
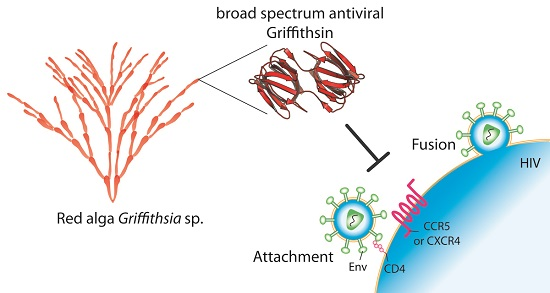
Griffithsin: An Antiviral Lectin with Outstanding Therapeutic Potential

Abstract
: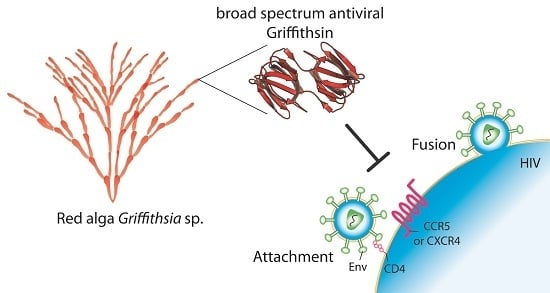
1. Introduction
2. Discovery and Recombinant Expression of Griffithsin
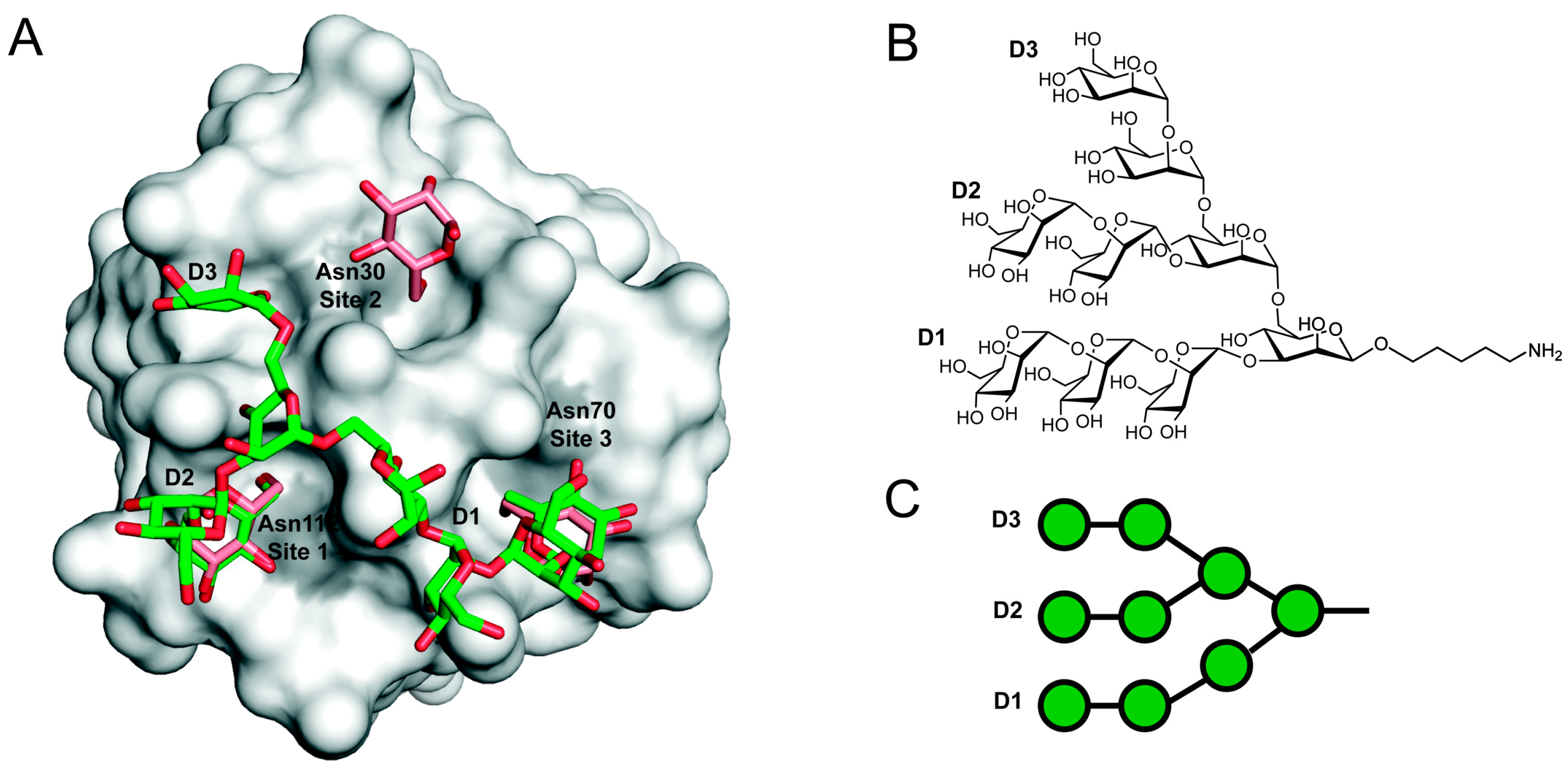
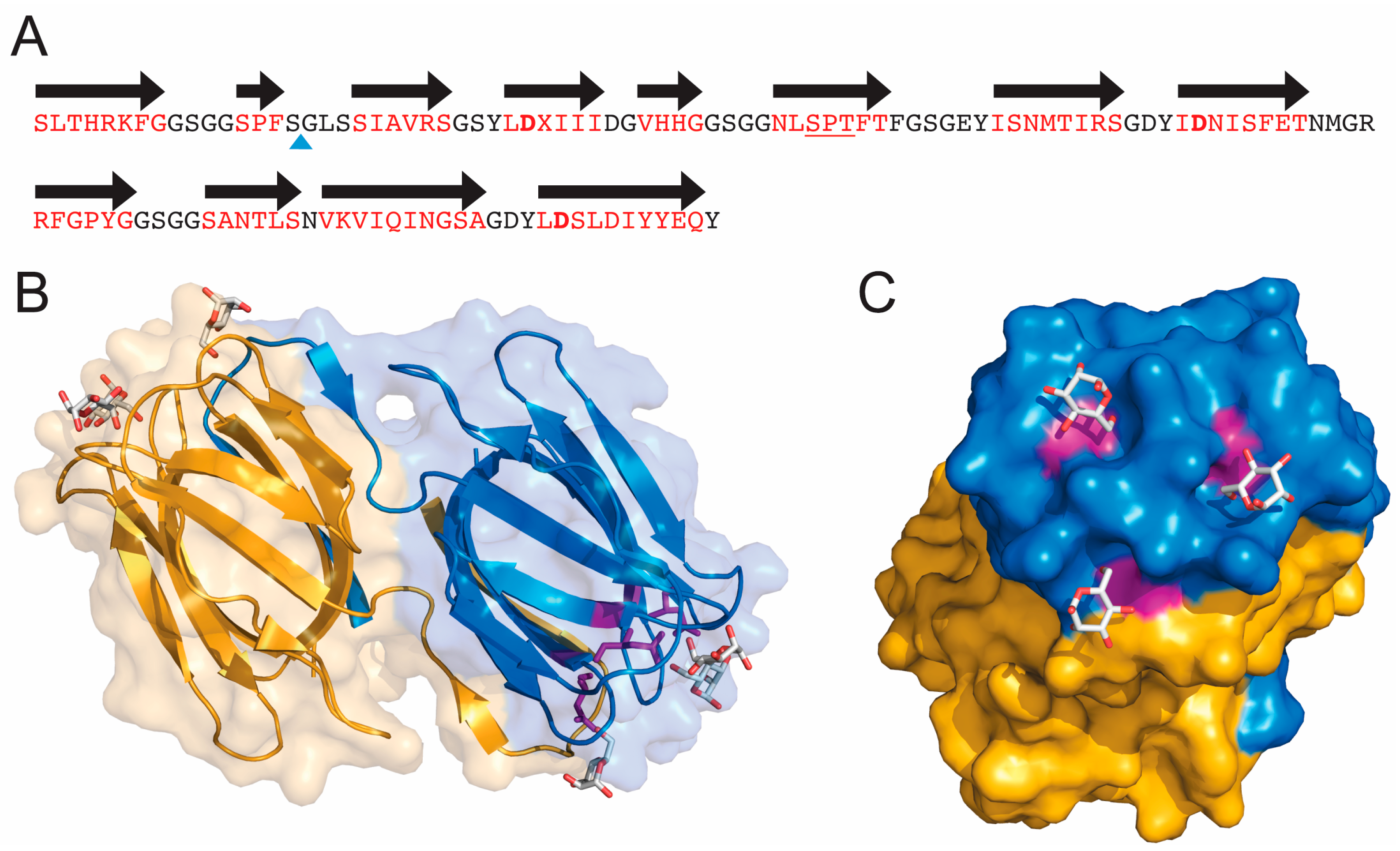
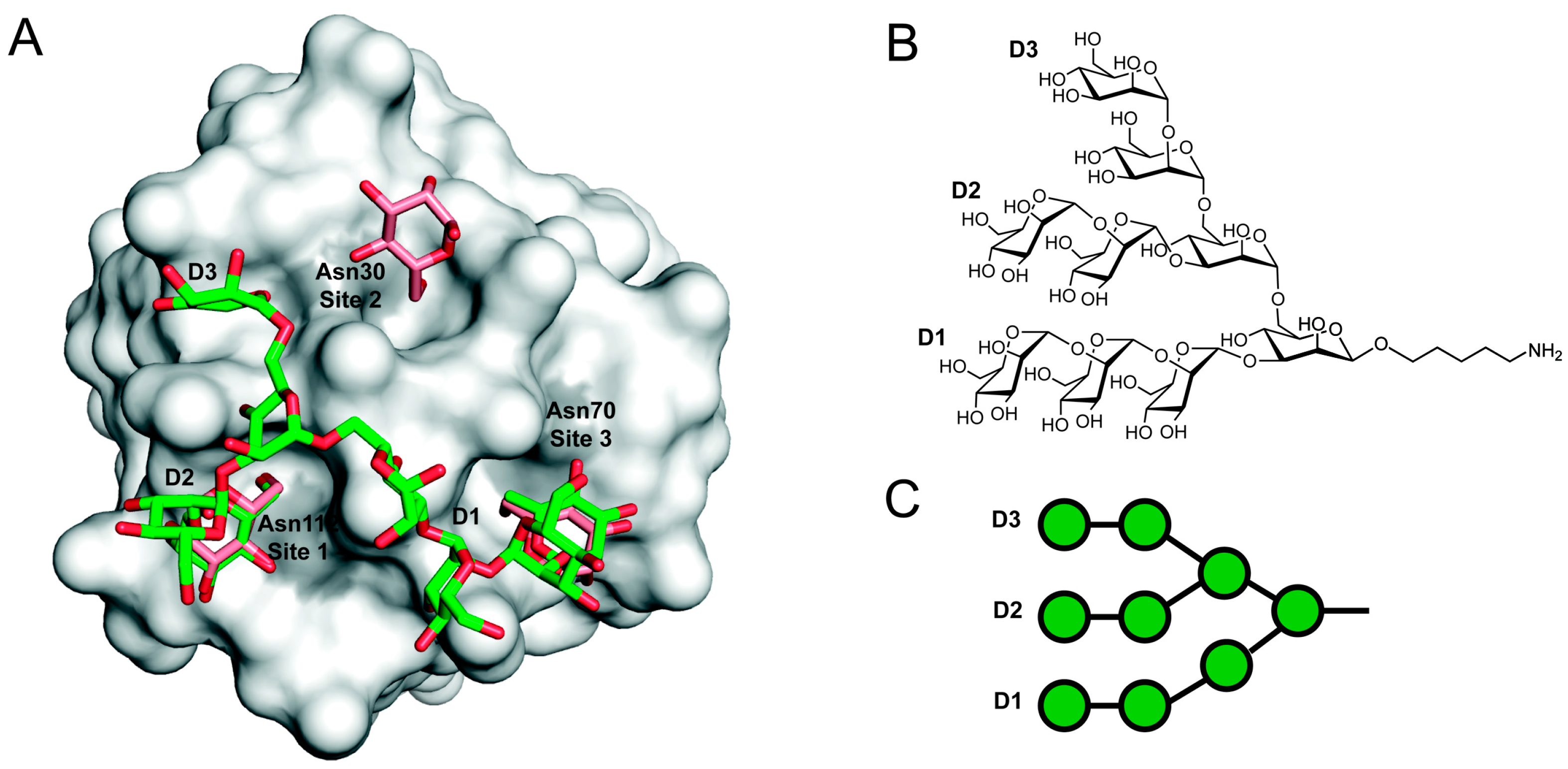
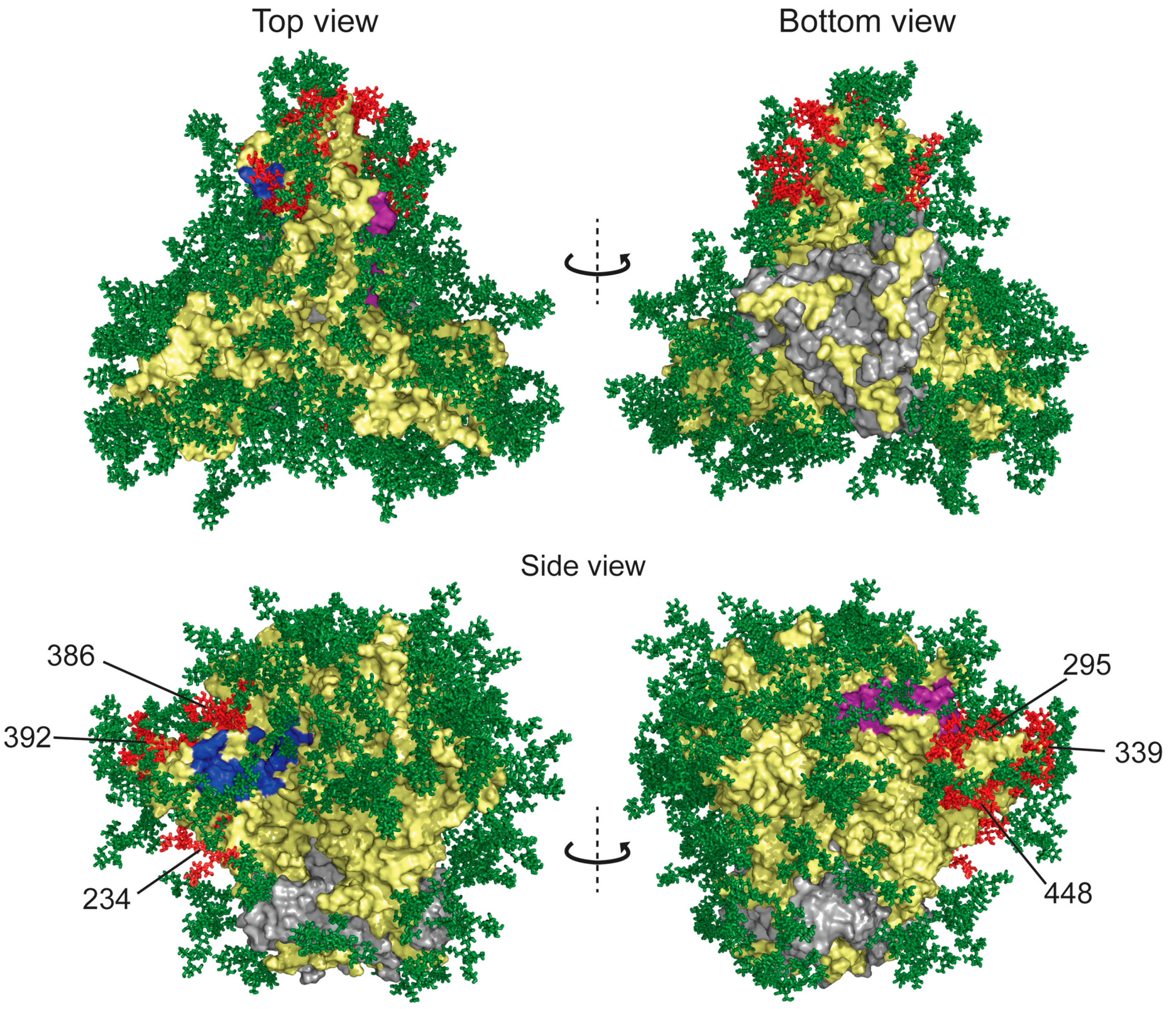
3. Three-Dimensional Structure
4. GRFT Fragments, Conjugates and Stability
4.1. Peptides Derived from GRFT
4.2. GRFT-C37
4.3. Tandemers
4.4. GRFT Stability
5. Carbohydrate-Mediated Crosslinking
6. Antiviral Activity
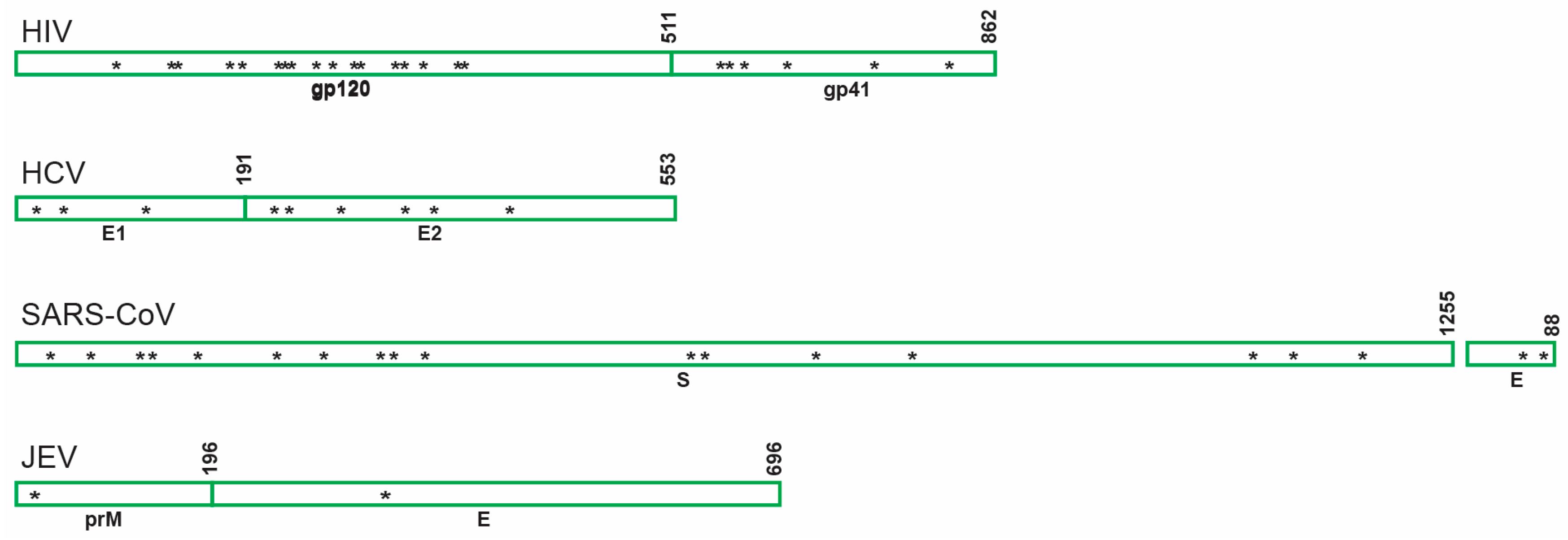
6.1. HIV
6.1.1. Mode of Action
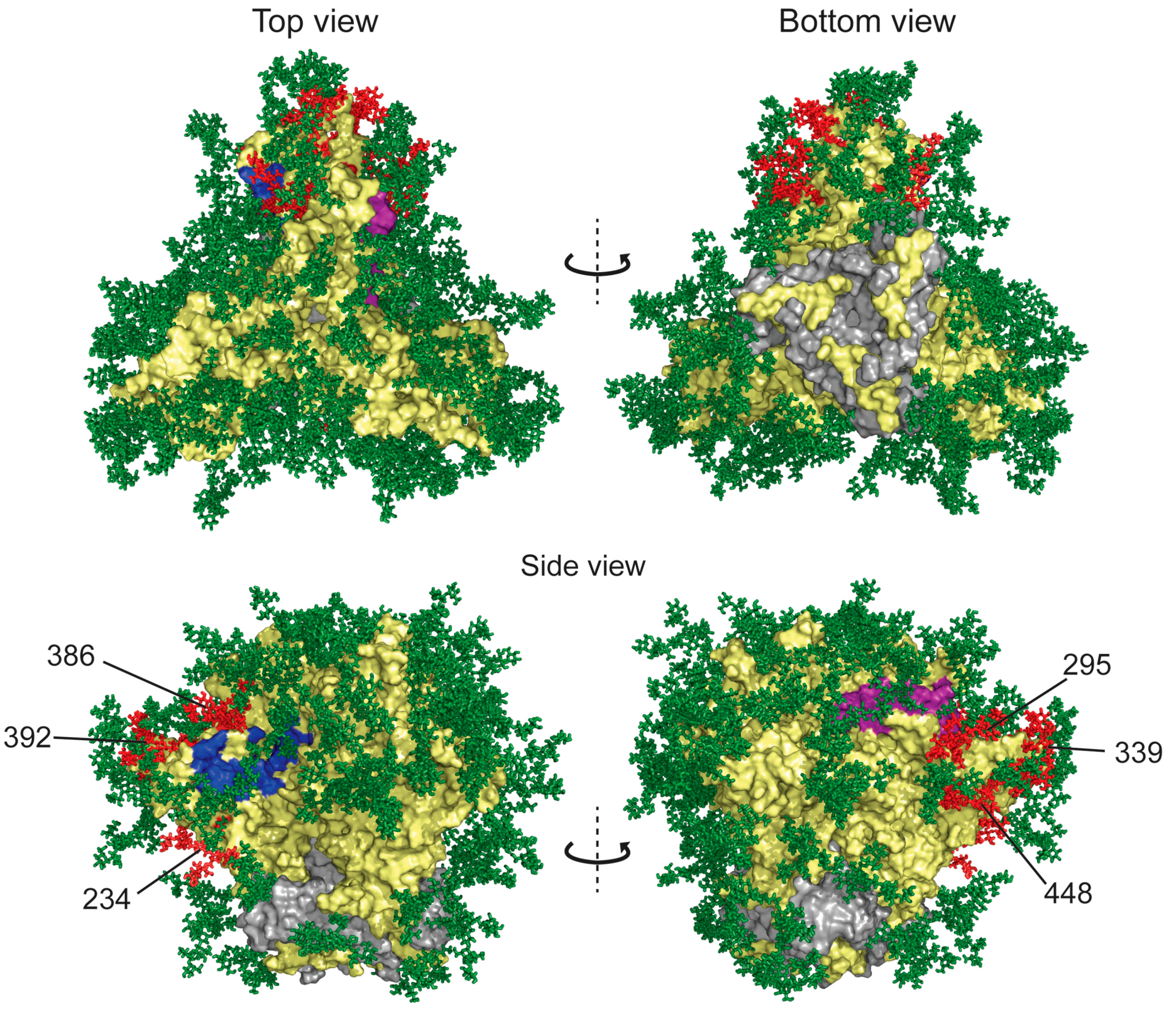
Binding to HIV-1 Envelope Glycoprotein gp120
Effects of GRFT on CD4 and Co-Receptor-Binding Sites
Inhibition of DC-SIGN Binding
6.1.2. HIV Resistance
6.1.3. Synergy Studies
Antibodies
Lectins
Tenofovir, Maraviroc and Enfuvitride
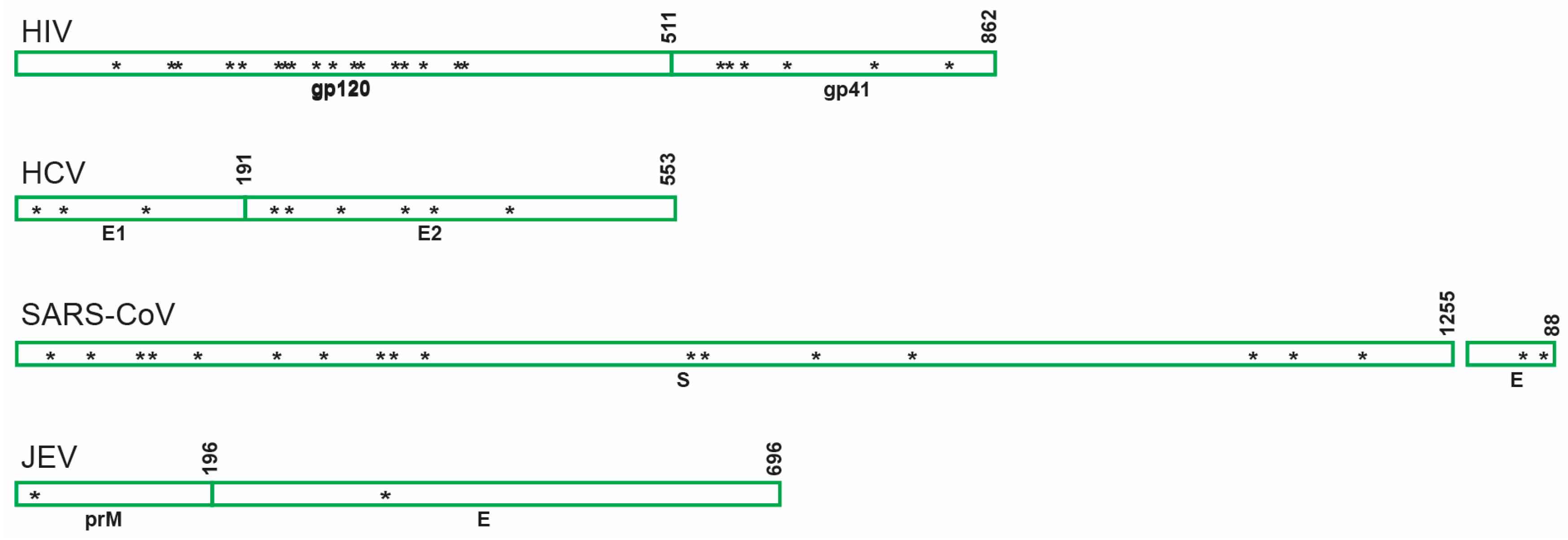
6.2. Other Viruses
6.2.1. Hepatitis C Virus (HCV)
6.2.2. Severe Acute Respiratory Syndrome-Related Coronavirus (SARS-CoV)
6.2.3. Japanese Encephalitis Virus (JEV)
6.2.4. Herpes Simplex Virus Type-2 (HSV-2)
6.2.5. Human Papilloma Virus (HPV)
6.2.6. Gamma- and Delta- Retroviruses
7. Activity against Protists
8. Toxicity and Immunogenicity of GRFT
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Doms, R.W.; Moore, J.P. HIV-1 membrane fusion: Targets of opportunity. J. Cell Biol. 2000, 151, F9–F14. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Poignard, P.; Stanfield, R.L.; Wilson, I.A. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 2012, 337, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.J. Current progress in development of hepatitis C virus vaccines. Nat. Med. 2013, 19, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Lusvarghi, S.; Bewley, C.A.; Kwong, P.D. HIV-1 gp120 as a therapeutic target: Navigating a moving labyrinth. Expert Opin. Ther. Targets 2015, 19, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J. Targeting the glycans of glycoproteins: A novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 2007, 5, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Akkouh, O.; Ng, T.B.; Singh, S.S.; Yin, C.; Dan, X.; Chan, Y.S.; Pan, W.; Cheung, R.C. Lectins with anti-HIV activity: A review. Molecules 2015, 20, 648–668. [Google Scholar] [CrossRef] [PubMed]
- Francois, K.O.; Balzarini, J. Potential of carbohydrate-binding agents as therapeutics against enveloped viruses. Med. Res. Rev. 2012, 32, 349–387. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; O’Keefe, B.R.; Sowder, R.C., 2nd; Bringans, S.; Gardella, R.; Berg, S.; Cochran, P.; Turpin, J.A.; Buckheit, R.W., Jr.; McMahon, J.B.; et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J. Biol. Chem. 2005, 280, 9345–9353. [Google Scholar] [CrossRef] [PubMed]
- Giomarelli, B.; Schumacher, K.M.; Taylor, T.E.; Sowder, R.C., 2nd; Hartley, J.L.; McMahon, J.B.; Mori, T. Recombinant production of anti-HIV protein, griffithsin, by auto-induction in a fermentor culture. Protein Expr. Purif. 2006, 47, 194–202. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, B.R.; Vojdani, F.; Buffa, V.; Shattock, R.J.; Montefiori, D.C.; Bakke, J.; Mirsalis, J.; d’Andrea, A.L.; Hume, S.D.; Bratcher, B.; et al. Scaleable manufacture of HIV-1 entry inhibitor griffithsin and validation of its safety and efficacy as a topical microbicide component. Proc. Natl. Acad. Sci. USA 2009, 106, 6099–6104. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Giritch, A.; Bartels, D.; Bortesi, L.; Gleba, Y. A novel and fully scalable Agrobacterium spray-based process for manufacturing cellulases and other cost-sensitive proteins in plants. Plant Biotechnol. J. 2015, 13, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Vamvaka, E.; Arcalis, E.; Ramessar, K.; Evans, A.; O’Keefe, B.R.; Shattock, R.J.; Medina, V.; Stoger, E.; Christou, P.; Capell, T. Rice endosperm is cost-effective for the production of recombinant griffithsin with potent activity against HIV. Plant Biotechnol. J. 2016, 14, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, J.L.; Wanga, V.; Palmer, K.E. Improving the large scale purification of the HIV microbicide, griffithsin. BMC Biotechnol. 2015, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Ziółkowska, N.E.; O’Keefe, B.R.; Mori, T.; Zhu, C.; Giomarelli, B.; Vojdani, F.; Palmer, K.E.; McMahon, J.B.; Wlodawer, A. Domain-swapped structure of the potent antiviral protein griffithsin and its mode of carbohydrate binding. Structure 2006, 14, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, N.E.; Shenoy, S.R.; O’Keefe, B.R.; McMahon, J.B.; Palmer, K.E.; Dwek, R.A.; Wormald, M.R.; Wlodawer, A. Crystallographic, thermodynamic, and molecular modeling studies of the mode of binding of oligosaccharides to the potent antiviral protein griffithsin. Proteins 2007, 67, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, N.E.; Shenoy, S.R.; O’Keefe, B.R.; Wlodawer, A. Crystallographic studies of the complexes of antiviral protein griffithsin with glucose and N-acetylglucosamine. Protein Sci. 2007, 16, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Gao, Y.; Hoorelbeke, B.; Kagiampakis, I.; Zhao, B.; Demeler, B.; Balzarini, J.; LiWang, P.J. The role of individual carbohydrate-binding sites in the function of the potent anti-HIV lectin griffithsin. Mol. Pharm. 2012, 9, 2613–2625. [Google Scholar] [CrossRef] [PubMed]
- Moulaei, T.; Shenoy, S.R.; Giomarelli, B.; Thomas, C.; McMahon, J.B.; Dauter, Z.; O’Keefe, B.R.; Wlodawer, A. Monomerization of viral entry inhibitor griffithsin elucidates the relationship between mulitivalent binding to carbohydrates and anti-HIV activity. Structure 2010, 18, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Hoorelbeke, B.; Xue, J.; LiWang, P.J.; Balzarini, J. Role of the carbohydrate-binding sites of griffithsin in the prevention of DC-SIGN-mediated capture and transmission of HIV-1. PLoS ONE 2013, 8, e64132. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Hoorelbeke, B.; Kagiampakis, I.; Demeler, B.; Balzarini, J.; Liwang, P.J. The griffithsin dimer is required for high-potency inhibition of HIV-1: Evidence for manipulation of the structure of gp120 as part of the griffithsin dimer mechanism. Antimicrob. Agents Chemother. 2013, 57, 3976–3989. [Google Scholar] [CrossRef] [PubMed]
- Micewicz, E.D.; Cole, A.L.; Jung, C.L.; Luong, H.; Phillips, M.L.; Pratikhya, P.; Sharma, S.; Waring, A.J.; Cole, A.M.; Ruchala, P. Grifonin-1: A small HIV-1 entry inhibitor derived from the algal lectin, Griffithsin. PLoS ONE 2010, 5, e14360. [Google Scholar] [CrossRef] [PubMed]
- Kagiampakis, I.; Gharibi, A.; Mankowski, M.K.; Snyder, B.A.; Ptak, R.G.; Alatas, K.; LiWang, P.J. Potent strategy to inhibit HIV-1 by binding both gp120 and gp41. Antimicrob. Agents Chemother. 2011, 55, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Moulaei, T.; Alexandre, K.B.; Shenoy, S.R.; Meyerson, J.R.; Krumpe, L.R.; Constantine, B.; Wilson, J.; Buckheit, R.W., Jr.; McMahon, J.B.; Subramaniam, S.; et al. Griffithsin tandemers: Flexible and potent lectin inhibitors of the human immunodeficiency virus. Retrovirology 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Moncla, B.J.; Pryke, K.; Rohan, L.C.; Graebing, P.W. Degradation of naturally occurring and engineered antimicrobial peptides by proteases. Adv. Biosci. Biotechnol. 2011, 2, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Lusvarghi, S.; Ghirlando, R.; Wong, C.H.; Bewley, C.A. Glycopeptide mimetics recapitulate high-mannose-type oligosaccharide binding and function. Angew. Chem. Int. Ed. 2015, 54, 5603–5608. [Google Scholar] [CrossRef] [PubMed]
- Lusvarghi, S.; Lohith, K.; Morin-Leisk, J.; Ghirlando, R.; Hinshaw, J.E.; Bewley, C.A. Binding Site Geometry and Subdomain Valency Control Effects of Neutralizing Lectins on HIV-1 Viral Particles. ACS Infect. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Emau, P.; Tian, B.; O’Keefe, B.R.; Mori, T.; McMahon, J.B.; Palmer, K.E.; Jiang, Y.; Bekele, G.; Tsai, C.C. Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti-HIV microbicide. J. Med. Primatol. 2007, 36, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Férir, G.; Huskens, D.; Palmer, K.E.; Boudreaux, D.M.; Swanson, M.D.; Markovitz, D.M.; Balzarini, J.; Schols, D. Combinations of griffithsin with other carbohydrate-binding agents demonstrate superior activity against HIV type 1, HIV type 2, and selected carbohydrate-binding agent-resistant HIV type 1 strains. AIDS Res. Hum. Retrovir. 2012, 28, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, B.R.; Giomarelli, B.; Barnard, D.L.; Shenoy, S.R.; Chan, P.K.; McMahon, J.B.; Palmer, K.E.; Barnett, B.W.; Meyerholz, D.K.; Wohlford-Lenane, C.L.; et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 2010, 84, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Albecka, A.; Belouzard, S.; Vercauteren, K.; Verhoye, L.; Wychowski, C.; Leroux-Roels, G.; Palmer, K.E.; Dubuisson, J. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob. Agents Chemother. 2011, 55, 5159–5167. [Google Scholar] [CrossRef] [PubMed]
- Takebe, Y.; Saucedo, C.J.; Lund, G.; Uenishi, R.; Hase, S.; Tsuchiura, T.; Kneteman, N.; Ramessar, K.; Tyrrell, D.L.; Shirakura, M.; et al. Antiviral lectins from red and blue-green algae show potent in vitro and in vivo activity against hepatitis C virus. PLoS ONE 2013, 8, e64449. [Google Scholar] [CrossRef] [PubMed]
- Ishag, H.Z.; Li, C.; Huang, L.; Sun, M.X.; Wang, F.; Ni, B.; Malik, T.; Chen, P.Y.; Mao, X. Griffithsin inhibits Japanese encephalitis virus infection in vitro and in vivo. Arch. Virol. 2013, 158, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Levendosky, K.; Mizenina, O.; Martinelli, E.; Jean-Pierre, N.; Kizima, L.; Rodriguez, A.; Kleinbeck, K.; Bonnaire, T.; Robbiani, M.; Zydowsky, T.M.; et al. Griffithsin and carrageenan combination to target Herpes Simplex Virus 2 and Human Papillomavirus. Antimicrob. Agents Chemother. 2015, 59, 7290–7298. [Google Scholar] [CrossRef] [PubMed]
- Turville, S.G.; Aravantinou, M.; Stossel, H.; Romani, N.; Robbiani, M. Resolution of de novo HIV production and trafficking in immature dendritic cells. Nat. Methods 2008, 5, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Gray, E.S.; Lambson, B.E.; Moore, P.L.; Choge, I.A.; Mlisana, K.; Karim, S.S.A.; McMahon, J.; O’Keefe, B.; Chikwamba, R.; et al. Mannose-rich glycosylation patterns on HIV-1 subtype C gp120 and sensitivity to the lectins, Griffithsin, Cyanovirin-N and Scytovirin. Virology 2010, 402, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Ferir, G. Griffithsin, alone and combined with all classes of antiretroviral drugs, potently inhibits HIV cell-cell transmission and destruction of CD4+ T cells. J. Antivir. Antiretrovir. 2012, 4, 103–112. [Google Scholar] [CrossRef]
- Hu, B.; Du, T.; Li, C.; Luo, S.; Liu, Y.; Huang, X.; Hu, Q. Sensitivity of transmitted and founder human immunodeficiency virus type 1 envelopes to carbohydrate-binding agents griffithsin, cyanovirin-N and Galanthus nivalis agglutinin. J. Gen. Virol. 2015, 96, 3660–3666. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Gray, E.S.; Pantophlet, R.; Moore, P.L.; McMahon, J.B.; Chakauya, E.; O’Keefe, B.R.; Chikwamba, R.; Morris, L. Binding of the mannose-specific lectin, griffithsin, to HIV-1 gp120 exposes the CD4-binding site. J. Virol. 2011, 85, 9039–9050. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Michael, E.; Eggink, D.; van Montfort, T.; Lasnik, A.B.; Palmer, K.E.; Sanders, R.W.; Moore, J.P.; Klasse, P.J. Occluding the mannose moieties on human immunodeficiency virus type 1 gp120 with griffithsin improves the antibody responses to both proteins in mice. AIDS Res. Hum. Retrovir. 2012, 28, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Gray, E.S.; Mufhandu, H.; McMahon, J.B.; Chakauya, E.; O’Keefe, B.R.; Chikwamba, R.; Morris, L. The lectins griffithsin, cyanovirin-N and scytovirin inhibit HIV-1 binding to the DC-SIGN receptor and transfer to CD4+ cells. Virology 2012, 423, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Nixon, B.; Stefanidou, M.; Mesquita, P.M.; Fakioglu, E.; Segarra, T.; Rohan, L.; Halford, W.; Palmer, K.E.; Herold, B.C. Griffithsin protects mice from genital herpes by preventing cell-to-cell spread. J. Virol. 2013, 87, 6257–6269. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jin, W.; Griffin, G.E.; Shattock, R.J.; Hu, Q. Removal of two high-mannose N-linked glycans on gp120 renders human immunodeficiency virus 1 largely resistant to the carbohydrate-binding agent griffithsin. J. Gen. Virol. 2011, 92, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Jones, G.B.E.; Soto, C.; Lemmin, T.; Chuang, G.Y.; Druz, A.; Kong, R.; Thomas, P.V.; Wagh, K.; Zhou, T.Q.; Behrens, A.J.; et al. Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell 2016, 165, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Moore, P.L.; Nonyane, M.; Gray, E.S.; Ranchobe, N.; Chakauya, E.; McMahon, J.B.; O’Keefe, B.R.; Chikwamba, R.; Morris, L. Mechanisms of HIV-1 subtype C resistance to GRFT, CV-N and SVN. Virology 2013, 446, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Ferir, G.; Huskens, D.; Noppen, S.; Koharudin, L.M.; Gronenborn, A.M.; Schols, D. Broad anti-HIV activity of the Oscillatoria agardhii agglutinin homologue lectin family. J. Antimicrob. Chemother. 2014, 69, 2746–2758. [Google Scholar] [CrossRef] [PubMed]
- Hamorsky, K.T.; Grooms-Williams, T.W.; Husk, A.S.; Bennett, L.J.; Palmer, K.E.; Matoba, N. Efficient single tobamoviral vector-based bioproduction of broadly neutralizing anti-HIV-1 monoclonal antibody VRC01 in Nicotiana benthamiana plants and utility of VRC01 in combination microbicides. Antimicrob. Agents Chemother. 2013, 57, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Ferir, G.; Palmer, K.E.; Schols, D. Synergistic activity profile of griffithsin in combination with tenofovir, maraviroc and enfuvirtide against HIV-1 clade C. Virology 2011, 417, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, L.; Oliveira, C.; Fournier, C.; Descamps, V.; Morel, V.; Dubuisson, J.; Brochot, E.; Francois, C.; Castelain, S.; Duverlie, G.; et al. Hepatitis C Virus resistance to carbohydrate-binding agents. PLoS ONE 2016, 11, e0149064. [Google Scholar] [CrossRef] [PubMed]
- Ishag, H.Z.; Li, C.; Wang, F.; Mao, X. Griffithsin binds to the glycosylated proteins (E and prM) of Japanese encephalitis virus and inhibit its infection. Virus Res. 2016, 215, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.M.; Ruscetti, F.W.; Rein, A.; Bertolette, D.C.; Saucedo, C.J.; O’Keefe, B.R.; Jones, K.S. Differential inhibitory effects of cyanovirin-N, griffithsin, and scytovirin on entry mediated by envelopes of gammaretroviruses and deltaretroviruses. J. Virol. 2014, 88, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Ratner, D.M.; Ryan, C.M.; Johnson, P.J.; O’Keefe, B.R.; Secor, W.E.; Anderson, D.J.; Robbins, P.W.; Samuelson, J. Anti-retroviral lectins have modest effects on adherence of trichomonas vaginalis to epithelial cells in vitro and on recovery of Tritrichomonas foetus in a mouse vaginal model. PLoS ONE 2015, 10, e0135340. [Google Scholar] [CrossRef] [PubMed]
- Kouokam, J.C.; Huskens, D.; Schols, D.; Johannemann, A.; Riedell, S.K.; Walter, W.; Walker, J.M.; Matoba, N.; O’Keefe, B.R.; Palmer, K.E. Investigation of griffithsin’s interactions with human cells confirms its outstanding safety and efficacy profile as a microbicide candidate. PLoS ONE 2011, 6, e22635. [Google Scholar] [CrossRef] [PubMed]
- Barton, C.; Kouokam, J.C.; Lasnik, A.B.; Foreman, O.; Cambon, A.; Brock, G.; Montefiori, D.C.; Vojdani, F.; McCormick, A.A.; O’Keefe, B.R.; et al. Activity of and effect of subcutaneous treatment with the broad-spectrum antiviral lectin griffithsin in two laboratory rodent models. Antimicrob. Agents Chemother. 2014, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression System | Yield | Recovery after Purification | Ref. |
|---|---|---|---|
| E. coli BL21(DE3)—shake flasks | 12 mg/L | [10] | |
| E. coli BL21(DE3)—fermenter | 819 mg/L | 542 mg/L | [10] |
| Tobacco leaves (Nicotiana benthamiana) | 1 g/kg leaf | 300 mg/kg | [11] |
| Rice seeds (Oryza sativa endosperm) | 301 mg/kg dry seed | 223 mg/kg | [13] |
| Sample | Virus a | Target Cell b | EC50 (nM) | Ref. |
|---|---|---|---|---|
| Native GRFT | HIV-1RF | CEM-SS | 0.043 | [9] |
| Recombinant GRFT | HIV-1RF | CEM-SS | 0.039 | [9] |
| Native GRFT | HIV-1RoJo | PBMC | 0.63 | [9] |
| Native GRFT | HIV-1ADA | Macrophage | 0.5 | [9] |
| Native GRFT | HIV-1Ba-L | Macrophage | 0.098 | [9] |
| Recombinant GRFT | HIV-1Ba-L | PMBC | 0.01 | [28] |
| Recombinant GRFT | HIV-1LAI | MT-4 | 0.01 | [28] |
| Recombinant GRFT | HIV-2ROD | MT-4 | 0.17 | [29] |
| Recombinant GRFT | SIV mac251 | CEMx174 | 0.35 | [28] |
| Recombinant GRFT | SARS-CoV200300592 | Vero 76 | 280 | [15] |
| Recombinant GRFT | SARS-CoV200300592 | Vero 76 | 960 | [15] |
| Recombinant GRFT | SARS-CoVUrbani | Vero 76 | 48 | [30] |
| Recombinant GRFT | SARS-CoVTor-II | Vero 76 | 48 | [30] |
| Recombinant GRFT | SARS-CoVCuHK | Vero 76 | 61 | [30] |
| Recombinant GRFT | SARS-CoVFrank | Vero 76 | 94 | [30] |
| Recombinant GRFT | HCVJFH1 | Huh-7 | 13.9 | [31] |
| Recombinant GRFT | HCVJFH-1 | Huh 7.5.1 | 0.4 | [32] |
| Recombinant GRFT | JEV | BHK-21 | 20 | [33] |
| Recombinant GRFT | HPV16 PsV | HeLa | 1390 | [34] |
| Recombinant GRFT | HPV18 PsV | HeLa | 428 | [34] |
| Recombinant GRFT | HPV45 PsV | HeLa | 928 | [34] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lusvarghi, S.; Bewley, C.A. Griffithsin: An Antiviral Lectin with Outstanding Therapeutic Potential. Viruses 2016, 8, 296. https://doi.org/10.3390/v8100296
Lusvarghi S, Bewley CA. Griffithsin: An Antiviral Lectin with Outstanding Therapeutic Potential. Viruses. 2016; 8(10):296. https://doi.org/10.3390/v8100296
Chicago/Turabian StyleLusvarghi, Sabrina, and Carole A. Bewley. 2016. "Griffithsin: An Antiviral Lectin with Outstanding Therapeutic Potential" Viruses 8, no. 10: 296. https://doi.org/10.3390/v8100296
APA StyleLusvarghi, S., & Bewley, C. A. (2016). Griffithsin: An Antiviral Lectin with Outstanding Therapeutic Potential. Viruses, 8(10), 296. https://doi.org/10.3390/v8100296