
Identification of Known and Novel Recurrent Viral Sequences in Data from Multiple Patients and Multiple Cancers

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Data Sets
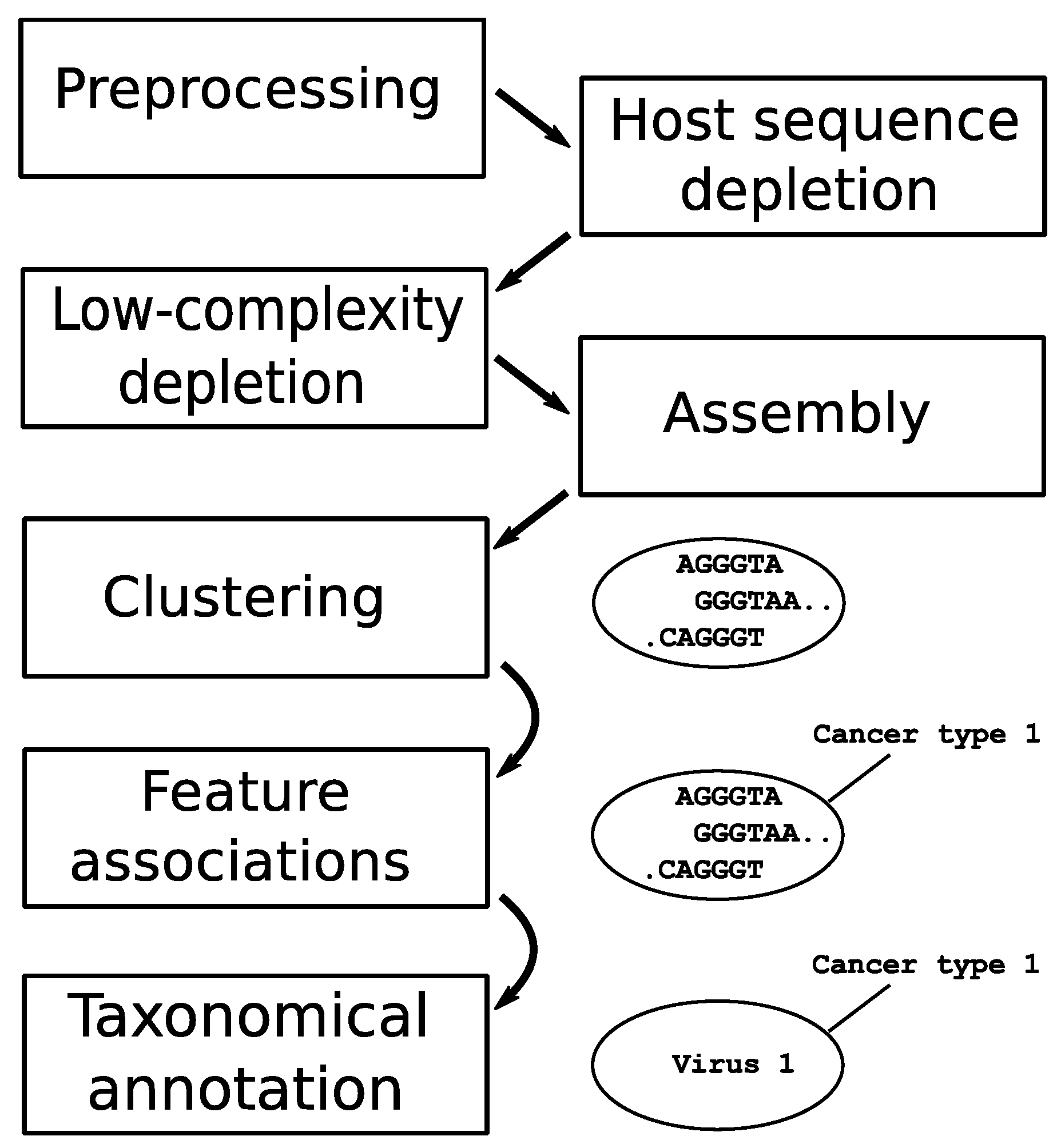
2.3. Constituents of the Software Pipeline and Execution Parameters
3. Results
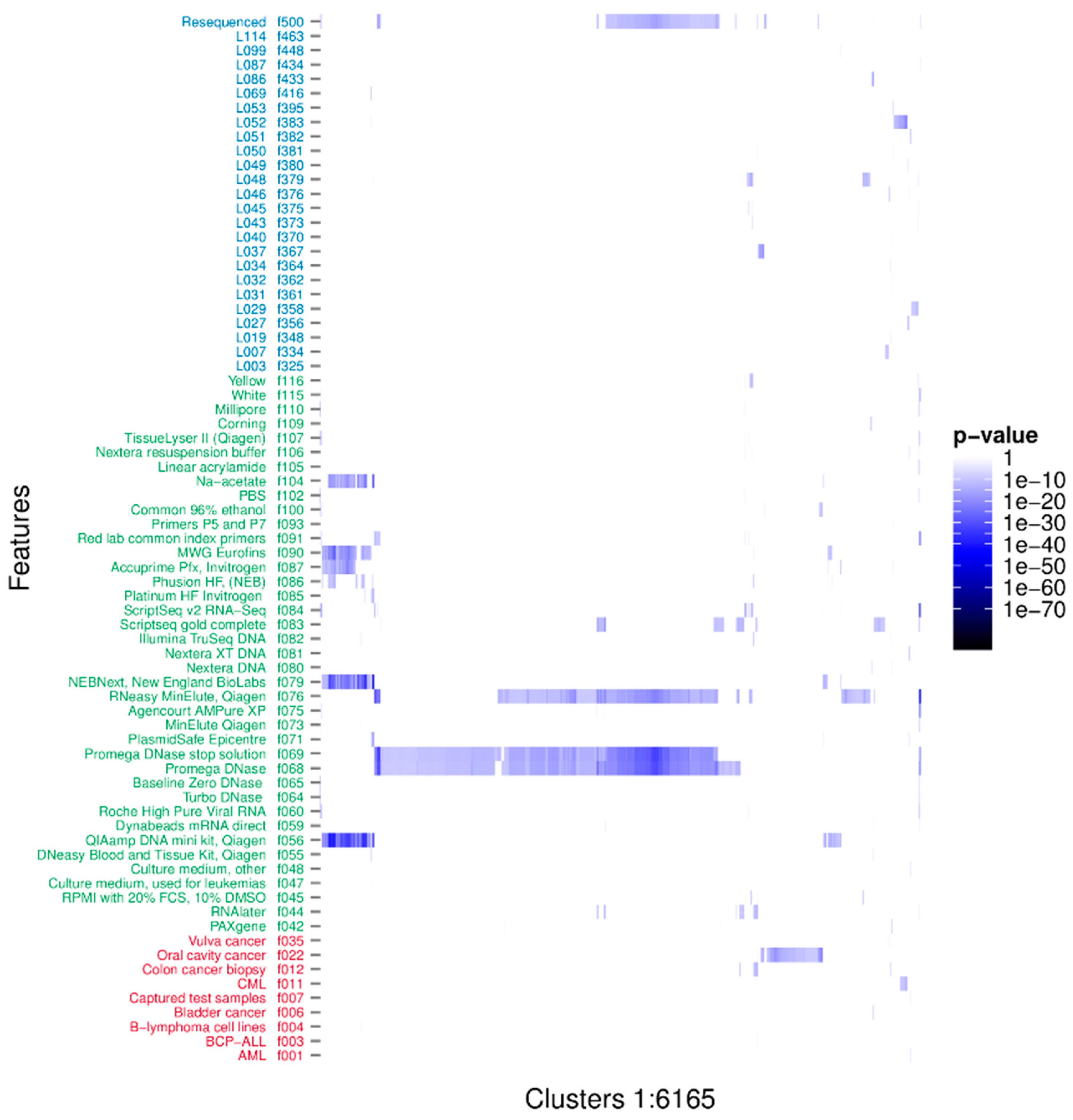
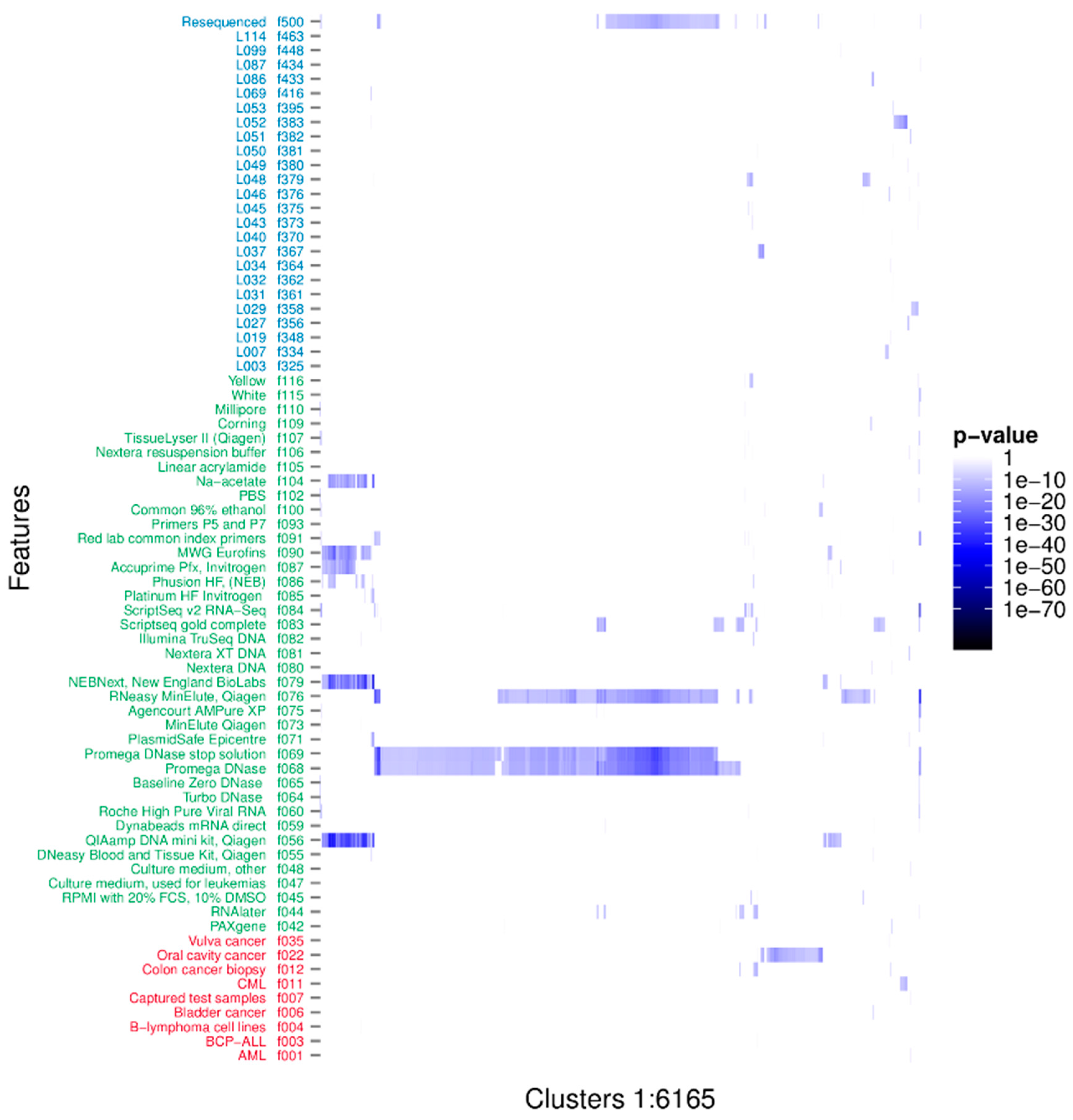
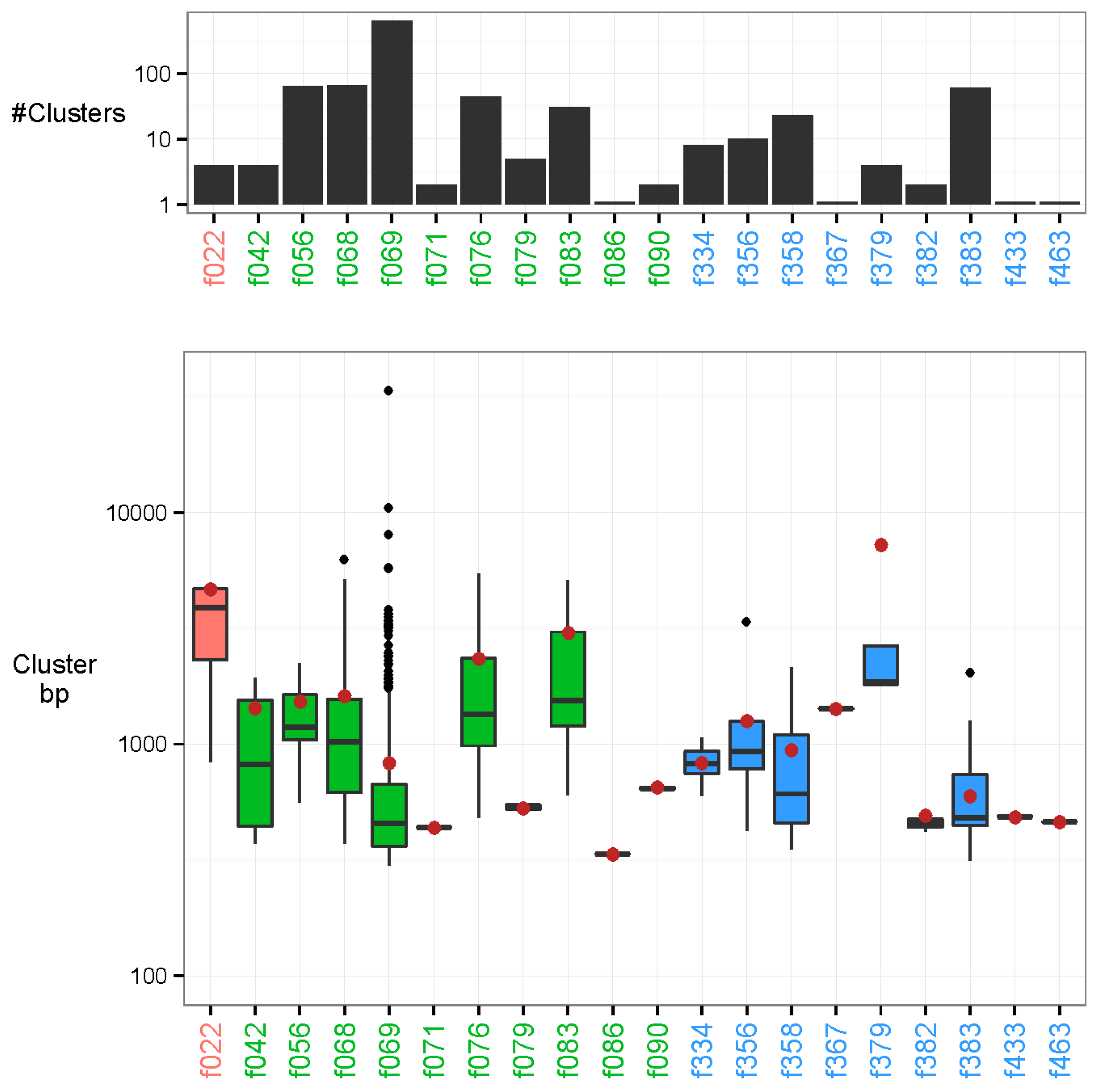
3.1. Clustering Identifies Recurrent Nucleotide Sequences across Samples
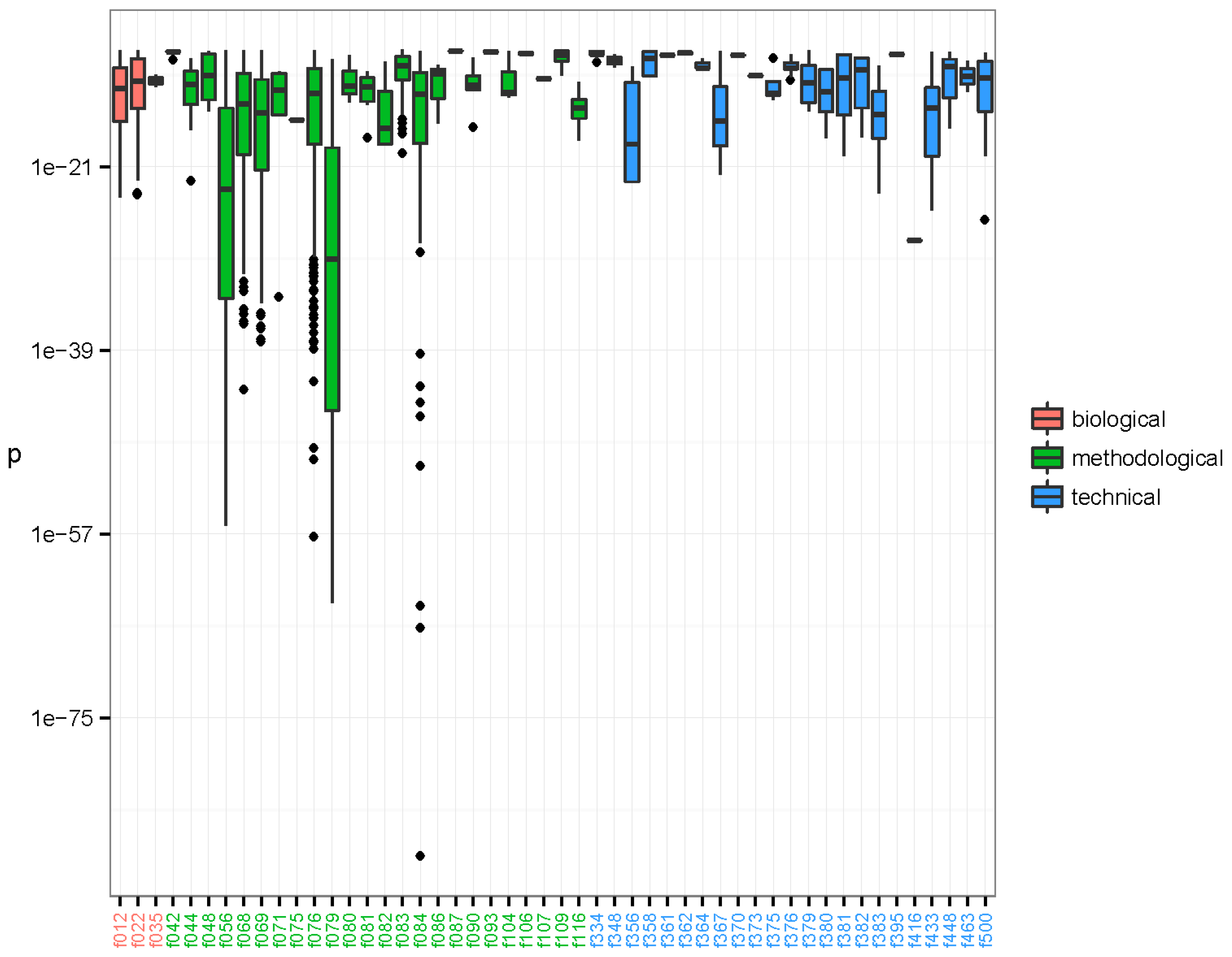
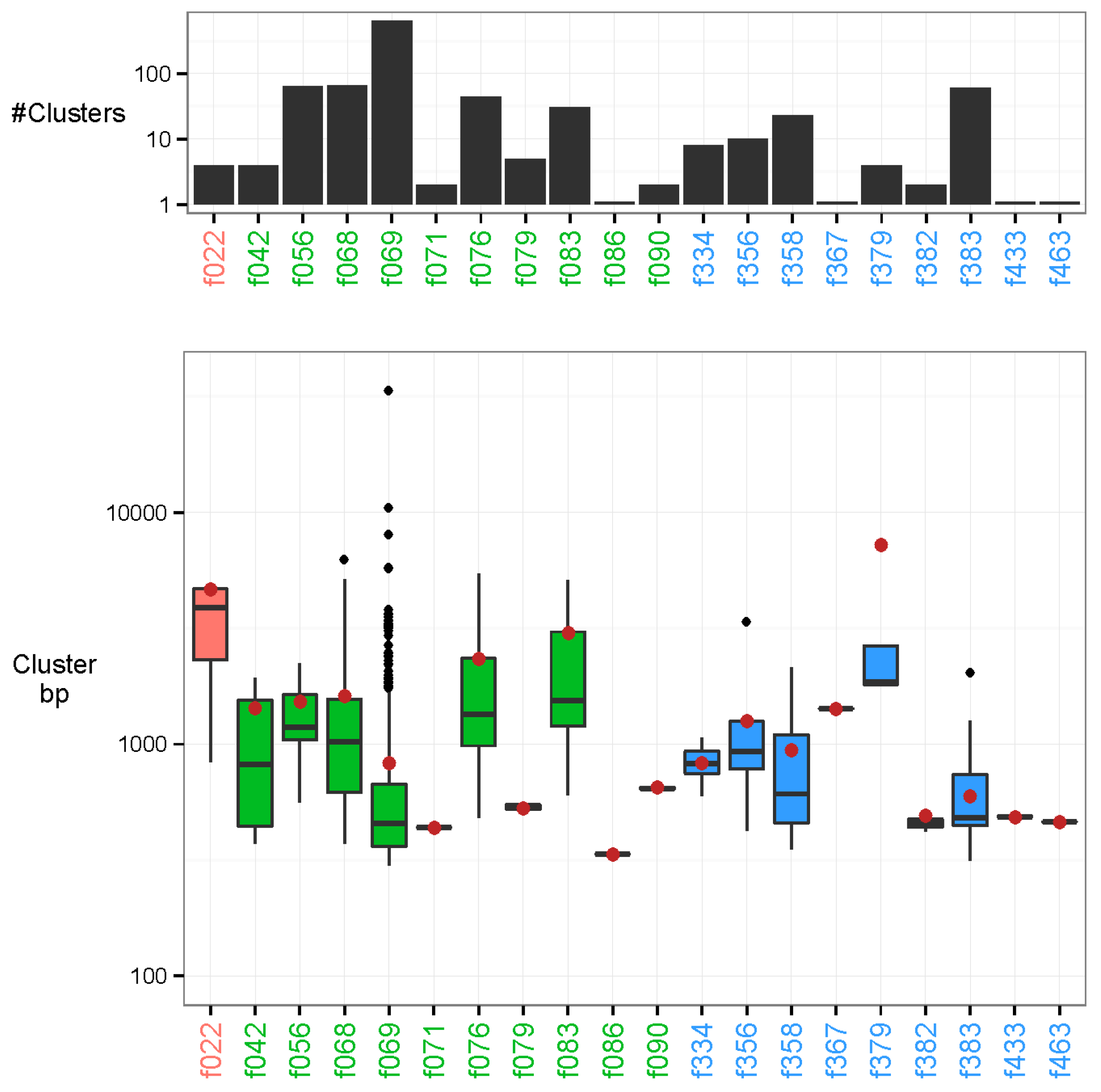
3.2. Characterisation of the Nature of the Recurrent Sequences
3.3. Taxonomic Characterisation
3.4. Identification of Novel Recurrent Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Van der Hoek, L. Identification of a New Human Coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a Human Parvovirus by Molecular Screening of Respiratory Tract Samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.S.; Kapoor, A.; Lukashov, V.V.; Simmonds, P.; Hecht, F.; Delwart, E. New DNA Viruses Identified in Patients with Acute Viral Infection Syndrome. J. Virol. 2005, 79, 8230–8236. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.; Chan, K.; Tsoi, H.; Huang, Y.; Wong, B.H.L.; Poon, R.W.S.; Cai, J.J.; Luk, W.; et al. Characterization and Complete Genome Sequence of a Novel Coronavirus, Coronavirus HKU1, from Patients with Pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Andreasson, K.; Gupta, S.; Bjerkner, A.; Bogdanovic, G.; Persson, M.A.A.; Dalianis, T.; Ramqvist, T.; Andersson, B. Identification of a Third Human Polyomavirus. J. Virol. 2007, 81, 4130–4136. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, A.M.; Nissen, M.D.; Whiley, D.M.; Mackay, I.M.; Lambert, S.B.; Wu, G.; Brennan, D.C.; Storch, G.A.; Sloots, T.P.; Wang, D. Identification of a Novel Polyomavirus from Patients with Acute Respiratory Tract Infections. PLoS Pathog. 2007, 3. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A Cornucopia of Human Polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Taylor, J.L.; Benos, P.V.; Newton, R.; Waddell, K.; Lucas, S.B.; Chang, Y.; Moore, P.S. Human Transcriptome Subtraction by Using Short Sequence Tags To Search for Tumor Viruses in Conjunctival Carcinoma. J. Virol. 2007, 81, 11332–11340. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Krishnamurthy, S.; Cai, Z.; Popov, V.L.; Travassos da Rosa, A.P.; Guzman, H.; Cao, S.; Virgin, H.W.; Tesh, R.B.; Wang, D. Identification of Novel Viruses Using VirusHunter -- an Automated Data Analysis Pipeline. PLoS ONE 2013, 8, e78470. [Google Scholar] [CrossRef] [PubMed]
- Borozan, I.; Wilson, S.; Blanchette, P.; Laflamme, P.; Watt, S.N.; Krzyzanowski, P.M.; Sircoulomb, F.; Rottapel, R.; Branton, P.E.; Ferretti, V. CaPSID: A Bioinformatics Platform for Computational Pathogen Sequence Identification in Human Genomes and Transcriptomes. BMC Bioinformatics 2012, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Ojesina, A.I.; Pedamallu, C.S.; Jung, J.; Verhaak, R.G.W.; Getz, G.; Meyerson, M. PathSeq: Software to Identify or Discover Microbes by Deep Sequencing of Human Tissue. Nat. Biotechnol. 2011, 29, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Petty, T.J.; Cordey, S.; Padioleau, I.; Docquier, M.; Turin, L.; Preynat-Seauve, O.; Zdobnov, E.M.; Kaiser, L. Comprehensive Human Virus Screening Using High-Throughput Sequencing with a User-Friendly Representation of Bioinformatics Analysis: A Pilot Study. J. Clin. Microbiol. 2014, 52, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Qu, K.; Lee, C.S.; Ungewickell, A.; Khavari, P.A. Rapid Identification of Non-Human Sequences in High-Throughput Sequencing Datasets. Bioinformatics 2012, 28, 1174–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, P.; Zhao, Z. VirusFinder: Software for Efficient and Accurate Detection of Viruses and Their Integration Sites in Host Genomes through Next Generation Sequencing Data. PLoS ONE 2013, 8, e64465. [Google Scholar]
- Lysholm, F.; Wetterbom, A.; Lindau, C.; Darban, H.; Bjerkner, A.; Fahlander, K.; Lindberg, A.M.; Persson, B.; Allander, T.; Andersson, B. Characterization of the Viral Microbiome in Patients with Severe Lower Respiratory Tract Infections, Using Metagenomic Sequencing. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mullighan, C.G.; Easton, J.; Roberts, S.; Heatley, S.L.; Ma, J.; Rusch, M.C.; Chen, K.; Harris, C.C.; Ding, L.; et al. CREST Maps Somatic Structural Variation in Cancer Genomes with Base-Pair Resolution. Nat. Methods 2011, 8, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, B.; Boeva, V.; Janoueix-Lerosey, I.; Loeillet, S.; Legoix-né, P.; Nicolas, A.; Delattre, O.; Barillot, E. SVDetect: A Tool to Identify Genomic Structural Variations from Paired-End and Mate-Pair Sequencing Data. Bioinformatics 2010, 26, 1895–1896. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.-C.; Enge, B.; et al. A Cloud-Compatible Bioinformatics Pipeline for Ultrarapid Pathogen Identification from next-Generation Sequencing of Clinical Samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Zaharia, M.; Bolosky, W.J.; Curtis, K.; Fox, A.; Patterson, D.; Shenker, S.; Stoica, I.; Karp, R.M.; Sittler, T. Faster and More Accurate Sequence Alignment with SNAP. 2011; arXiv:1111.5572. [Google Scholar]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A Fast and Memory-Efficient Protein Similarity Search Tool for next-Generation Sequencing Data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, J.; Durbin, R. Mapping Short DNA Sequencing Reads and Calling Variants Using Mapping Quality Scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M.; Oude Munnink, B.; Canuti, M.; Deijs, M.; Watson, S.J.; Kellam, P.; van der Hoek, L. Full Genome Virus Detection in Fecal Samples Using Sensitive Nucleic Acid Preparation, Deep Sequencing, and a Novel Iterative Sequence Classification Algorithm. PLoS ONE 2014, 9, e93269. [Google Scholar]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative Analysis of Environmental Sequences Using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A New Arenavirus in a Cluster of Fatal Transplant-Associated Diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and Future Perspectives in Virus Discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A Highly Abundant Bacteriophage Discovered in the Unknown Sequences of Human Faecal Metagenomes. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Malboeuf, C.M.; Yang, X.; Charlebois, P.; Qu, J.; Berlin, A.M.; Casali, M.; Pesko, K.N.; Boutwell, C.L.; DeVincenzo, J.P.; Ebel, G.D.; et al. Complete Viral RNA Genome Sequencing of Ultra-Low Copy Samples by Sequence-Independent Amplification. Nucleic Acids Res. 2012, gks794. [Google Scholar] [CrossRef] [PubMed]
- Whitacre, L.K.; Tizioto, P.C.; Kim, J.; Sonstegard, T.S.; Schroeder, S.G.; Alexander, L.J.; Medrano, J.F.; Schnabel, R.D.; Taylor, J.F.; Decker, J.E. What’s in Your next-Generation Sequence Data? An Exploration of Unmapped DNA and RNA Sequence Reads from the Bovine Reference Individual. bioRxiv 2015, 022731. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M.; Møller Aarestrup, F.; Lund, O. In Silico Prediction of Human Pathogenicity in the γ-Proteobacteria. PLoS ONE 2010, 5, e13680. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, D.N.; Relman, D.A. Sequence-Based Identification of Microbial Pathogens: A Reconsideration of Koch’s Postulates. Clin. Microbiol. Rev. 1996, 9, 18–33. [Google Scholar]
- Simmons, G.; Glynn, S.A.; Komaroff, A.L.; Mikovits, J.A.; Tobler, L.H.; Hackett, J.; Tang, N.; Switzer, W.M.; Heneine, W.; Hewlett, I.K.; et al. Failure to Confirm XMRV/MLVs in the Blood of Patients with Chronic Fatigue Syndrome: A Multi-Laboratory Study. Science 2011, 334, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. The Perils of Pathogen Discovery: Origin of a Novel Parvovirus-Like Hybrid Genome Traced to Nucleic Acid Extraction Spin Columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed]
- Smuts, H.; Kew, M.; Khan, A.; Korsman, S. Novel Hybrid Parvovirus-Like Virus, NIH-CQV/PHV, Contaminants in Silica Column-Based Nucleic Acid Extraction Kits. J. Virol. 2014, 88, 1398–1398. [Google Scholar] [CrossRef] [PubMed]
- Kjartansdóttir, K.R.; Friis-Nielsen, J.; Asplund, M.; Mollerup, S.; Mourier, T.; Jensen, R.H.; Hansen, T.A.; Rey-Iglesia, A.; Richter, S.R.; Alquezar-Planas, D.E.; et al. Traces of ATCV-1 Associated with Laboratory Component Contamination. Proc. Natl. Acad. Sci. 2015, 112, E925–E926. [Google Scholar] [CrossRef] [PubMed]
- Vinner, L.; Mourier, T.; Friis-Nielsen, J.; Gniadecki, R.; Dybkaer, K.; Rosenberg, J.; Langhoff, J.L.; Cruz, D.F.S.; Fonager, J.; Izarzugaza, J.M.G.; et al. Investigation of Human Cancers for Retrovirus by Low-Stringency Target Enrichment and High-Throughput Sequencing. Sci. Rep. 2015, 5, 13201. [Google Scholar] [CrossRef] [PubMed]
- Rosseel, T. False-Positive Results in Metagenomic Virus Discovery: A Strong Case for Follow-Up Diagnosis. Transbound. Emerg. Dis. 2014, 61, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Zhi, N.; Hu, G.; Wan, Z.; Zheng, X.; Liu, X.; Wong, S.; Kajigaya, S.; Zhao, K.; Mao, Q.; et al. Hybrid DNA Virus in Chinese Patients with Seronegative Hepatitis Discovered by Deep Sequencing. Proc. Natl. Acad. Sci. 2013, 110, 10264–10269. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Kelso, J. High-Throughput DNA Sequencing – Concepts and Limitations. BioEssays 2010, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.H.; Mollerup, S.; Mourier, T.; Hansen, T.A.; Fridholm, H.; Nielsen, L.P.; Willerslev, E.; Hansen, A.J.; Vinner, L. Target-Dependent Enrichment of Virions Determines the Reduction of High-Throughput Sequencing in Virus Discovery. PLoS ONE 2015, 10, e0122636. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.A.; Fridholm, H.; Frøslev, T.G.; Kjartansdóttir, K.R.; Willerslev, E.; Nielsen, L.P.; Hansen, A.J. New Type of Papillomavirus and Novel Circular Single Stranded DNA Virus Discovered in Urban Rattus Norvegicus Using Circular DNA Enrichment and Metagenomics. PLoS ONE 2015, 10, e0141952. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S. AdapterRemoval: Easy Cleaning of Next Generation Sequencing Reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. 2013; arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAM tools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Gertz, E.M.; Schäffer, A.A.; Agarwala, R. A Fast and Symmetric DUST Implementation to Mask Low-Complexity DNA Sequences. J. Comput. Biol. 2006, 13, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de Novo Assembler for Single-Cell and Metagenomic Sequencing Data with Highly Uneven Depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mulder, C.P.H.; Bazeley-white, E.; Dimitrakopoulos, P.G.; Hector, A.; Scherer-lorenzen, M.; Schmid, B. Species evenness and productivity in experimental plant communities. Oikos 2004, 107, 50–63. [Google Scholar] [CrossRef]
- Perbal, B. Avian Myeoloblastosis Virus (AMV): Only One Side of the Coin. Retrovirology 2008, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A Mathematical Theory of Communication. SIGMOBILE Mob. Comput. Commun. Rev. 2001, 5, 3–55. [Google Scholar] [CrossRef]
- Noble, W.S. How Does Multiple Testing Correction Work? Nat. Biotechnol. 2009, 27, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Gevers, D.; Petrosino, J.F.; Abubucker, S.; Badger, J.H.; et al. A Framework for Human Microbiome Research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s Conserved Domain Database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Seguritan, V.; Alves, N.; Arnoult, M.; Raymond, A.; Lorimer, D.; Burgin, A.B.; Salamon, P.; Segall, A.M. Artificial Neural Networks Trained to Detect Viral and Phage Structural Proteins. PLoS Comput. Biol. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature type | BLASTn | BLASTx | Unmapped | Total |
|---|---|---|---|---|
| Biological | 593 | 5 | 4 | 602 |
| Methodological | 2662 | 1515 | 868 | 5045 |
| Technical | 298 | 110 | 110 | 518 |
| Total | 3553 | 1630 | 982 | 6165 |
| Feature | Cluster annotation (species) | #sig. | p-value | HMP body site |
|---|---|---|---|---|
| Colon cancer biopsy | Bacteroides fragilis | 2 | 2.43e-20 | Gastrointestinal tract |
| Colon cancer biopsy | Faecalibacterium prausnitzii | 3 | 1.60e-20 | Gastrointestinal tract |
| Colon cancer biopsy | Eubacterium rectale | 1 | 2.92e-17 | Gastrointestinal tract |
| Colon cancer biopsy | Alistipes shahii | 1 | 1.34e-13 | Gastrointestinal tract |
| Oral cavity cancer | Prevotella melaninogenica | 292 | 1.74e-24 | Oral |
| Oral cavity cancer | Streptococcus agalactiae | 2 | 4.60e-23 | Oral |
| Oral cavity cancer | Prevotella veroralis | 8 | 1.73e-21 | Oral |
| Oral cavity cancer | Prevotella histicola | 1 | 5.37e-16 | Oral |
| Oral cavity cancer | Streptococcus oralis | 22 | 2.31e-15 | Oral |
| Oral cavity cancer | Prevotella dentalis | 7 | 2.31e-15 | Oral |
| Oral cavity cancer | Porphyromonas gingivalis | 2 | 4.49e-14 | Oral |
| Oral cavity cancer | Solobacterium moorei | 1 | 1.34e-13 | Oral |
| Oral cavity cancer | Treponema denticola | 2 | 8.26e-13 | Oral |
| Oral cavity cancer | Campylobacter rectus | 1 | 2.60e-12 | Oral |
| Oral cavity cancer | Filifactor alocis | 2 | 4.12e-11 | Oral |
| Oral cavity cancer | Streptococcus dysgalactiae | 1 | 4.12e-11 | Oral |
| Oral cavity cancer | Prevotella sp. oral taxon 306 | 1 | 4.85e-11 | Oral |
| Vulva cancer | Campylobacter ureolyticus | 1 | 1.03e-12 | Urogenital tract |
| Cluster representative | BLASTn | BLASTx | CCD |
|---|---|---|---|
| 1789 bp | - | Prevotella veroralis (WP_026284690.1) 92% / 42% | - |
| 3246 bp | Prevotella fusca JCM 17724 (CP012075.1) 76% / 18% | Prevotella veroralis (WP_004384161.1) 90% / 56% | DUF4280 super family (cl16620) TauE super family (cl21514) |
| 4661 bp | Prevotella fusca JCM 17724 (CP012075.1) 91% / 87% | Prevotella fusca (WP_050696472.1) 85% / 66% | Peptidase_M23 (pfam01551) lysozyme_like super family (cl00222) DUF4280 (pfam14107) Fil_haemagg_2 (pfam13332) Phage_base_V (pfam04717) |
| 4720 bp | Eubacterium sulci ATCC 35585 (CP012068.1) 71% / 21% | Peptostreptococcus anaerobius CAG:621 (CCY47489.1) 72% / 36% | Acyl_transf_3 super family (cl21495) ND5 (MTH00095) rve (pfam00665) ND2 super family (cl10157) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friis-Nielsen, J.; Kjartansdóttir, K.R.; Mollerup, S.; Asplund, M.; Mourier, T.; Jensen, R.H.; Hansen, T.A.; Rey-Iglesia, A.; Richter, S.R.; Nielsen, I.B.; et al. Identification of Known and Novel Recurrent Viral Sequences in Data from Multiple Patients and Multiple Cancers. Viruses 2016, 8, 53. https://doi.org/10.3390/v8020053
Friis-Nielsen J, Kjartansdóttir KR, Mollerup S, Asplund M, Mourier T, Jensen RH, Hansen TA, Rey-Iglesia A, Richter SR, Nielsen IB, et al. Identification of Known and Novel Recurrent Viral Sequences in Data from Multiple Patients and Multiple Cancers. Viruses. 2016; 8(2):53. https://doi.org/10.3390/v8020053
Chicago/Turabian StyleFriis-Nielsen, Jens, Kristín Rós Kjartansdóttir, Sarah Mollerup, Maria Asplund, Tobias Mourier, Randi Holm Jensen, Thomas Arn Hansen, Alba Rey-Iglesia, Stine Raith Richter, Ida Broman Nielsen, and et al. 2016. "Identification of Known and Novel Recurrent Viral Sequences in Data from Multiple Patients and Multiple Cancers" Viruses 8, no. 2: 53. https://doi.org/10.3390/v8020053
APA StyleFriis-Nielsen, J., Kjartansdóttir, K. R., Mollerup, S., Asplund, M., Mourier, T., Jensen, R. H., Hansen, T. A., Rey-Iglesia, A., Richter, S. R., Nielsen, I. B., Alquezar-Planas, D. E., Olsen, P. V. S., Vinner, L., Fridholm, H., Nielsen, L. P., Willerslev, E., Sicheritz-Pontén, T., Lund, O., Hansen, A. J., ... Brunak, S. (2016). Identification of Known and Novel Recurrent Viral Sequences in Data from Multiple Patients and Multiple Cancers. Viruses, 8(2), 53. https://doi.org/10.3390/v8020053