyces sp. oral taxon 171 | 122 | 122 | 64% | 10−30 | 29% | WP_009394707.1 | ||
| ORF_L292 | coiled-coil and C2 domain-containing protein 1A isoform X5 | Papio anubis | 3 | 112 | 112 | 62% | 10−25 | 29% | XP_009191945.1 |
| coiled-coil and C2 domain-containing protein 1A isoform X8 | Cercocebus atys | 112 | 112 | 62% | 10−25 | 29% | XP_011949500.1 | ||
| ORF_L300 | ATP-dependent helicase | Oenococcus oeni | 3 | 120 | 120 | 96% | 3 × 10−28 | 20% | WP_002822412.1 |
| ATP-dependent helicase/nuclease subunit A | Fructobacillus ficulneus | 117 | 117 | 94% | 7 × 10−28 | 14% | GAO99721.1 | ||
| ORF_R303 | glycoside hydrolase family 9 | Ruminiclostridium thermocellum | 4 | 104 | 104 | 93% | 4 × 10−23 | 26% | WP_023062725.1 |
| glycosyl hydrolase | Ruminiclostridium thermocellum | 103 | 103 | 93% | 6 × 10−23 | 24% | WP_020457778.1 | ||
| ORF_R304 | aggrecan core protein | Callorhinchus milii | 3 | 118 | 696 | 100% | 10−27 | 24% | XP_007906559.1 |
| aggrecan core protein | Corvus brachyrhynchos | 103 | 926 | 100% | 2 × 10−22 | 30% | XP_008638374.1 | ||
| ORF_L309 | peptide synthetase | Microcoleus sp. PCC 7113 | 6 | 424 | 424 | 97% | 2 × 10−136 | 13% | WP_041780594.1 |
| ORF_L324 | N-acetylneuraminic acid mutarotase | Vibrio variabilis | 3 | 212 | 212 | 78% | 6 × 10−63 | 15% | WP_038216942.1 |
| N-acetylneuraminic acid mutarotase | Vibrio sinaloensis | 211 | 211 | 78% | 2 × 10−62 | 15% | WP_039481213.1 | ||
| ORF_L337 | cytochrome C | Coleofasciculus chthonoplastes | 3 | 178 | 178 | 99% | 2 × 10−50 | 17% | WP_006101072.1 |
| cytochrome C | Mastigocladopsis repens | 177 | 177 | 99% | 5 × 10−50 | 20% | WP_017318476.1 | ||
| ORF_R351 | extracellular dioxygenase | Aspergillus kawachii | 3 | 151 | 151 | 61% | 4 × 10−39 | 25% | CCX09620.1 |
| Intradiol ring-cleavage dioxygenase | Penicillium expansum | 143 | 143 | 67% | 3 × 10−36 | 17% | KGO45757.1 | ||
| ORF_L367 | regulator of telomere elongation helicase 1 | Cavia porcellus | 3 | 170 | 170 | 94% | 10−45 | 17% | XP_013000054.1 |
| regulator of telomere elongation helicase 1 | Charadrius vociferus | 169 | 169 | 94% | 2 × 10−45 | 18% | KGM00023.1 | ||
| ORF_L375 | putative protein binding surface, polypeptide binding | Albugo laibachii Nc14 | 4 | 137 | 137 | 90% | 2 × 10−36 | 25% | CCA16909.1 |
| ORF_R485 | ephrin type-B receptor 4 | Nomascus leucogenys | 3 | 115 | 115 | 58% | 7 × 10−27 | 21% | XP_012352012.1 |
| ephrin type-B receptor 4 | Microcebus murinus | 115 | 115 | 59% | 9 × 10−27 | 21% | XP_012614574.1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dornas, F.P.; Assis, F.L.; Aherfi, S.; Arantes, T.; Abrahão, J.S.; Colson, P.; La Scola, B. A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae. Viruses 2016, 8, 76. https://doi.org/10.3390/v8030076
Dornas FP, Assis FL, Aherfi S, Arantes T, Abrahão JS, Colson P, La Scola B. A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae. Viruses. 2016; 8(3):76. https://doi.org/10.3390/v8030076
Chicago/Turabian StyleDornas, Fábio P., Felipe L. Assis, Sarah Aherfi, Thalita Arantes, Jônatas S. Abrahão, Philippe Colson, and Bernard La Scola. 2016. "A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae" Viruses 8, no. 3: 76. https://doi.org/10.3390/v8030076
APA StyleDornas, F. P., Assis, F. L., Aherfi, S., Arantes, T., Abrahão, J. S., Colson, P., & La Scola, B. (2016). A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae. Viruses, 8(3), 76. https://doi.org/10.3390/v8030076