
Analyses of Evolutionary Characteristics of the Hemagglutinin-Esterase Gene of Influenza C Virus during a Period of 68 Years Reveals Evolutionary Patterns Different from Influenza A and B Viruses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Data
2.2. Phylogenetic Tree
2.3. Phylodynamics
2.4. Hemagglutination Inhibition Test
2.5. Selection Pressure
3. Results
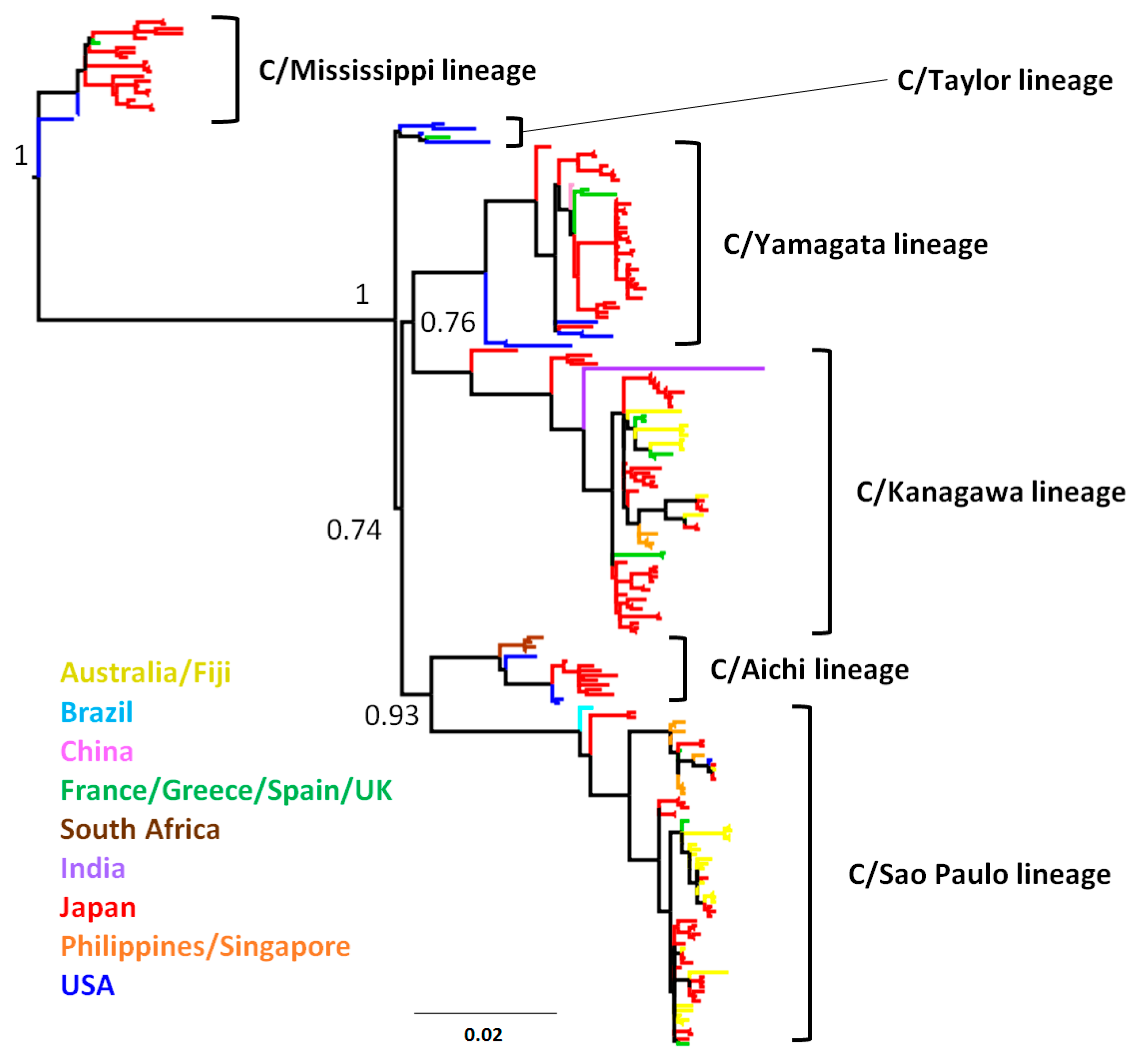
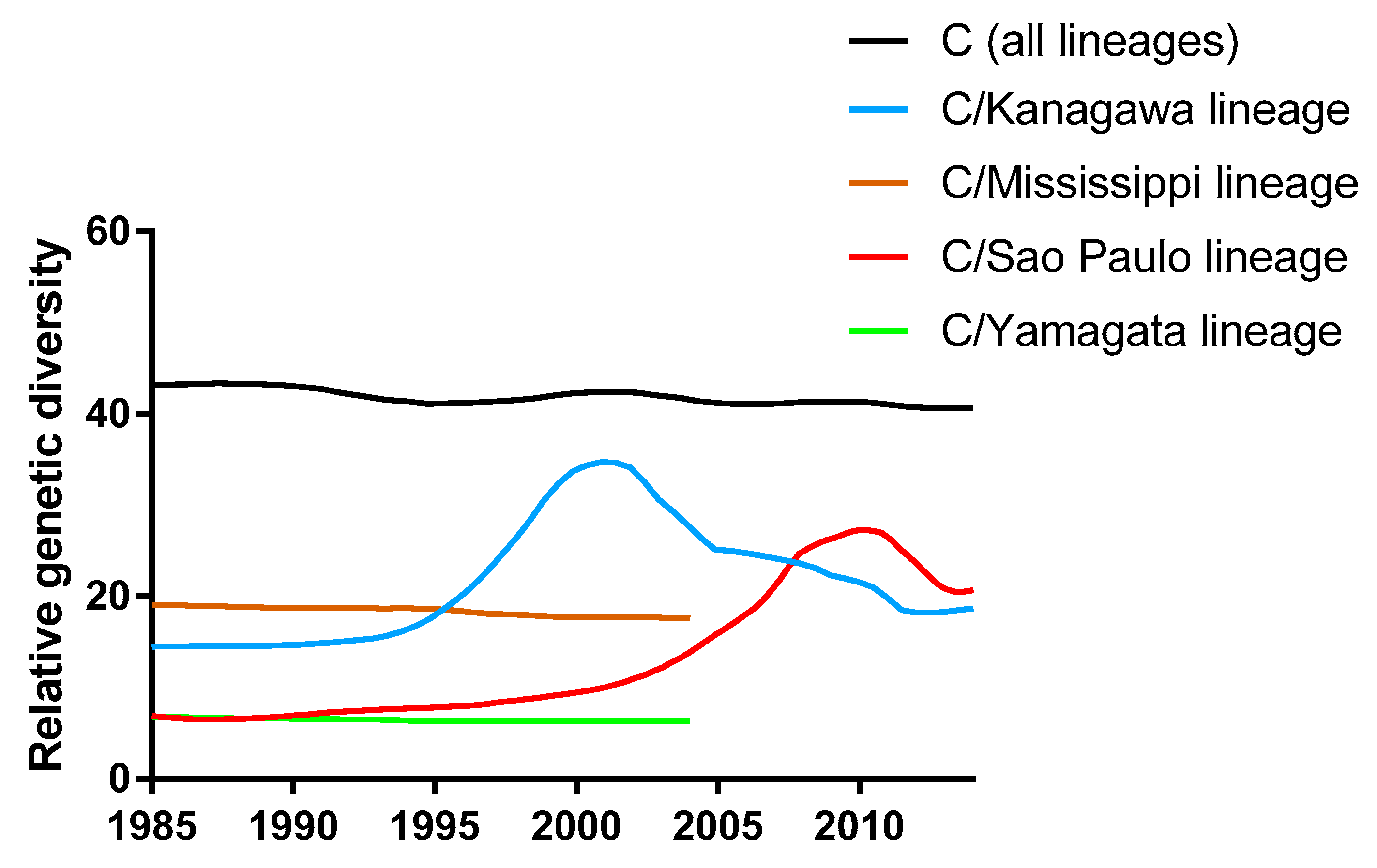
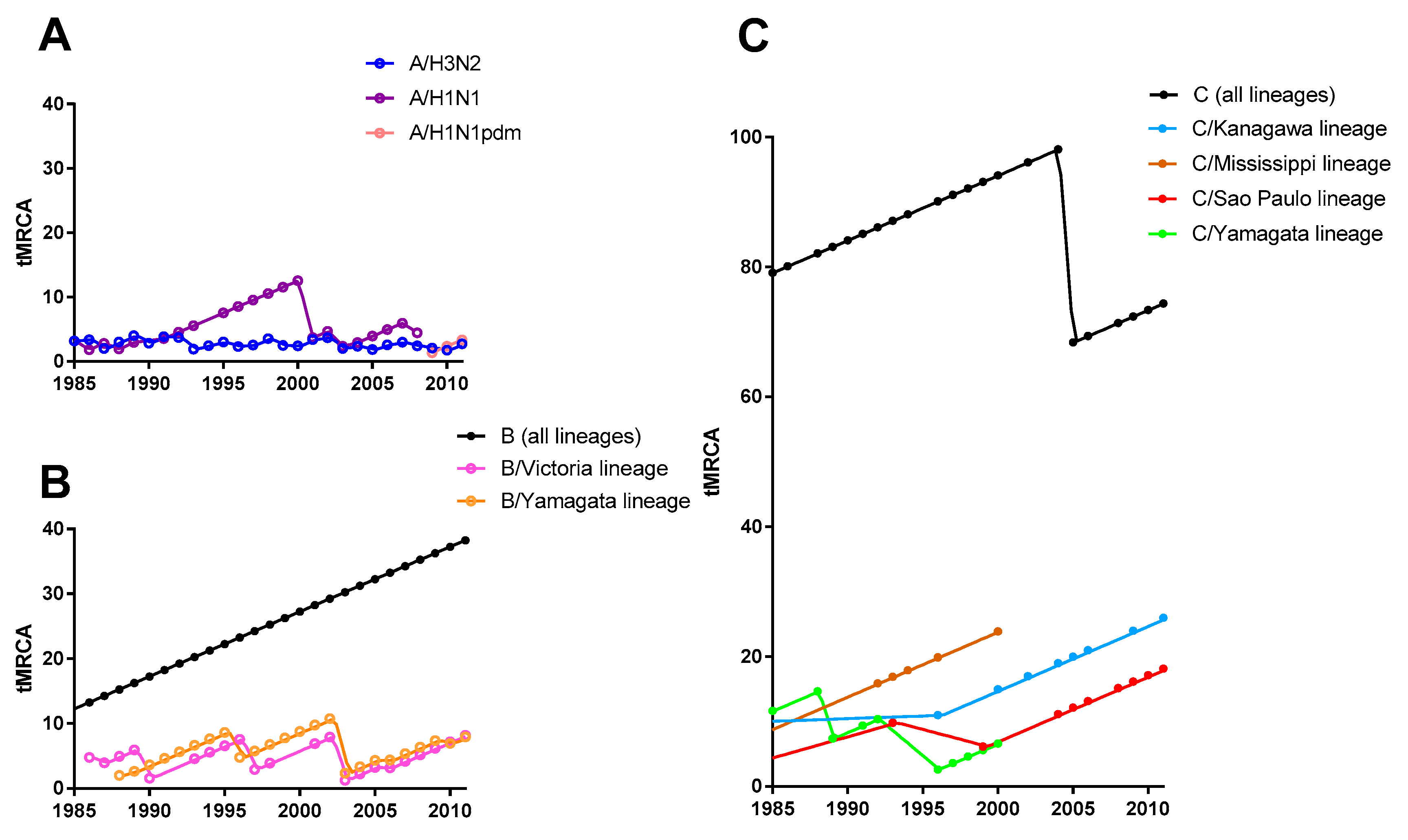
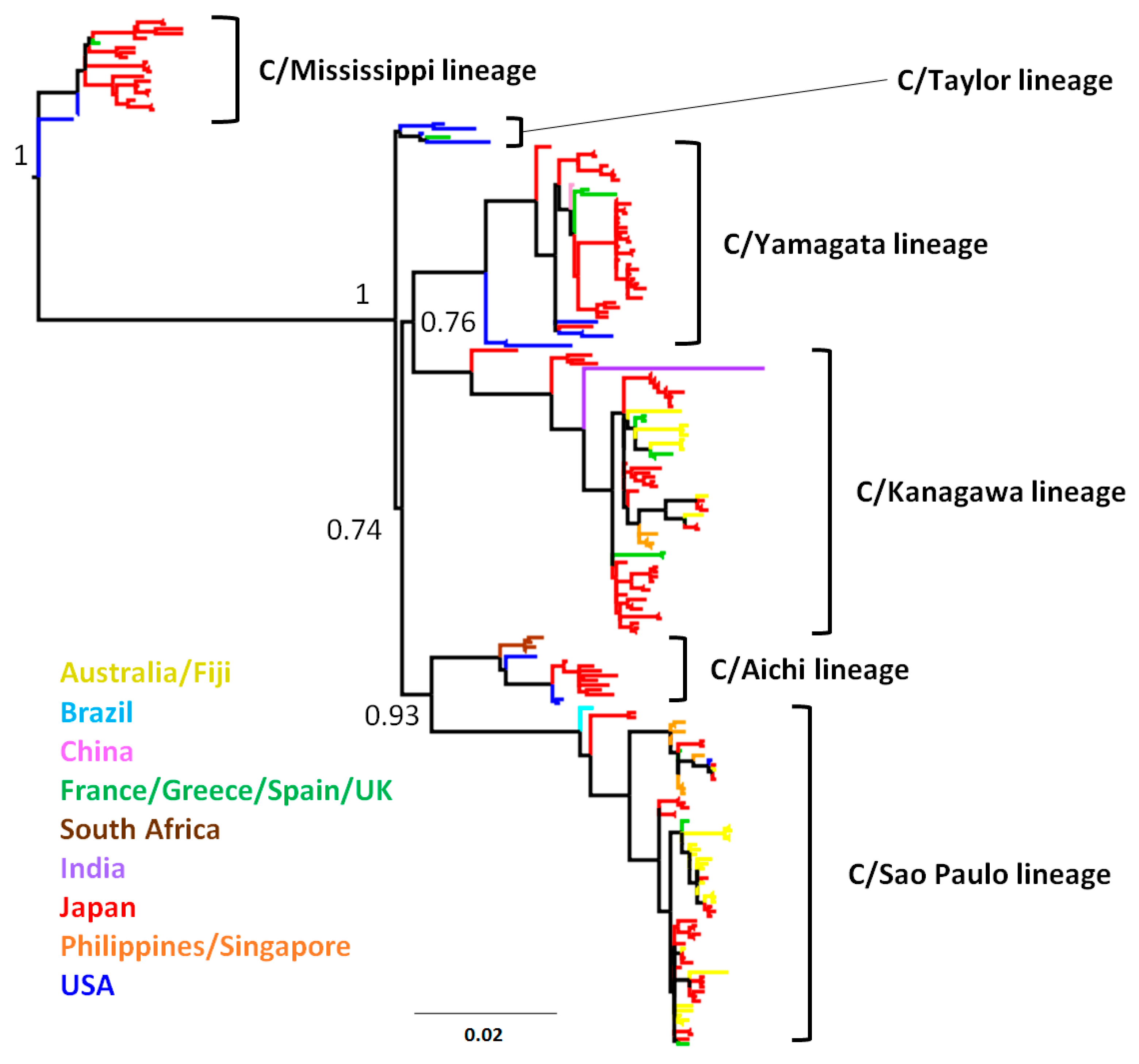
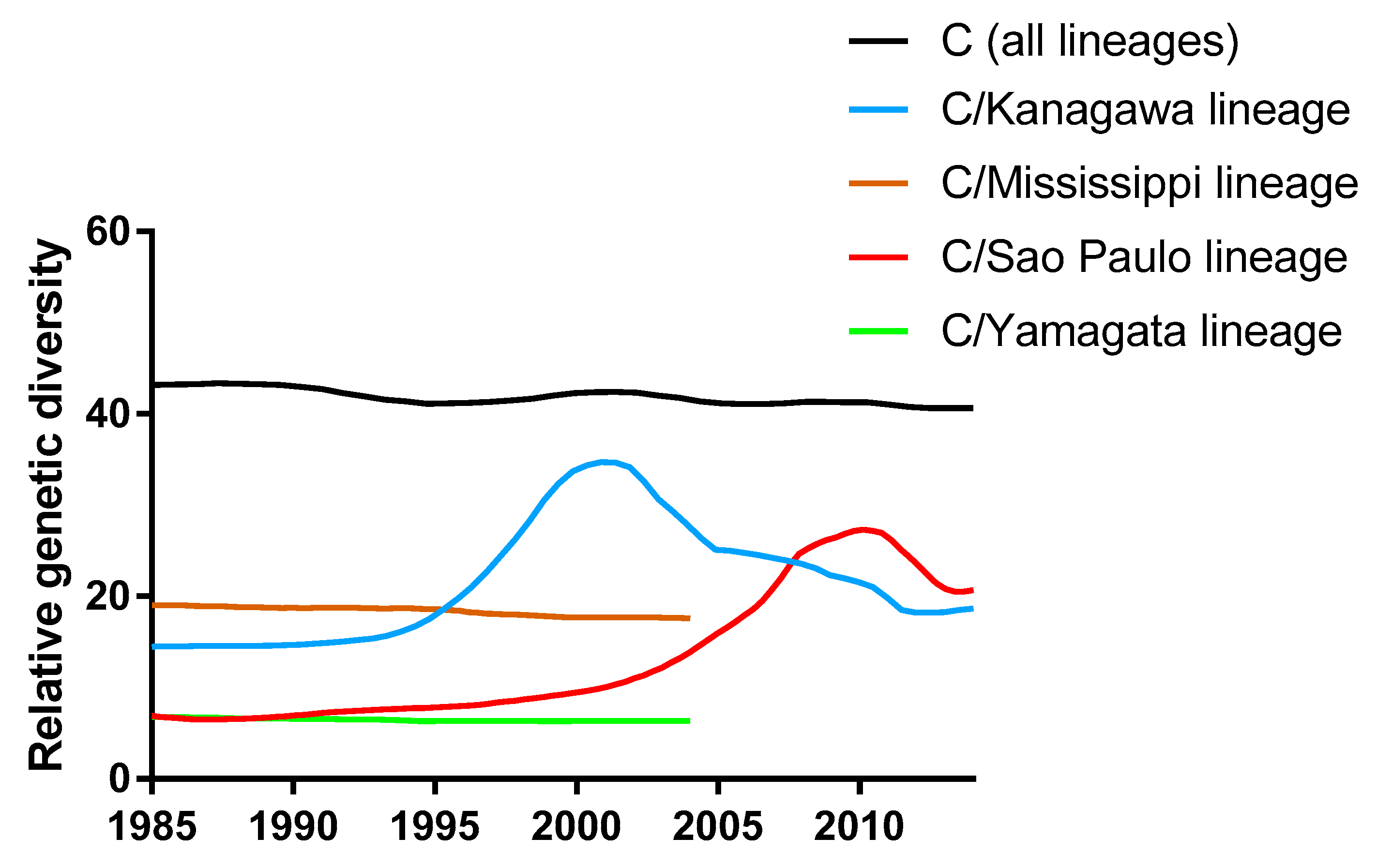
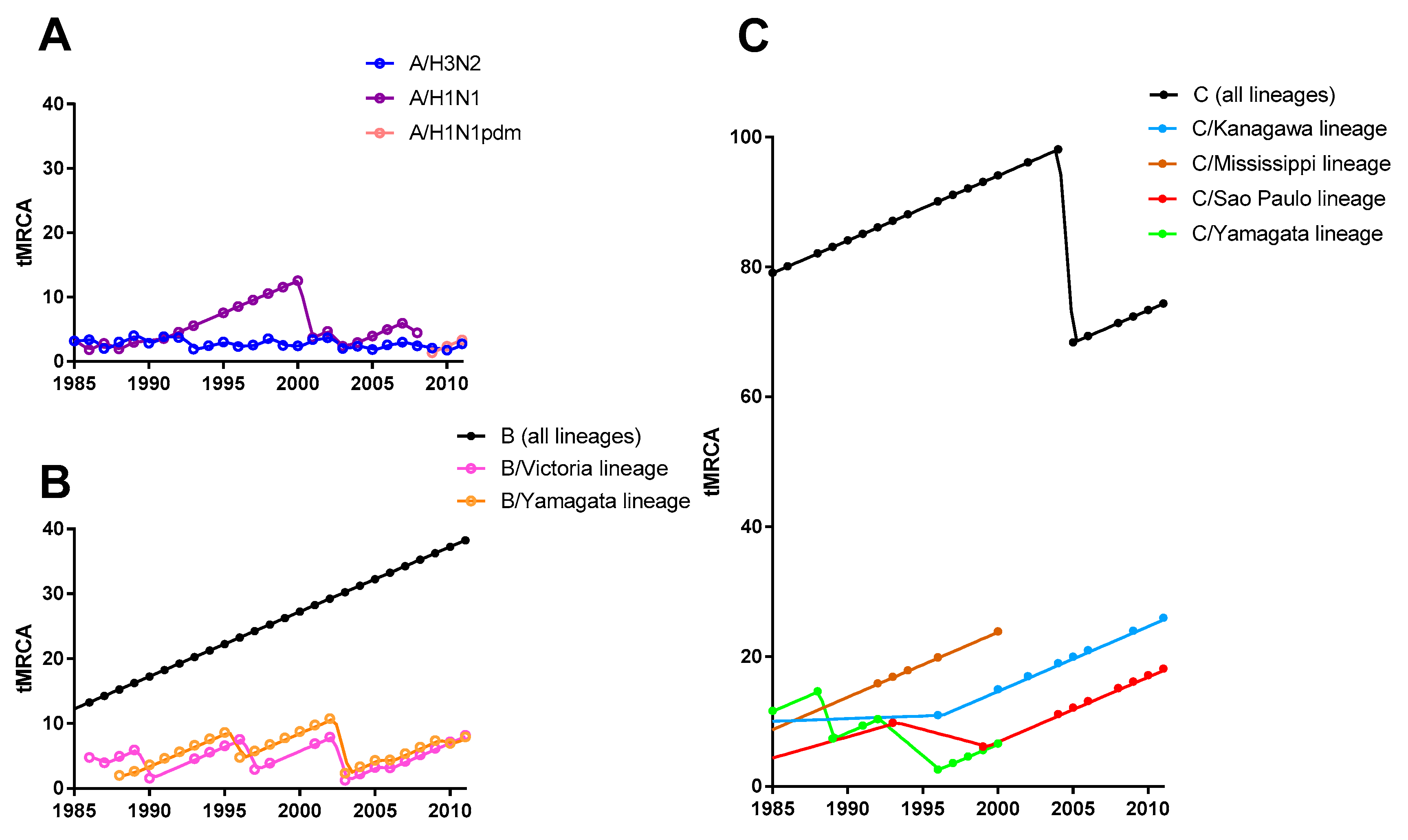
3.1. Phylogenetic Tree and Population Dynamics of Influenza C Virus
3.2. Selective Bottleneck
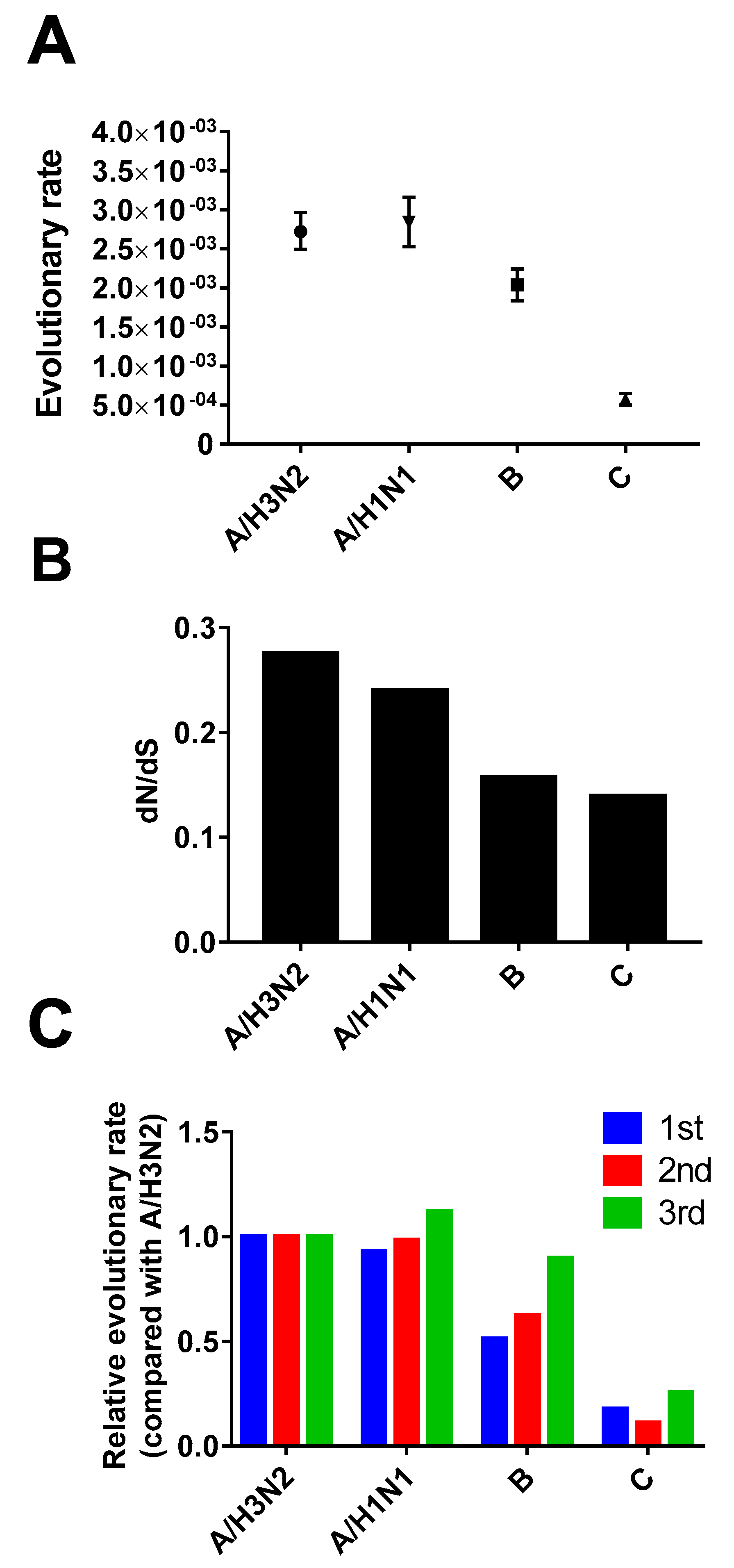
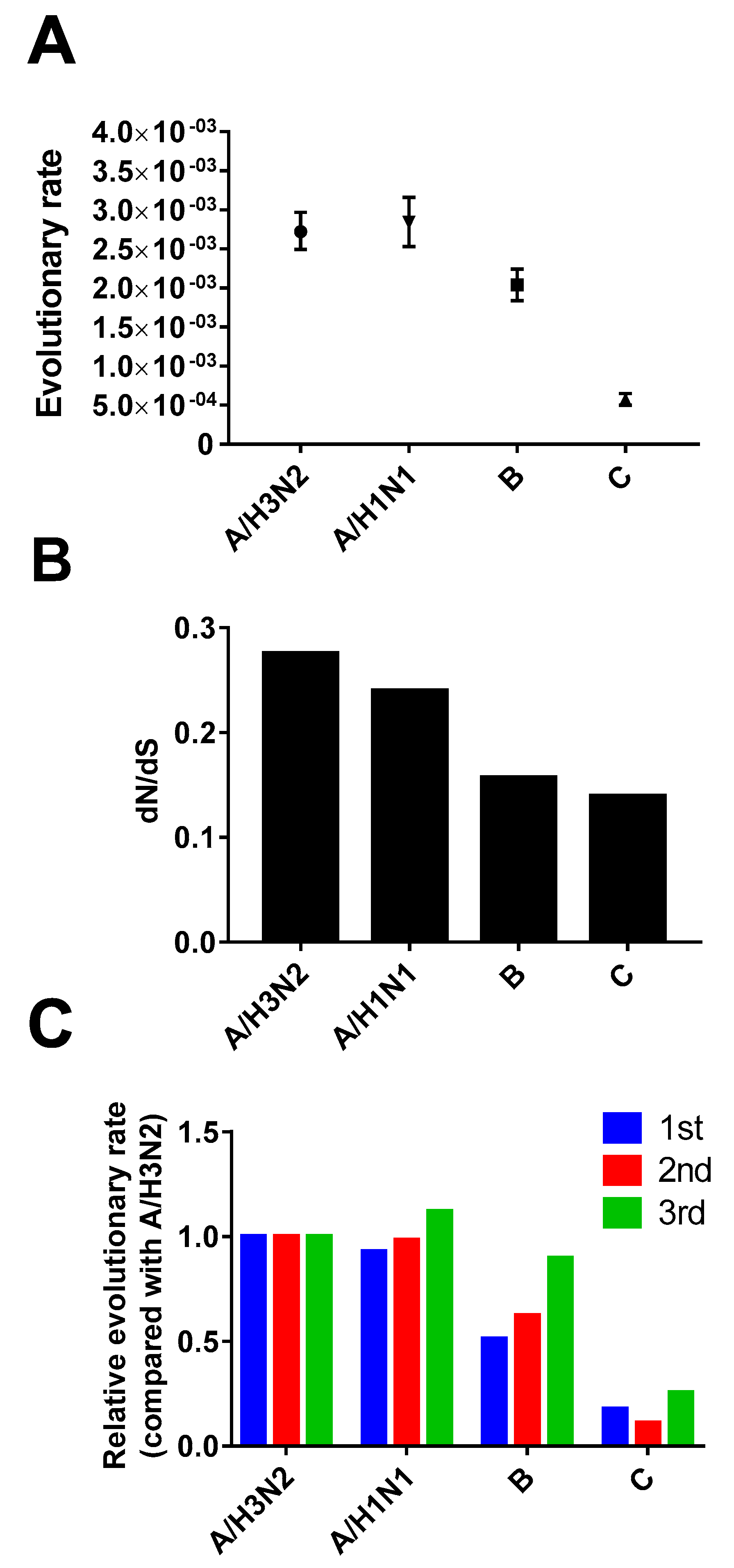
3.3. Evolutionary Rate and Selection Pressure
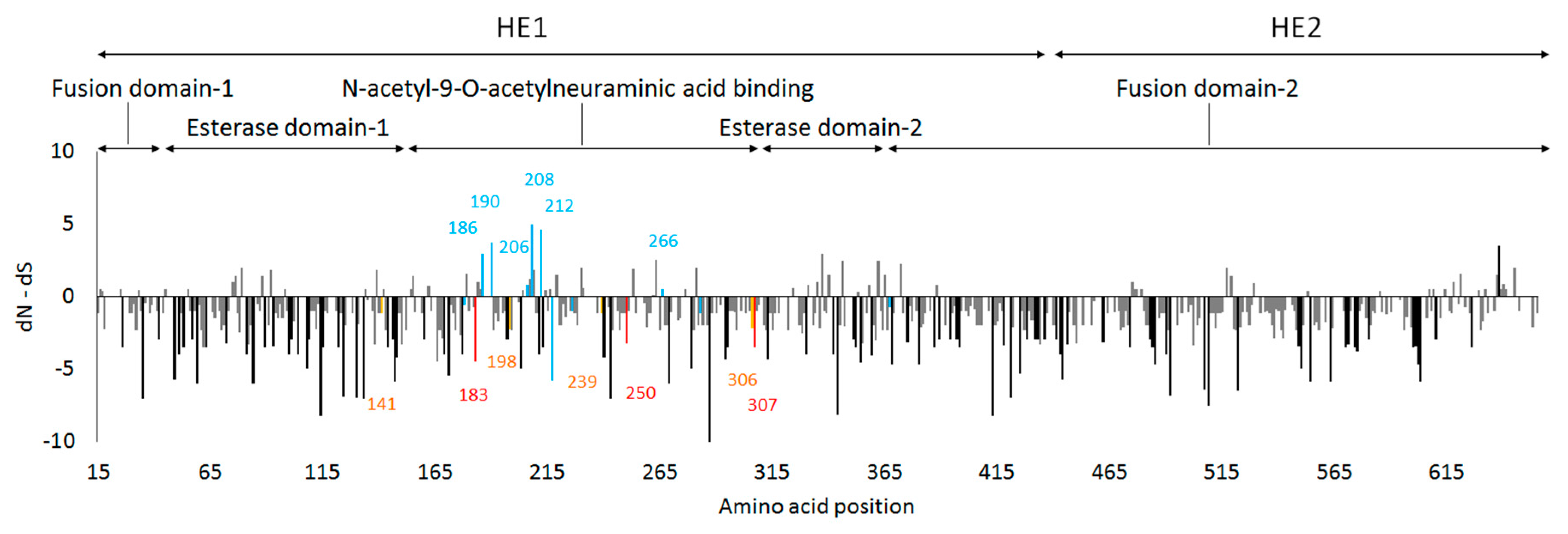
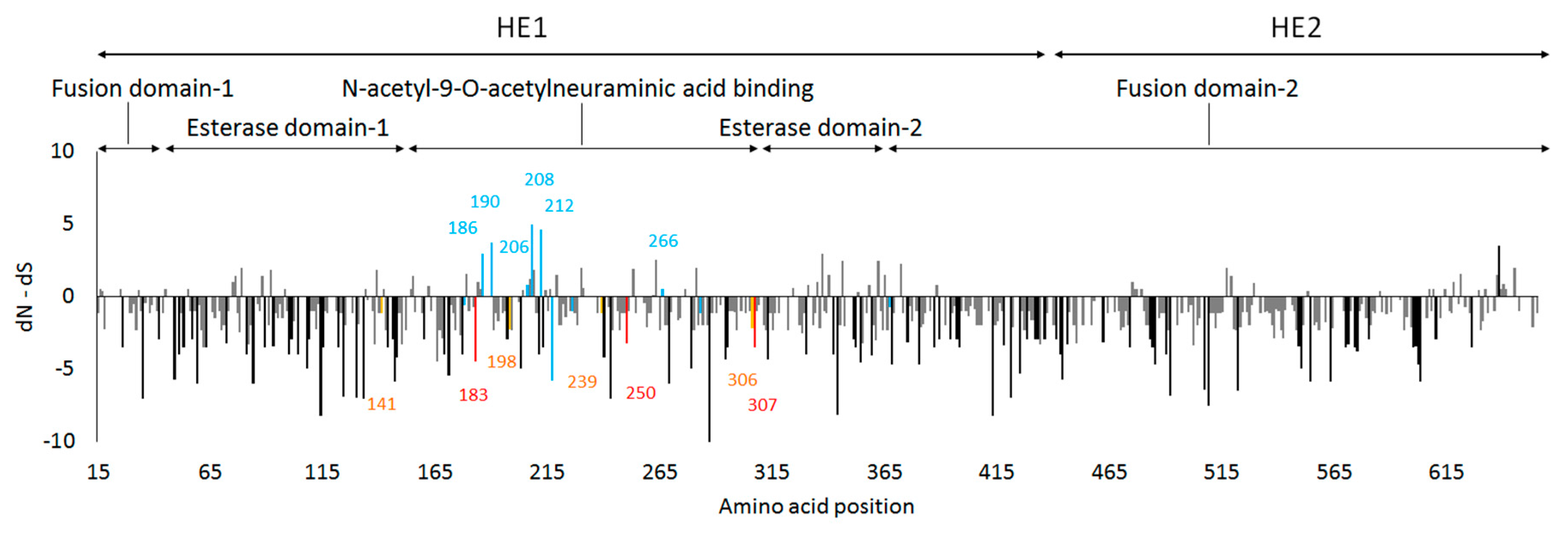
3.4. Site-by-Site Selective Pressure on the HE Gene
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Matsuzaki, Y.; Katsushima, N.; Nagai, Y.; Shoji, M.; Itagaki, T.; Sakamoto, M.; Kitaoka, S.; Mizuta, K.; Nishimura, H. Clinical features of influenza C virus infection in children. J. Infect. Dis. 2006, 193, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Dykes, A.C.; Cherry, J.D.; Nolan, C.E. A clinical, epidemiologic, serologic, and virologic study of influenza C virus infection. Arch. Intern. Med. 1980, 140, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, R.J.; Gohd, R.S.; Labat, D.D. Human antibody to influenza C virus: Its age-related distribution and distinction from receptor analogs. Infect. Immun. 1980, 30, 500–505. [Google Scholar] [PubMed]
- Homma, M.; Ohyama, S.; Katagiri, S. Age distribution of the antibody to type C influenza virus. Microbiol. Immunol. 1982, 26, 639–642. [Google Scholar] [PubMed]
- Salez, N.; Mélade, J.; Pascalis, H.; Aherfi, S.; Dellagi, K.; Charrel, R.N.; Carrat, F.; de Lamballerie, X. Influenza C virus high seroprevalence rates observed in 3 different population groups. J. Infect. 2014, 69, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Yuanji, G.; Fengen, J.; Ping, W. Isolation of influenza C virus from pigs and experimental infection of pigs with influenza C virus. J. Gen. Virol. 1983, 64, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Abiko, C.; Peng, G.; Muraki, Y.; Sugawara, K.; Hongo, S.; Kitame, F.; Mizuta, K.; Numazaki, Y.; Suzuki, H.; et al. Interspecies transmission of influenza C virus between humans and pigs. Virus Res. 1997, 48, 71–79. [Google Scholar] [CrossRef]
- Ohwada, K.; Kitame, F.; Sugawara, K.; Nishimura, H.; Homma, M.; Nakamura, K. Distribution of the antibody to influenza C virus in dogs and pigs in Yamagata Prefecture, Japan. Microbiol. Immunol. 1987, 31, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Herrler, G.; Compans, R.W.; Meier-Ewert, H. A precursor glycoprotein in influenza C virus. Virology 1979, 99, 49–56. [Google Scholar] [CrossRef]
- Sugawara, K.; Kitame, F.; Nishimura, H.; Nakamura, K. Operational and topological analyses of antigenic sites on influenza C virus glycoprotein and their dependence on glycosylation. J. Gen. Virol. 1988, 69 Pt 3, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Sugawara, K.; Adachi, K.; Hongo, S.; Nishimura, H.; Kitame, F.; Nakamura, K. Location of neutralizing epitopes on the hemagglutinin-esterase protein of influenza C virus. Virology 1992, 189, 79–87. [Google Scholar] [CrossRef]
- Sugawara, K.; Nishimura, H.; Hongo, S.; Muraki, Y.; Kitame, F.; Nakamura, K. Construction an antigenic map of the haemagglutinin-esterase protein of influenza C virus. J. Gen. Virol. 1993, 74, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.N.; Herrler, G.; Paulson, J.C.; Klenk, H.D. Influenza C virus uses 9-O-acetyl-N-acetylneuraminic acid as a high affinity receptor determinant for attachment to cells. J. Biol. Chem. 1986, 261, 5947–5951. [Google Scholar] [PubMed]
- Vlasak, R.; Krystal, M.; Nacht, M.; Palese, P. The influenza C virus glycoprotein (HE) exhibits receptor-binding (hemagglutinin) and receptor-destroying (esterase) activities. Virology 1987, 160, 419–425. [Google Scholar] [CrossRef]
- Ohuchi, M.; Ohuchi, R.; Mifune, K. Demonstration of hemolytic and fusion activities of influenza C virus. J. Virol. 1982, 42, 1076–1079. [Google Scholar] [PubMed]
- Taylor, R.M. Studies on survival of influenza virus between epidemics and antigenic variants of the virus. Am. J. Public Health Nations Health 1949, 39, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Sugawara, K.; Abiko, C.; Ikeda, T.; Aoki, Y.; Mizuta, K.; Katsushima, N.; Katsushima, F.; Katsushima, Y.; Itagaki, T.; et al. Epidemiological information regarding the periodic epidemics of influenza C virus in Japan (1996–2013) and the seroprevalence of antibodies to different antigenic groups. J. Clin. Virol. 2014, 61, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Sugawara, K.; Furuse, Y.; Shimotai, Y.; Hongo, S.; Oshitani, H.; Mizuta, K.; Nishimura, H. Genetic lineage and reassortment of influenza C viruses circulating between 1947 and 2014. J. Virol. 2016, 90, 8251–8265. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.A.; Jones, T.C.; Barr, I.G.; Cox, N.J.; Garten, R.J.; Gregory, V.; Gust, I.D.; Hampson, A.W.; Hay, A.J.; Hurt, A.C.; et al. The global circulation of seasonal influenza A (H3N2) viruses. Science 2008, 320, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Furuse, Y.; Suzuki, A.; Kamigaki, T.; Mpolya, E.A.; Khandaker, I.; Oshitani, H. Viruses that cross borders: Factors responsible for global dissemination of viral infections. Intervirology 2011, 54, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Lapedes, A.S.; de Jong, J.C.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Mapping the antigenic and genetic evolution of influenza virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Furuse, Y.; Odagiri, T.; Tamaki, R.; Kamigaki, T.; Otomaru, H.; Opinion, J.; Santo, A.; Dolina-Lacaba, D.; Daya, E.; Okamoto, M.; et al. Local persistence and global dissemination play a significant role in the circulation of influenza B viruses in Leyte Island, Philippines. Virology 2016, 492, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Bedford, T.; Cobey, S.; Beerli, P.; Pascual, M. Global migration dynamics underlie evolution and persistence of human influenza A (H3N2). PLoS Pathog. 2010, 6, e1000918. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [PubMed]
- Bush, R.M.; Fitch, W.M.; Bender, C.A.; Cox, N.J. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol. Biol. Evol. 1999, 16, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Bush, R.M.; Bender, C.A.; Subbarao, K.; Cox, N.J.; Fitch, W.M. Predicting the evolution of human influenza A. Science 1999, 286, 1921–1925. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Natural selection on the influenza virus genome. Mol. Biol. Evol. 2006, 23, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Furuse, Y.; Shimabukuro, K.; Odagiri, T.; Sawayama, R.; Okada, T.; Khandaker, I.; Suzuki, A.; Oshitani, H. Comparison of selection pressures on the HA gene of pandemic (2009) and seasonal human and swine influenza A H1 subtype viruses. Virology 2010, 405, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gaschen, B.; Blay, W.; Foley, B.; Haigwood, N.; Kuiken, C.; Korber, B. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 2004, 14, 1229–1246. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Takashita, E.; Sugawara, K.; Matsuzaki, Y.; Muraki, Y.; Hongo, S. Effect of the addition of oligosaccharides on the biological activities and antigenicity of influenza A/H3N2 virus hemagglutinin. J. Virol. 2004, 78, 9605–9611. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Kirk, B.D.; Ma, J.; Wang, Q. Diversifying selective pressure on influenza B virus hemagglutinin. J. Med. Virol. 2009, 81, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Krystal, M.; Fitch, W.M.; Palese, P. Influenza B virus evolution: Co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology 1988, 163, 112–122. [Google Scholar] [CrossRef]
- Muraki, Y.; Hongo, S.; Sugawara, K.; Kitame, F.; Nakamura, K. Evolution of the haemagglutinin-esterase gene of influenza C virus. J. Gen. Virol. 1996, 77, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Buonagurio, D.A.; Nakada, S.; Desselberger, U.; Krystal, M.; Palese, P. Noncumulative sequence changes in the hemagglutinin genes of influenza C virus isolates. Virology 1985, 146, 221–232. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Mizuta, K.; Kimura, H.; Sugawara, K.; Tsuchiya, E.; Suzuki, H.; Hongo, S.; Nakamura, K. Characterization of antigenically unique influenza C virus strains isolated in Yamagata and Sendai Cities, Japan, during 1992–1993. J. Gen. Virol. 2000, 81, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Jelley, L.; Levy, A.; Deng, Y.-M.; Spirason, N.; Lang, J.; Buettner, I.; Druce, J.; Blyth, C.; Effler, P.; Smith, D.; et al. Influenza C infections in Western Australia and Victoria from 2008 to 2014. Influenza Other Respir. Viruses 2016, 10, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Gaunt, E.R.; Digard, P.; Templeton, K.; Simmonds, P. Detection of influenza C virus but not influenza D virus in Scottish respiratory samples. J. Clin. Virol. 2016, 74, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Influenza Virus Resource. Available online: http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html (accessed on 7 May 2016).
- GISAID. Available online: http://platform.gisaid.org (accessed on 7 July 2016).
- Furuse, Y.; Suzuki, A.; Kamigaki, T.; Oshitani, H. Evolution of the M gene of the influenza A virus in different host species: Large-scale sequence analysis. Virol. J. 2009, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Desselberger, U.; Palese, P. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature 1978, 274, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Holmes, E.C.; Joseph, U.; Fourment, M.; Su, Y.C.F.; Halpin, R.; Lee, R.T.C.; Deng, Y.-M.M.; Gunalan, V.; Lin, X.; et al. The contrasting phylodynamics of human influenza B viruses. eLife 2015, 4, e05055. [Google Scholar] [CrossRef] [PubMed]
- Zinder, D.; Bedford, T.; Gupta, S.; Pascual, M. The roles of competition and mutation in shaping antigenic and genetic diversity in influenza. PLoS Pathog. 2013, 9, e1003104. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Sugawara, K.; Mizuta, K.; Tsuchiya, E.; Muraki, Y.; Hongo, S.; Suzuki, H.; Nakamura, K. Antigenic and genetic characterization of influenza C viruses which caused two outbreaks in Yamagata City, Japan, in 1996 and 1998. J. Clin. Microbiol. 2002, 40, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Takao, S.; Shimada, S.; Mizuta, K.; Sugawara, K.; Takashita, E.; Muraki, Y.; Hongo, S.; Nishimura, H. Characterization of antigenically and genetically similar influenza C viruses isolated in Japan during the 1999–2000 season. Epidemiol. Infect. 2004, 132, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Abiko, C.; Mizuta, K.; Sugawara, K.; Takashita, E.; Muraki, Y.; Suzuki, H.; Mikawa, M.; Shimada, S.; Sato, K.; et al. A nationwide epidemic of influenza C virus infection in Japan in 2004. J. Clin. Microbiol. 2007, 45, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Barclay, G.R.; Leader-Williams, L.K.; Flewett, T.H. Aspects of influenza C virus replication. J. Hyg. 1971, 69, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Hechtfischer, A.; Marschall, M.; Helten, A.; Böswald, C.; Meier-Ewert, H. A highly cytopathogenic influenza C virus variant induces apoptosis in cell culture. J. Gen. Virol. 1997, 78, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, K.; Tashiro, M.; Nakamura, K. Influenza C virus infection in rats. Microbiol. Immunol. 1990, 34, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, P.B.; Zhang, X.; Formanowski, F.; Fitz, W.; Wong, C.H.; Meier-Ewert, H.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 1998, 396, 92–96. [Google Scholar] [PubMed]
- Ferguson, N.M.; Galvani, A.P.; Bush, R.M. Ecological and immunological determinants of influenza evolution. Nature 2003, 422, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, V.; Sasaki, A. Shaping the phylogenetic tree of influenza by cross-immunity. Theor. Popul. Biol. 2006, 70, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Omori, R.; Adams, B.; Sasaki, A. Coexistence conditions for strains of influenza with immune cross-reaction. J. Theor. Biol. 2010, 262, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Manuguerra, J.C.; Hannoun, C.; Aymard, M. Influenza C virus infection in France. J. Infect. 1992, 24, 91–99. [Google Scholar] [CrossRef]
- Sauerbrei, A.; Langenhan, T.; Brandstädt, A.; Schmidt-Ott, R.; Krumbholz, A.; Girschick, H.; Huppertz, H.; Kaiser, P.; Liese, J.; Streng, A.; et al. Prevalence of antibodies against influenza A and B viruses in children in Germany, 2008 to 2010. Eurosurveillance 2014, 19, pii: 20687. [Google Scholar] [CrossRef]
- Otomaru, H.; Kamigaki, T.; Tamaki, R.; Opinion, J.; Santo, A.; Daya, E.; Okamoto, M.; Saito, M.; Tallo, V.; Lupisan, S.; et al. Influenza and other respiratory viruses detected by influenza-like illness surveillance in Leyte Island, the Philippines, 2010–2013. PLoS ONE 2015, 10, e0123755. [Google Scholar] [CrossRef] [PubMed]
- Calvo, C.; Garcia-Garcia, M.L.; Borrell, B.; Pozo, F.; Casas, I. Prospective study of influenza C in hospitalized children. Pediatr. Infect. Dis. J. 2013, 32, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Bedford, T.; Riley, S.; Barr, I.G.; Broor, S.; Chadha, M.; Cox, N.J.; Daniels, R.S.; Gunasekaran, C.P.; Hurt, A.C.; Kelso, A.; et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 2015, 523, 217–220. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigenic Group | Reference Virus | Amino Acid at Position 190 | Amino Acid at Position 212 | HI Titer of Monoclonal Antibodies | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| J14 | J9 | Q5 | U4 | U9 | U1 | U2 | MS22 | ||||
| KA | C/Kanagawa/1/76 | D | E | 256,000 | <20 | 1280 | 20 | 80 | <20 | <20 | <20 |
| AO | C/Aomori/74 | D | K | 1,024,000 | <20 | 640 | <20 | 40 | 3200 | 640 | <20 |
| MI | C/Miyagi/77 | N | E | 1,024,000 | <20 | 6400 | 2560 | 64,000 | <20 | <20 | <20 |
| KA | AO | MI | Undetermined | Total | |
|---|---|---|---|---|---|
| 1996 | 1 | 0 | 0 | 0 | 1 |
| 2000 | 6 | 0 | 0 | 0 | 6 |
| 2001 | 2 | 0 | 0 | 0 | 2 |
| 2002 | 31 | 17 | 3 | 1 | 52 |
| 2003 | 0 | 0 | 0 | 0 | 0 |
| 2004 | 59 | 10 | 9 | 0 | 78 |
| 2005 | 0 | 7 | 0 | 0 | 7 |
| 2006 | 0 | 3 | 2 | 0 | 5 |
| 2007 | 0 | 0 | 0 | 0 | 0 |
| 2008 | 0 | 0 | 0 | 0 | 0 |
| 2009 | 0 | 0 | 0 | 0 | 0 |
| 2010 | 0 | 0 | 0 | 0 | 0 |
| 2011 | 0 | 0 | 0 | 0 | 0 |
| 2012 | 8 | 0 | 0 | 0 | 8 |
| 2013 | 0 | 0 | 0 | 0 | 0 |
| 2014 | 21 | 0 | 0 | 0 | 21 |
| Total | 128 | 37 | 14 | 1 | 180 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuse, Y.; Matsuzaki, Y.; Nishimura, H.; Oshitani, H. Analyses of Evolutionary Characteristics of the Hemagglutinin-Esterase Gene of Influenza C Virus during a Period of 68 Years Reveals Evolutionary Patterns Different from Influenza A and B Viruses. Viruses 2016, 8, 321. https://doi.org/10.3390/v8120321
Furuse Y, Matsuzaki Y, Nishimura H, Oshitani H. Analyses of Evolutionary Characteristics of the Hemagglutinin-Esterase Gene of Influenza C Virus during a Period of 68 Years Reveals Evolutionary Patterns Different from Influenza A and B Viruses. Viruses. 2016; 8(12):321. https://doi.org/10.3390/v8120321
Chicago/Turabian StyleFuruse, Yuki, Yoko Matsuzaki, Hidekazu Nishimura, and Hitoshi Oshitani. 2016. "Analyses of Evolutionary Characteristics of the Hemagglutinin-Esterase Gene of Influenza C Virus during a Period of 68 Years Reveals Evolutionary Patterns Different from Influenza A and B Viruses" Viruses 8, no. 12: 321. https://doi.org/10.3390/v8120321
APA StyleFuruse, Y., Matsuzaki, Y., Nishimura, H., & Oshitani, H. (2016). Analyses of Evolutionary Characteristics of the Hemagglutinin-Esterase Gene of Influenza C Virus during a Period of 68 Years Reveals Evolutionary Patterns Different from Influenza A and B Viruses. Viruses, 8(12), 321. https://doi.org/10.3390/v8120321