
HTLV-1 Rex Tunes the Cellular Environment Favorable for Viral Replication


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
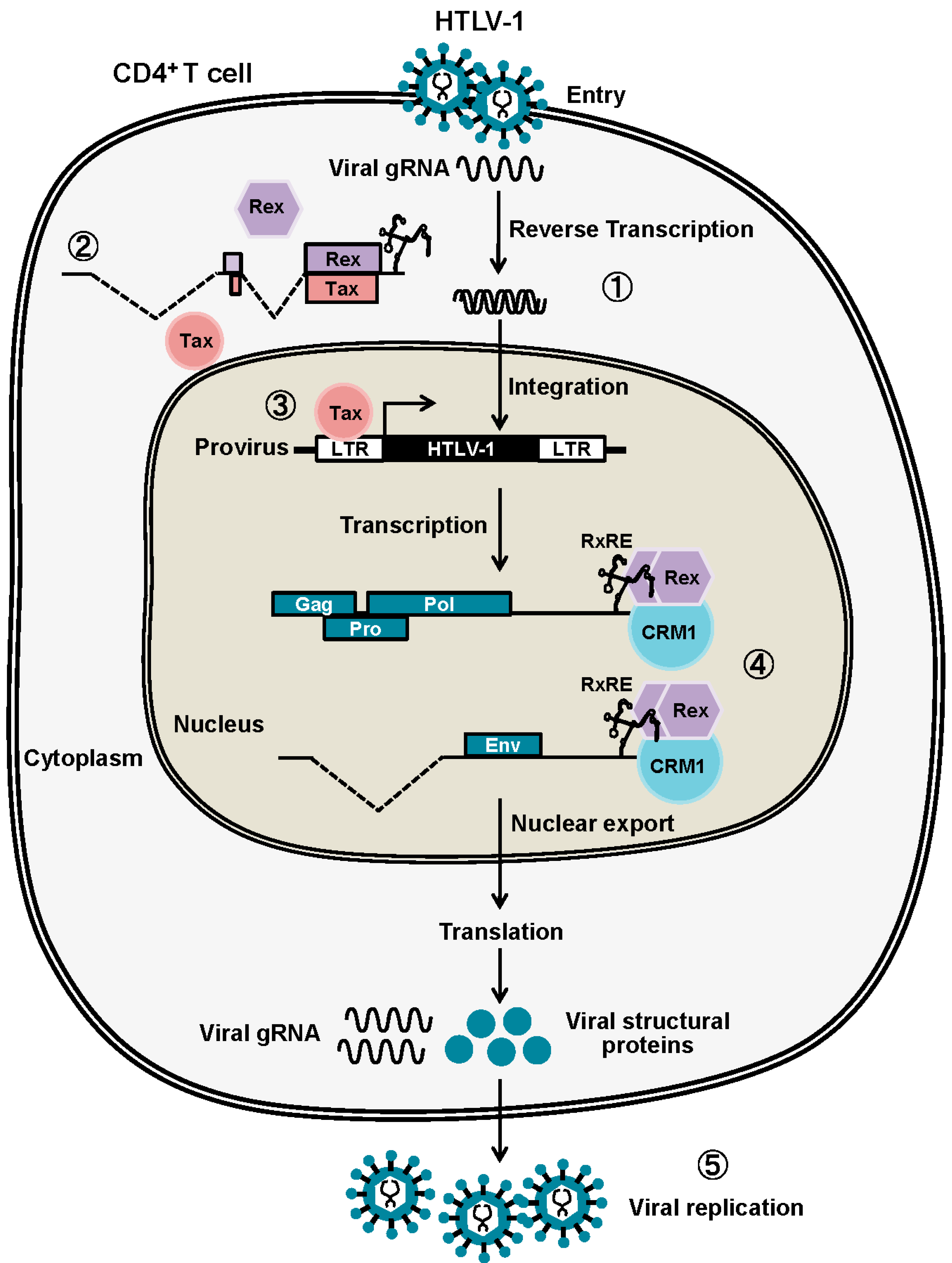
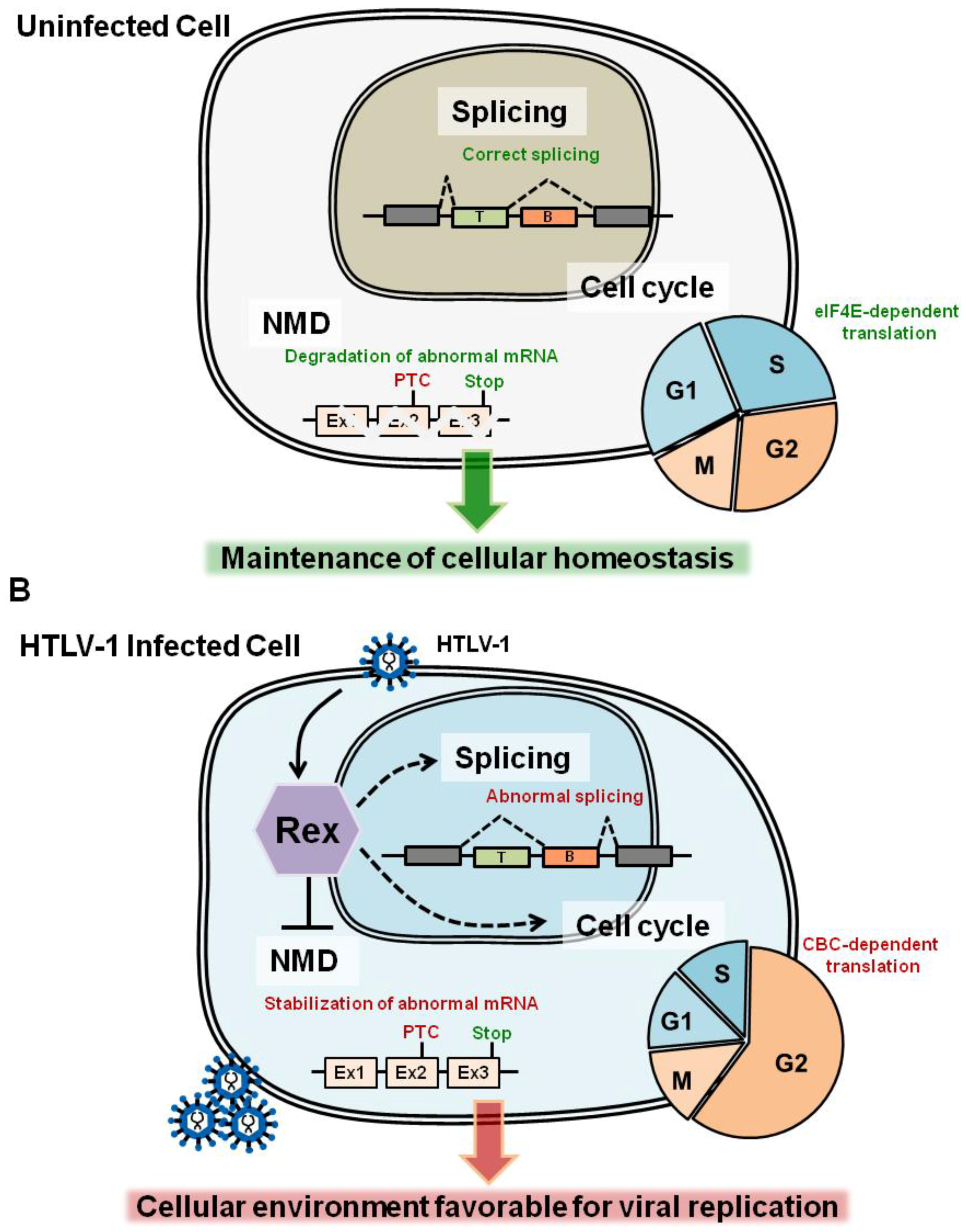
:1. Molecular Events in the Host Cell Caused by HTLV-1 Infection
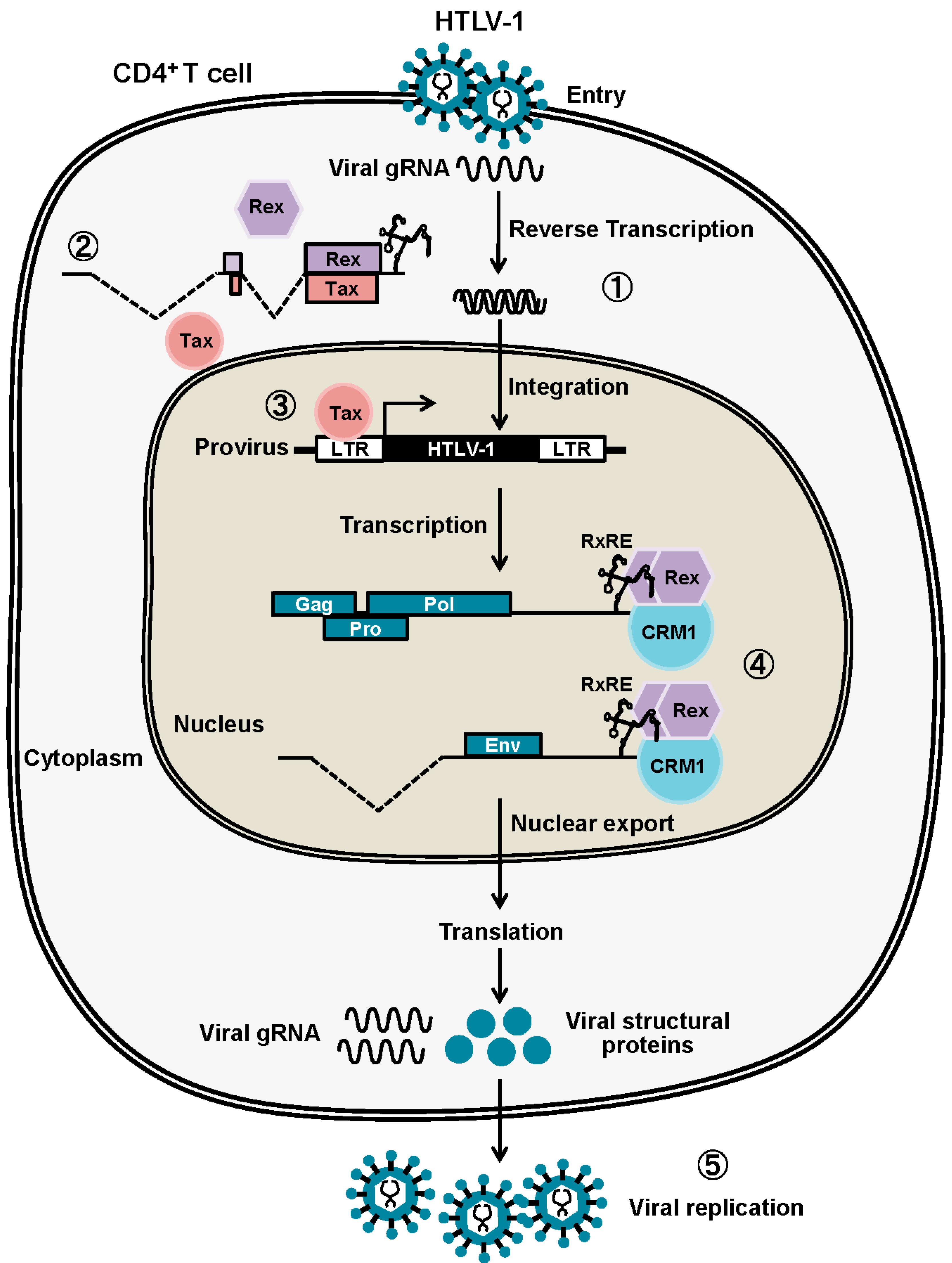
2. Canonical Rex Function as a Post-Transcriptional Regulator of Viral Expression
2.1. Rex-Dependent Nuclear Export of Viral mRNAs
2.2. Primary Structure of Rex and Its Function
2.3. Rex Activity and Phosphorylation
2.4. Regulation of Rex by Other HTLV-1 Viral Proteins
3. Non-Canonical Functions of Rex: Exploring New Aspects of Rex
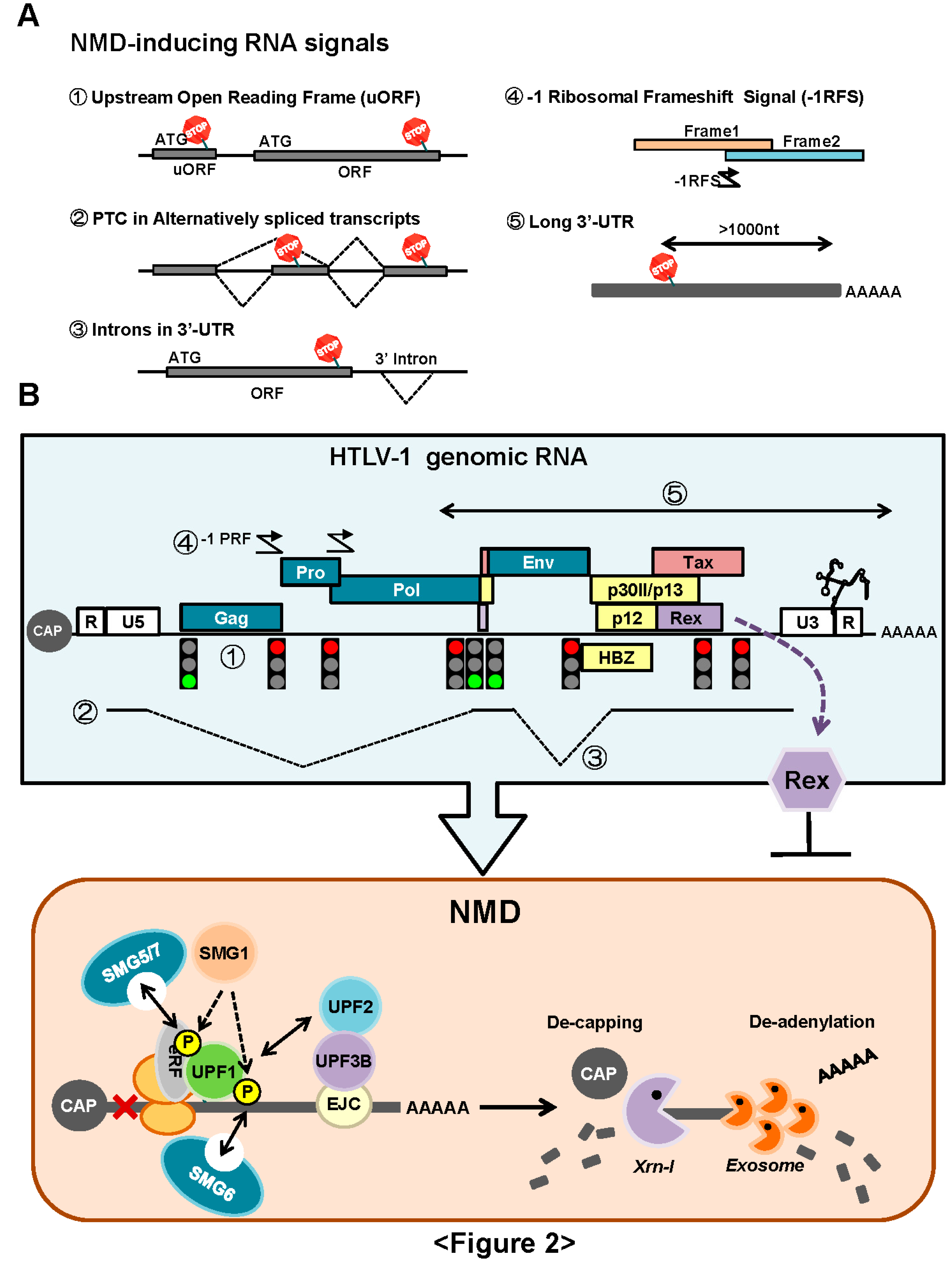
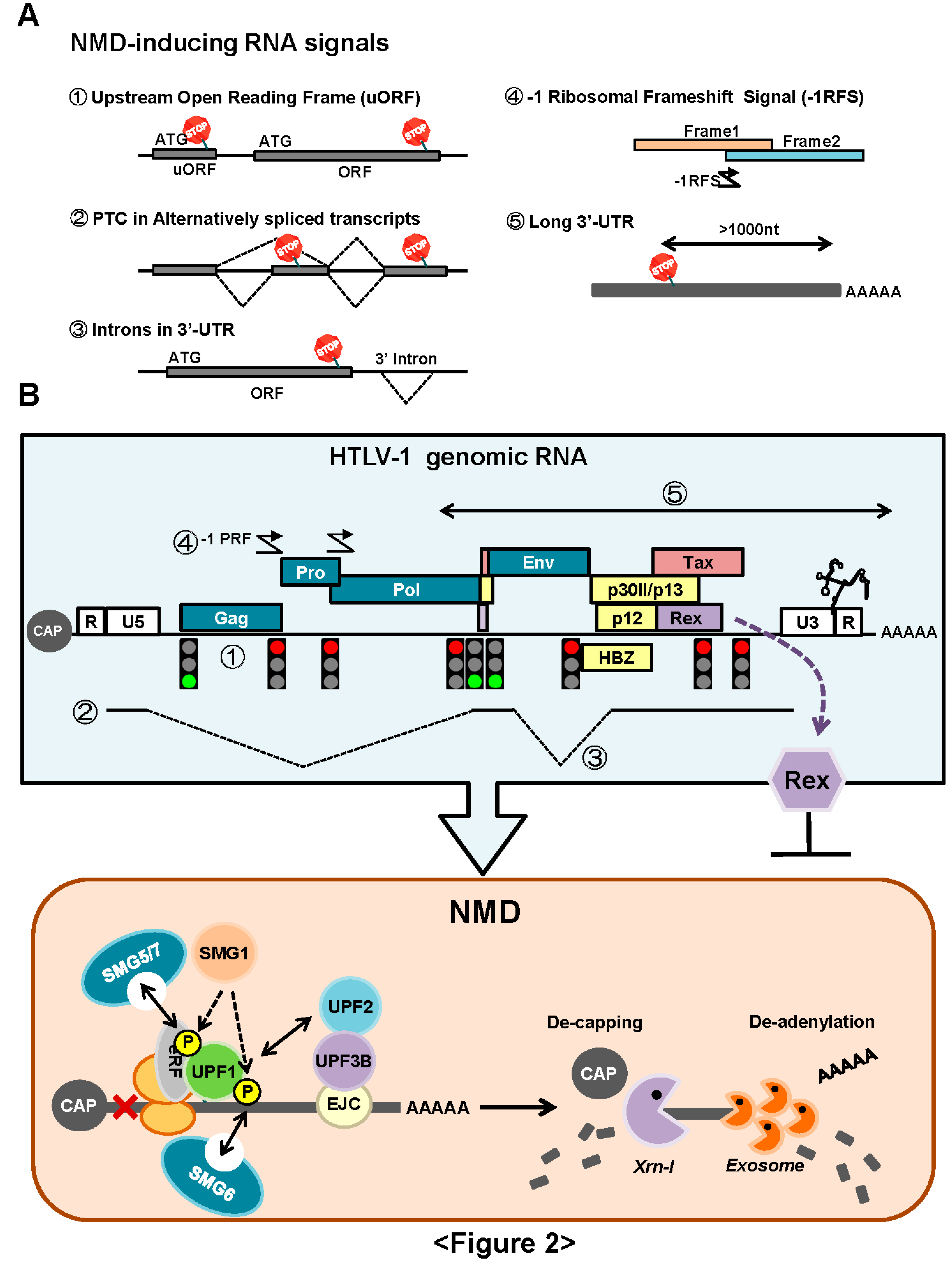
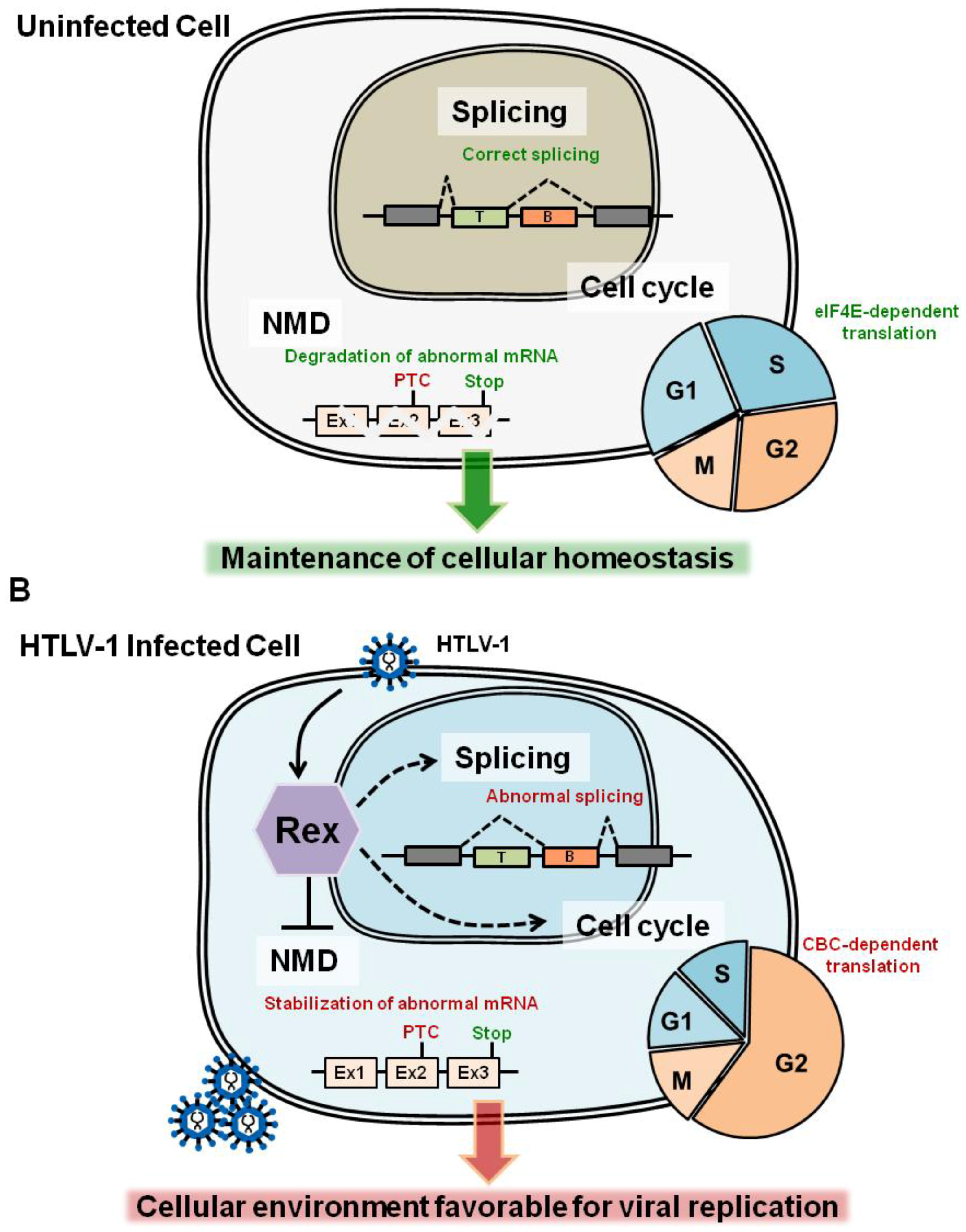
3.1. NMD Inhibition by Rex
3.1.1. Rex Stabilizes HTLV-1 Genomic RNA by Inhibition of NMD
3.1.2. How Does Rex Protect Viral mRNAs from NMD in the Cytoplasm?
3.2. Regulation of mRNA Splicing Machinery by Rex
3.3. Cell-Cycle Regulation: Does Rex Interfere the Host Cell-Cycle Regulation?
4. Function of Rex and the Viral Pathogenesis
4.1. Do Rex-1/Rex-2 Functions Relate to the Pathogenesities of HTLV-1/HTLV-2?
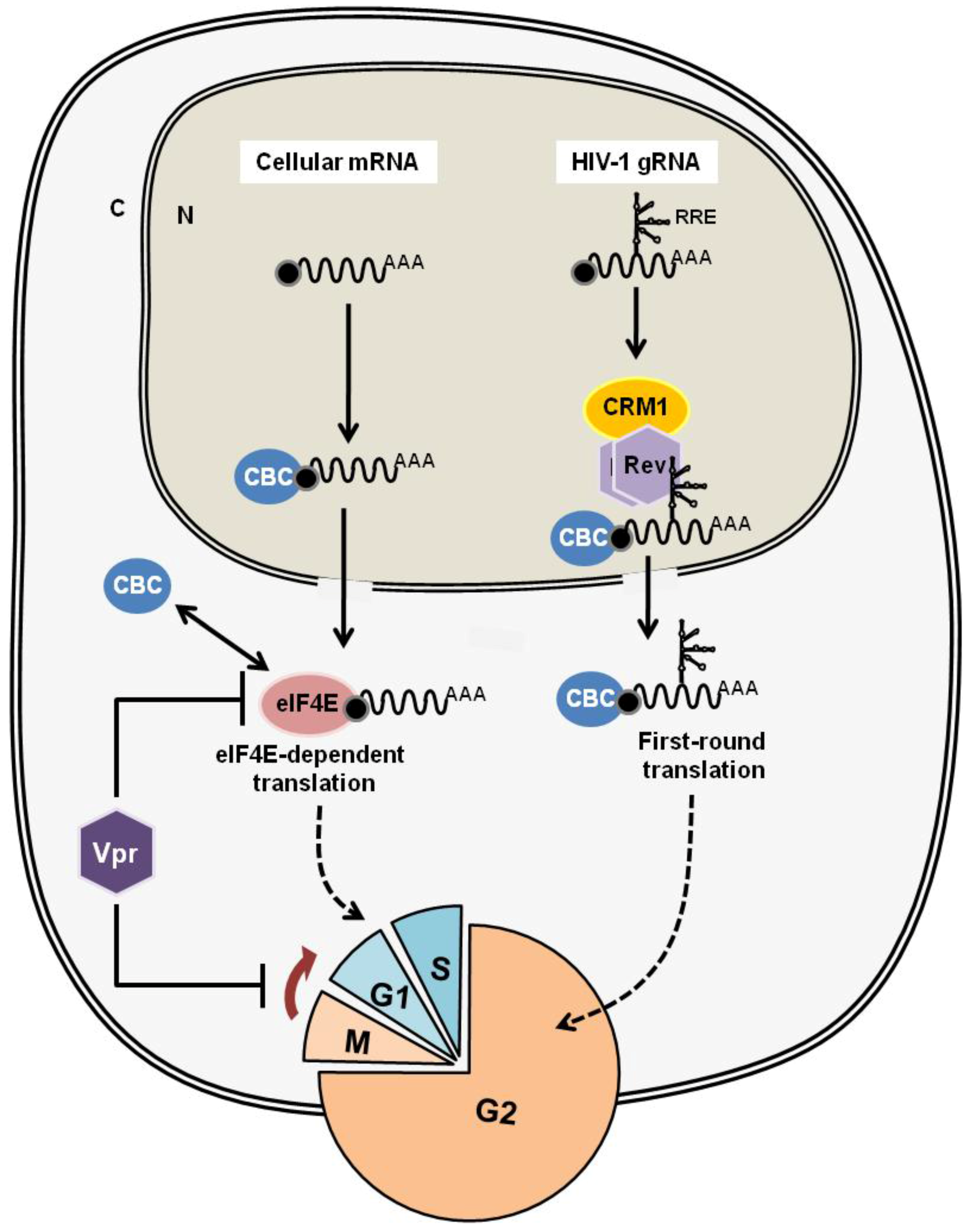
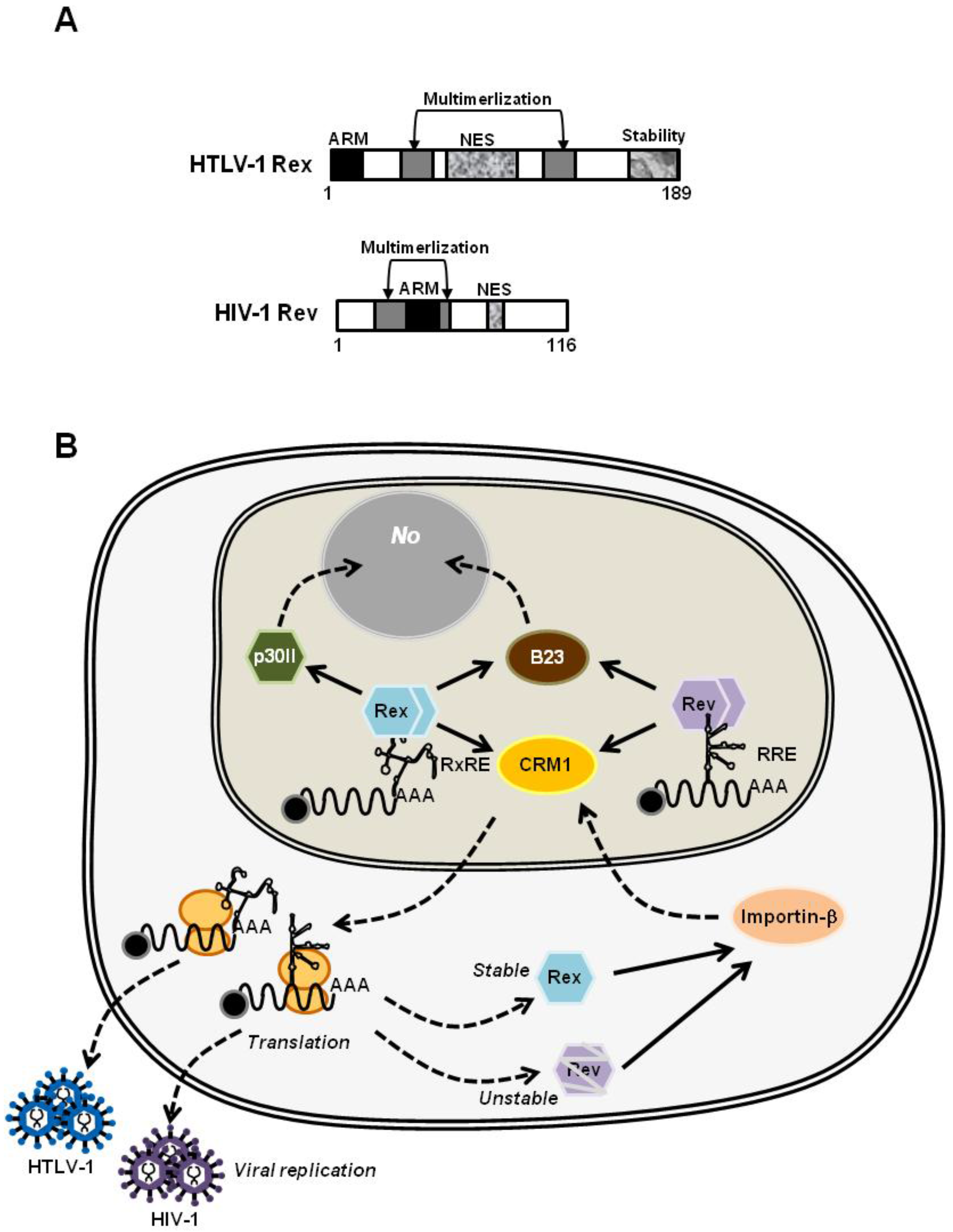
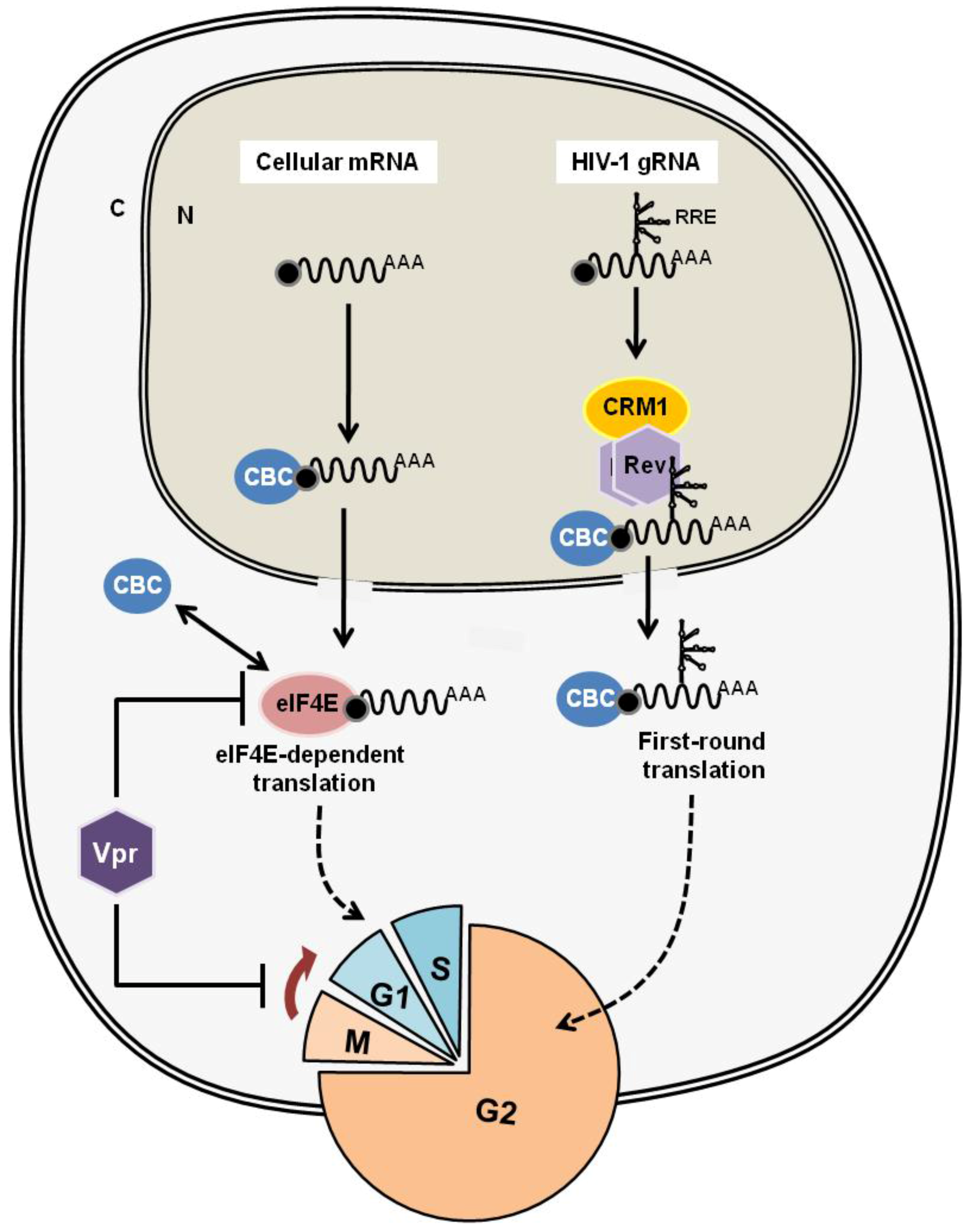
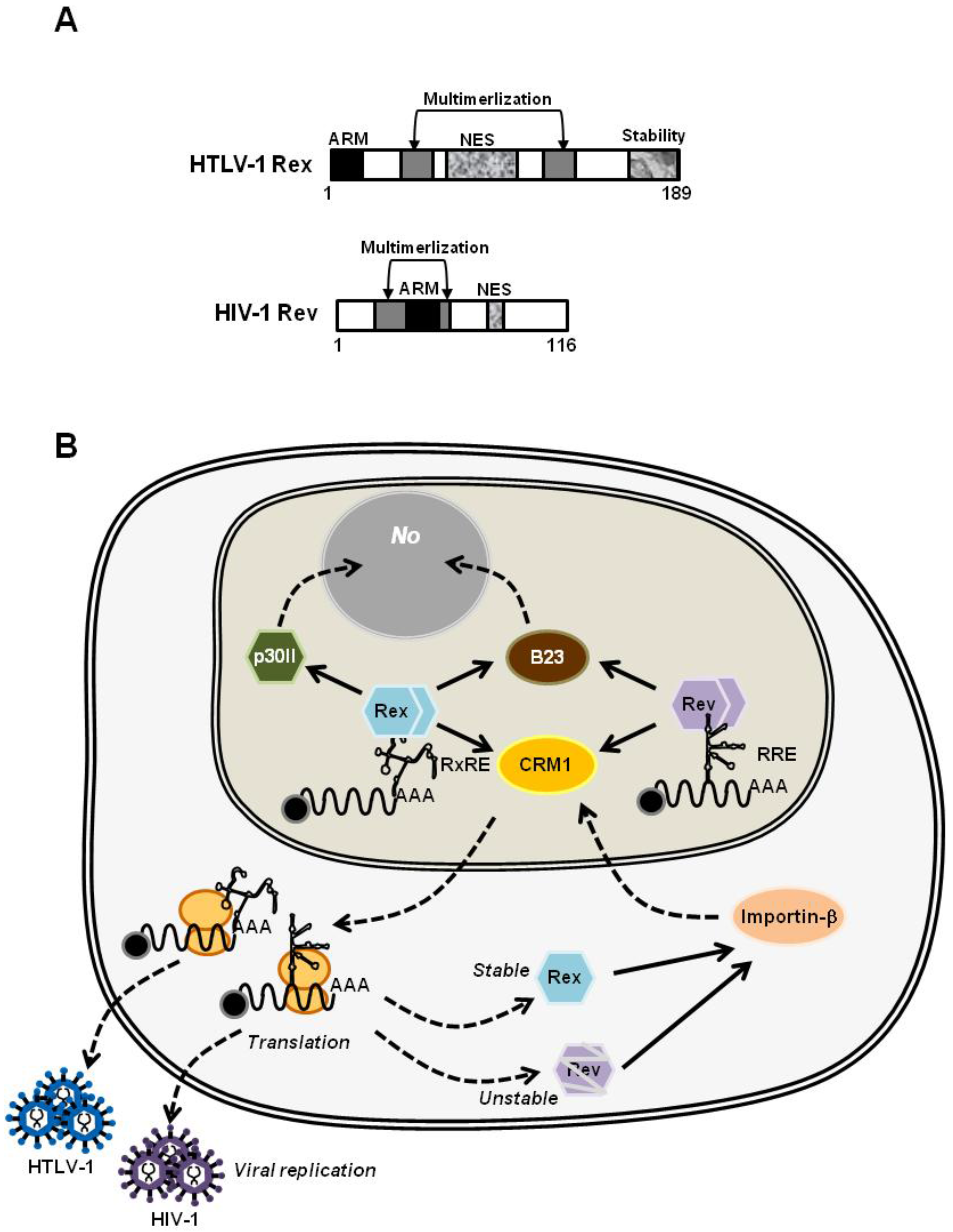
4.2. HTLV-1 Rex and HIV-1 Rev: Are They Similar or Different?
4.2.1. HIV-1 Rev, the Molecular Counterpart of HTLV-1 Rex
4.2.2. The Structural Biology of HTLV-1 Rex; Learning from that of HIV-1 Rev
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gallo, R.C. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology 2005, 7, 1–7. [Google Scholar]
- Takatsuki, K. Discovery of adult T-cell leukemia. Retrovirology 2005, 3, 1–3. [Google Scholar]
- Seiki, M.; Hattori, S.; Hirayama, Y.; Yoshida, M. Human adult T-cell leukemia virus: Complete nucleotide sequence of the provirus genome integrated in leukemia cell DNA. Proc. Natl. Acad. Sci. USA 1983, 80, 3618–3622. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.M.; Harrod, R.; Franchini, G. Molecular biology and pathogenesis of the human T-cell leukaemia/lymphotropic virus Type-1 (HTLV-1). Int. J. Exp. Pathol. 2001, 82, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Franchini, G.; Fukumoto, R.; Fullen, J.R. T-cell control by human T-cell leukemia/lymphoma virus type 1. Int. J. Hematol. 2003, 78, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Kashanchi, F.; Brady, J.N. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene 2005, 24, 5938–5951. [Google Scholar] [CrossRef] [PubMed]
- Kannian, P.; Green, P.L. Human T Lymphotropic Virus Type 1 (HTLV-1): Molecular Biology and Oncogenesis. Viruses 2010, 2, 2037–2077. [Google Scholar] [CrossRef] [PubMed]
- Balvay, L.; Lastra, M.L.; Sargueil, B.; Darlix, J.-L.; Ohlmann, T. Translational control of retroviruses. Nat. Rev. Microbiol. 2007, 5, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Corradin, A.; DI Camillo, B.; Rende, F.; Ciminale, V.; Toffolo, G.M.; Cobelli, C. Retrovirus HTLV-1 gene circuit: A potential oscillator for eukaryotes. Pac. Symp. Biocomput. 2010, 432, 421–432. [Google Scholar]
- Li, M.; Kesic, M.; Yin, H.; Yu, L.; Green, P.L. Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J. Virol. 2009, 83, 3788–3797. [Google Scholar] [CrossRef] [PubMed]
- Green, P.L.; Chen, I.S.Y. Regulation of human T cell leukemia virus expression. FASEB J. 1990, 4, 169–174. [Google Scholar] [PubMed]
- Rende, F.; Cavallari, I.; Corradin, A.; Silic-Benussi, M.; Toulza, F.; Toffolo, G.M.; Tanaka, Y.; Jacobson, S.; Taylor, G.P.; D’Agostino, D.M.; et al. Kinetics and intracellular compartmentalization of HTLV-1 gene expression: Nuclear retention of HBZ mRNAs. Blood 2011, 117, 4855–4859. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Watanabe, T. HTLV-1 Rex: The courier of viral messages making use of the host vehicle. Front. Microbiol. 2012, 3, 330. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Y.F.; Gilmartin, G.M.; Hanly, S.M.; Nevins, J.R.; Greene, W.C. The HTLV-I Rex response element mediates a novel form of mRNA polyadenylation. Cell 1991, 64, 727–737. [Google Scholar] [CrossRef]
- Ahmed, Y.F.; Hanly, S.M.; Malim, M.H.; Cullen, B.R.; Greene, W.C. Structure-function analyses of the HTLV-I Rex and HIV-1 Rev RNA response elements: Insights into the mechanism of Rex and Rev action. Genes Dev. 1990, 4, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, M.; Inoue, J.; Yoshida, M.; Seiki, M. Post-transcriptional regulator (rex) of HTLV-1 initiates expression of viral structural proteins but suppresses expression of regulatory proteins. EMBO J. 1988, 7, 519–523. [Google Scholar] [PubMed]
- Bai, X.T.; Sinha-Datta, U.; Ko, N.L.; Bellon, M.; Nicot, C. Nuclear export and expression of HTLV-I tax/rex mRNA is RxRE/Rex-dependent. J. Virol. 2012, 86, 4559–4565. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, I.; Rende, F.; Bona, M.K.; Sztuba-solinska, J.; Silic-benussi, M.; Tognon, M.; Legrice, S.F.J.; Franchini, G.; Ciminale, V. Expression of alternatively spliced human T-cell leukemia virus type 1 mRNAs is influenced by mitosis and by a novel cis-acting regulatory sequence. J. Virol. 2016, 90, 1486–1498. [Google Scholar]
- Philip, S.; Zahoor, M.A.; Zhi, H.; Ho, Y.-K.; Giam, C.-Z. Regulation of human T-lymphotropic virus type I latency and reactivation by HBZ and Rex. PLoS Pathog. 2014, 10, e1004040. [Google Scholar] [CrossRef] [PubMed]
- Hakata, Y.; Umemoto, T.; Matsushita, S.; Shida, H. Involvement of human CRM1 (exportin 1) in the export and multimerization of the Rex protein of human T-cell leukemia virus type 1. J. Virol. 1998, 72, 6602–6607. [Google Scholar] [PubMed]
- Hakata, Y.; Yamada, M.; Shida, H. Rat CRM1 is responsible for the poor activity of human T-cell leukemia virus type 1 Rex protein in rat cells. J. Virol. 2001, 75, 11515–11525. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Bellon, M.; Nicot, C. HTLV-1 Yin and Yang: Rex and p30 master regulators of viral mRNA trafficking. AIDS Rev. 2008, 10, 195–204. [Google Scholar] [PubMed]
- Younis, I.; Green, P.L. The human T-cell leukemia virus Rex protein. Front. Biosci. 2005, 1, 431–445. [Google Scholar] [CrossRef]
- Kesic, M.; Doueiri, R.; Ward, M.; Semmes, O.J.; Green, P.L. Phosphorylation regulates human T-cell leukemia virus type 1 Rex function. Retrovirology 2009, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Kesic, M.; Ward, M.; Semmes, O.J.; Green, P.L. Site-specific phosphorylation regulates human T-cell leukemia virus type 2 Rex function in vivo. J. Virol. 2009, 83, 8859–8868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, L.; Kesic, M.; Yamamoto, B.; Li, M.; Younis, I.; Lairmore, M.D.; Green, P.L. Human T-cell leukemia virus type 2 Rex carboxy terminus is an inhibitory/stability domain that regulates Rex functional activity and viral replication. J. Virol. 2009, 83, 5232–5243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adachi, Y.; Nosaki, T.; Hatanaka, M. Protein kinase inhibitor H-7 blocks accumulation of unspliced mRNA of human T-cell leukemia virus type I (HTLV-I). Biochem. Biophys. Res. Commun. 1990, 169, 469–475. [Google Scholar] [CrossRef]
- Adachi, Y.; Copeland, T.D.; Takahashi, C.; Nosaka, T.; Ahmed, A.; Oroszlan, S.; Hatanaka, M. Phosphorylation of the Rex protein of human T-cell leukemia virus type I. J. Biol. Chem. 1992, 267, 21977–21981. [Google Scholar] [PubMed]
- Bai, X.T.; Baydoun, H.H.; Nicot, C. HTLV-I p30: A versatile protein modulating virus replication and pathogenesis. Mol. Aspects Med. 2010, 31, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Ghorbel, S.; Sinha-Datta, U.; Dundr, M.; Brown, M.; Franchini, G.; Nicot, C. Human T-cell leukemia virus type I p30 nuclear/nucleolar retention is mediated through interactions with RNA and a constituent of the 60 S ribosomal subunit. J. Biol. Chem. 2006, 281, 37150–37158. [Google Scholar] [CrossRef] [PubMed]
- Nicot, C.; Dundr, M.; Johnson, J.M.; Fullen, J.R.; Alonzo, N.; Fukumoto, R.; Princler, G.L.; Derse, D.; Misteli, T.; Franchini, G. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat. Med. 2004, 10, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Sinha-Datta, U.; Datta, A.; Ghorbel, S.; Dodon, M.D.; Nicot, C. Human T-cell lymphotrophic virus type I rex and p30 interactions govern the switch between virus latency and replication. J. Biol. Chem. 2007, 282, 14608–14615. [Google Scholar] [CrossRef] [PubMed]
- Berneman, Z.N.; Gartenhaus, R.B.; Reitz, M.S.; Blattner, W.A.; Manns, A.; Hanchard, B.; Ikehara, O.; Gallo, R.C.; Klotman, M.E. Expression of alternatively spliced human T-lymphotropic virus type I pX mRNA in infected cell lines and in primary uncultured cells from patients with adult T-cell leukemia/lymphoma and healthy carriers. Proc. Natl. Acad. Sci. USA 1992, 89, 3005–3009. [Google Scholar] [CrossRef] [PubMed]
- Orita, S.; Takagi, S.; Saiga, A.; Minoura, N.; Araki, K.; Kinoshita, K.; Kondo, T.; Hinuma, Y.; Igarashi, H. Human T cell leukaemia virus type 1 p21X mRNA: Constitutive expression in peripheral blood mononuclear cells of patients with adult T cell leukaemia. J. Gen. Virol. 1992, 73 Pt 9, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Saiga, A.; Aono, Y.; Imai, J.; Kinoshita, K.; Orita, S.; Igarashi, H. Presence of antibodies to p21X and/or p27rex proteins in sera from human T-cell leukemia virus type I-infected individuals. J. Virol. Methods 1996, 57, 157–168. [Google Scholar] [CrossRef]
- Kiyokawa, T.; Seiki, M.; Iwashita, S.; Imagawa, K.; Shimizu, F.; Yoshida, M. T-cell leukemia virus type I p27X-III and p21x-III, proteins encoded by the pX sequence of human T-cell leukemia virus type I. Proc. Natl. Acad. Sci. USA 1985, 82, 8359–8363. [Google Scholar] [CrossRef] [PubMed]
- Orita, S.; Kobayashi, H.; Aono, Y.; Saiga, A.; Maeda, M.; Igarashi, H. p21X mRNA is expressed as a singly spliced pX transcript from defective provirus genomes having a partial delection of the pol-env region in human T-cell leukemia virus type 1-infected cells. Nucleic Acids Res. 1993, 21, 3799–3807. [Google Scholar] [CrossRef] [PubMed]
- Dickson, A.M.; Wilusz, J. Strategies for viral RNA stability: Live long and prosper. Trends Genet. 2011, 27, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Garcia, S.; Voinnet, O. Nonsense-Mediated Decay Serves as a General Viral Restriction Mechanism in Plants. Cell Host Microbe 2014, 16, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Mühlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host Microbe 2014, 16, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Quek, B.L.; Beemon, K. Retroviral strategy to stabilize viral RNA. Curr. Opin. Microbiol. 2014, 18, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.R.; Kumar, G.R.; Verschueren, E.; Johnson, J.R.; Von Dollen, J.; Johnson, T.; Newton, B.; Shah, P.; Horner, J.; Krogan, N.J.; et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 2015, 57, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.; Maquat, L. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet. 2013, 47, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Kervestin, S.; Jacobson, A. NMD: A multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012, 13, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Ando, T.; Yamagishi, M.; Yokoyama, K.; Ishida, T.; Ohsugi, T.; Tanaka, Y.; Brighty, D.W.; Watanabe, T. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: Implications for retroviral replication. Microbes Infect. 2013, 15, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Kodama, T.; Matsumoto, A.; Itakura, H.; Yazaki, Y. Stabilization od interleukin-2 receptor a chain mRNA by HTLV-1 Rex in mouse L cells: Lower amounts of Rex do not stabilize the mRNA. Biochem. Biophys. Res. Commun. 1994, 198, 243–250. [Google Scholar]
- White, K.N.; Nosaka, T.; Kanamori, H.; Hatanaka, M.; Honjo, T. The nucleolar localisation signal of the HTLV-1 protein p27Rex is important for stabilisation of IL-2 receptor α subunit mRNA by p27Rex. Biochem. Biophys. Res. Commun. 1991, 175, 98–103. [Google Scholar] [CrossRef]
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Gröne, M.; Koch, C.; Grassmann, R. The HTLV-1 Rex protein induces nuclear accumulation of unspliced viral RNA by avoiding intron excision and degradation. Virology 1996, 218, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Princler, G.; Julias, J.G.; Hughes, S.H.; Derse, D. Roles of viral and cellular proteins in the expression of alternatively spliced HTLV-1 pX mRNAs. Virology 2003, 317, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.M.; Amaral, M.C.; Wu, J.Y.; Maniatis, T.; Greene, W.C. HIV Rev-dependent binding of SF2/ASF to the Rev response element: Possible role in Rev-mediated inhibition of HIV RNA splicing. Proc. Natl. Acad. Sci. USA 1997, 94, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Tange, T.O.; Jensen, T.H.; Kjems, J. In vitro interaction between human immunodeficiency virus type 1 Rev protein and splicing factor ASF/SF2-associated protein, p32. J. Biol. Chem. 1996, 271, 10066–10072. [Google Scholar] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Hamaia, S.; Casse, H.; Gazzolo, L.; Duc Dodon, M. The human T-cell leukemia virus type 1 Rex regulatory protein exhibits an impaired functionality in human lymphoblastoid Jurkat T cells. J. Virol. 1997, 71, 8514–8521. [Google Scholar] [PubMed]
- Dodon, M.D.; Hamaia, S.; Martin, J.; Gazzolo, L. Heterogeneous nuclear ribonucleoprotein A1 interferes with the binding of the human T cell leukemia virus type 1 Rex regulatory protein to its response element. J. Biol. Chem. 2002, 277, 18744–18752. [Google Scholar] [CrossRef] [PubMed]
- Kress, E.; Baydoun, H.H.; Bex, F.; Gazzolo, L.; Dodon, M.D. Critical role of hnRNP A1 in HTLV-1 replication in human transformed T lymphocytes. Retrovirology 2005, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Levraud, J.P.; Dodon, M.D.; Bessia, C.; Hazan, U.; Kourilsky, P.; Israël, A. Altered expression of tyrosine kinases of the Src and Syk families in human T-cell leukemia virus type 1-infected T-cell lines. J. Virol. 1999, 73, 3709–3717. [Google Scholar] [PubMed]
- Picard, C.; Gabert, J.; Olive, D.; Collette, Y. Altered splicing in hematological malignancies reveals a tissue-specific translational block of the Src-family tyrosine kinase fyn brain isoform expression. Leukemia 2004, 18, 1737–1739. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Elder, R.T. Viral infections and cell cycle G2/M regulation. Cell Res. 2005, 15, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; Le Rouzic, E.; Planelles, V. HIV-1 Vpr: Mechanisms of G2 arrest and apoptosis. Exp. Mol. Pathol. 2008, 85, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Guenzel, C.A.; Hérate, C.; Benichou, S. HIV-1 Vpr-a still “enigmatic multitasker”. Front. Microbiol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, W.; Macurek, L.; Freire, R.; Lindqvist, A.; Medema, R.H. Bora and Aurora-A continue to activate Plk1 in mitosis. J. Cell Sci. 2014, 127, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.C.; Manel, N.; Emerman, M. The human immunodeficiency virus Vpr protein binds Cdc25C: Implications for G2 arrest. Virology 2004, 318, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Zimmerman, E.S.; Planelles, V.; Chen, J.; Irol, J.V. Activation of the ATR pathway by human immunodeficiency virus type 1 Vpr involves its direct binding to chromatin in vivo. J. Virol. 2005, 79, 15443–15451. [Google Scholar] [CrossRef] [PubMed]
- De Noronha, C.M.C.; Sherman, M.P.; Lin, H.W.; Cavrois, M.V.; Moir, R.D.; Goldman, R.D.; Greene, W.C. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 2001, 294, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, S.; Batisse, J.; Libre, C.; Bernacchi, S.; Marquet, R.; Paillart, J.-C. HIV-1 replication and the cellular eukaryotic translation apparatus. Viruses 2015, 7, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yilmaz, A.; Marsh, K.; Cochrane, A.; Boris-Lawrie, K. Thriving under stress: Selective translation of HIV-1 structural protein mRNA during Vpr-mediated impairment of eIF4E translation activity. PLoS Pathog. 2012, 8, e1002612. [Google Scholar] [CrossRef] [PubMed]
- Brégnard, C.; Benkirane, M.; Laguette, N. DNA damage repair machinery and HIV escape from innate immune sensing. Front. Microbiol. 2014, 5, 176. [Google Scholar] [PubMed]
- Haoudi, A.; Daniels, R.C.; Wong, E.; Kupfer, G.; Semmes, O.J. Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J. Biol. Chem. 2003, 278, 37736–37744. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Guo, X.; Durkin, S.S.; Fryrear, K.F.; Ward, M.D.; Semmes, O.J. Human T-cell leukemia virus type 1 Tax oncoprotein prevents DNA damage-induced chromatin egress of hyperphosphorylated Chk2. J. Biol. Chem. 2007, 282, 29431–29440. [Google Scholar] [CrossRef] [PubMed]
- Park, H.U.; Jeong, J.-H.; Chung, J.H.; Brady, J.N. Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene 2004, 23, 4966–4974. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-X.; Kuo, Y.-L.; Liu, B.-Y.; Jeang, K.-T.; Giam, C.-Z. Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J. Biol. Chem. 2003, 278, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Anupam, R.; Datta, A.; Kesic, M.; Green-Church, K.; Shkriabai, N.; Kvaratskhelia, M.; Lairmore, M.D. Human T-lymphotropic virus type 1 p30 interacts with REGgamma and modulates ATM (ataxia telangiectasia mutated) to promote cell survival. J. Biol. Chem. 2011, 286, 7661–7668. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, V.O.; Laskey, R. a Control of mammalian gene expression by selective mRNA export. Nat. Rev. Mol. Cell Biol. 2015, 16, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Topisirovic, I.; Siddiqui, N.; Lapointe, V.L.; Trost, M.; Thibault, P.; Bangeranye, C.; Piñol-Roma, S.; Borden, K.L.B. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J. 2009, 28, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Culjkovic, B.; Topisirovic, I.; Skrabanek, L.; Ruiz-Gutierrez, M.; Borden, K.L.B. eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3’UTR. J. Cell Biol. 2005, 169, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Culjkovic, B.; Topisirovic, I.; Skrabanek, L.; Ruiz-Gutierrez, M.; Borden, K.L.B. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J. Cell Biol. 2006, 175, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, A.-M.; Salemi, M.; Desmyter, J. The simian origins of the pathogenic human T-cell lymphotropic virus type I. Trends Microbiol. 1998, 6, 477–483. [Google Scholar] [CrossRef]
- Rende, F.; Cavallari, I.; Romanelli, M.G.; Diani, E.; Bertazzoni, U.; Ciminale, V. Comparison of the genetic organization, expression strategies and oncogenic potential of HTLV-1 and HTLV-2. Leuk. Res. Treat. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Suhasini, M.; Reddy, T.R. Cellular proteins and HIV-1 Rev function. Curr. HIV Res. 2009, 7, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Copeland, T.D.; Hatanakall, M.; Oroszlansii, S. Nucleolar targeting signal of Rex protein of human T-cell leukemia virus type I specifically binds to nucleolar shuttle protein B-23. J. Biol. Chem. 1993, 268, 13930–13934. [Google Scholar] [PubMed]
- Palmeri, D.; Malim, M.H. Importin β can mediate the nuclear import of an arginine-rich nuclear localization signal in the absence of importin α. Mol. Cell. Biol. 1999, 19, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Truant, R.; Cullen, B.R. The Arginine-rich domains present in human immunodeficiency virus type 1 Tat and Rev function as direct importin β -dependent nuclear localization signals. Mol. Cell. Biol. 1999, 19, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, Y. Nucleocytoplasmic protein trafic and its significance to cell function. Genes Cells 2000, 5, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Zapp, M.L.; Hope, T.J.; Parslow, T.G.; Green, M.R. Oligomerization and RNA binding domains of the type 1 human immunodeficiency virus Rev protein: A dual function for an arginine-rich binding motif. Proc. Natl. Acad. Sci. USA 1991, 88, 7734–7738. [Google Scholar] [CrossRef] [PubMed]
- Zemmel, R.W.; Kelley, A.C.; Karn, J.; Butler, P.J. Flexible regions of RNA structure facilitate co-operative Rev assembly on the Rev-response element. J. Mol. Biol. 1996, 258, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Gorin, A.; Hu, W.; Majumdar, A.; Baskerville, S.; Xu, W.; Ellington, A.; Patel, D.J. Anchoring an extended HTLV-1 Rex peptide within an RNA major groove containing junctional base triples. Structure 1999, 7, 1461–1472. [Google Scholar] [CrossRef]
- Daugherty, M.D.; D’Orso, I.; Frankel, A.D. A Solution to limited genomic capacity: Using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol. Cell 2008, 31, 824–834. [Google Scholar] [CrossRef] [PubMed]
- DiMattiaa, M.A.; Wattsc, N.R.; Stahlc, S.J.; Raderd, C.; Wingfieldc, P.T.; Stuarta, D.I.; Stevenb, A.C.; Grimes, J.M. Implications of the HIV-1 Rev dimer structure at 3.2 Å resolution for multimeric binding to the Rev response element. Proc. Natl. Acad. Sci. USA 2010, 107, 5810–5814. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, M.; Liu, B.; Frankel, A. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat. Struct. Mol. Biol. 2010, 17, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, J.; O’Carroll, I.P.; Mitchell, M.; Zuo, X.; Wang, Y.; Yu, P.; Liu, Y.; Rausch, J.W.; Dyba, M.A.; et al. An unusual topological structure of the HIV-1 rev response element. Cell 2013, 155, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Nawroth, I.; Mueller, F.; Basyuk, E.; Beerens, N.; Rahbek, U.L.; Darzacq, X.; Bertrand, E.; Kjems, J.; Schmidt, U. Stable assembly of HIV-1 export complexes occurs cotranscriptionally. RNA 2014, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Tambe, A.; Zhou, K.; Doudna, J.A.; States, U.; Division, P.B.; Berkeley, L. RNA-guided assembly of Rev-RRE nuclear export complexes. Elife 2014, 3, e03656. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.S.; Cheng, Y.; Frankel, A.D. The export receptor Crm1 forms a dimer to promote nuclear export of HIV RNA. Elife 2014, 3, e04121. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, B.; Crosby, D.C.; Homer, C.; Ribeiro, I.; Mavor, D.; Frankel, A.D. RNA-directed remodeling of the HIV-1 protein Rev orchestrates assembly of the Rev-Rev response element complex. Elife 2014, 3, e04120. [Google Scholar] [CrossRef] [PubMed]
- Simonis, N.; Rual, J.-F.; Lemmens, I.; Boxus, M.; Hirozane-Kishikawa, T.; Gatot, J.-S.; Dricot, A.; Hao, T.; Vertommen, D.; Legros, S.; et al. Host-pathogen interactome mapping for HTLV-1 and 2 retroviruses. Retrovirology 2012, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Suzuki, H.; Nishitsuji, H.; Shida, H.; Takaku, H. Interaction of human T-cell lymphotropic virus type I Rex protein with Dicer suppresses RNAi silencing. FEBS Lett. 2010, 584, 4313–4318. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakano, K.; Watanabe, T. HTLV-1 Rex Tunes the Cellular Environment Favorable for Viral Replication. Viruses 2016, 8, 58. https://doi.org/10.3390/v8030058
Nakano K, Watanabe T. HTLV-1 Rex Tunes the Cellular Environment Favorable for Viral Replication. Viruses. 2016; 8(3):58. https://doi.org/10.3390/v8030058
Chicago/Turabian StyleNakano, Kazumi, and Toshiki Watanabe. 2016. "HTLV-1 Rex Tunes the Cellular Environment Favorable for Viral Replication" Viruses 8, no. 3: 58. https://doi.org/10.3390/v8030058
APA StyleNakano, K., & Watanabe, T. (2016). HTLV-1 Rex Tunes the Cellular Environment Favorable for Viral Replication. Viruses, 8(3), 58. https://doi.org/10.3390/v8030058