
The Microtubule Inhibitor Podofilox Inhibits an Early Entry Step of Human Cytomegalovirus

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Antibodies, Viruses and Chemicals
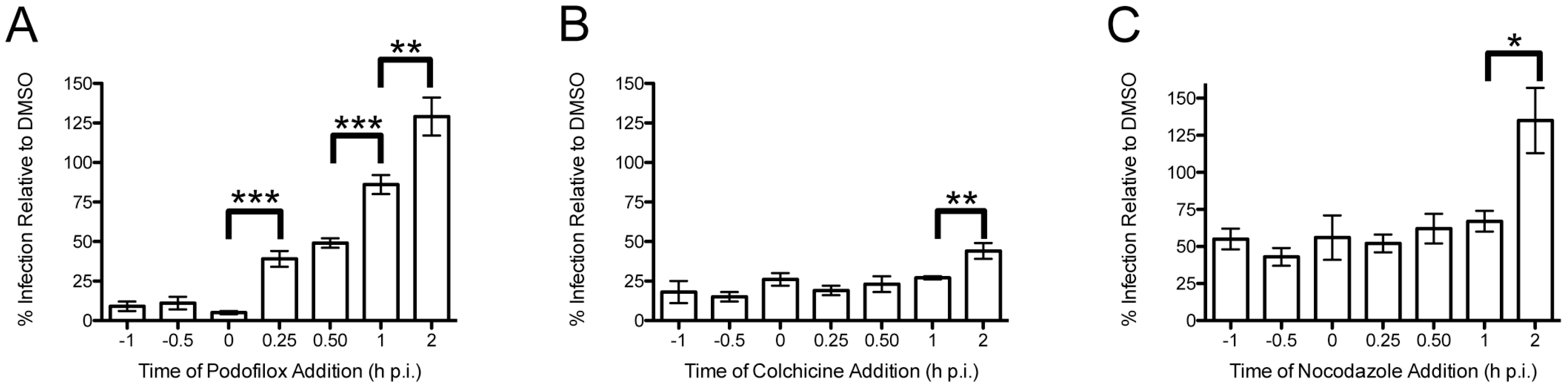
2.2. Time-of-Addition Experiments
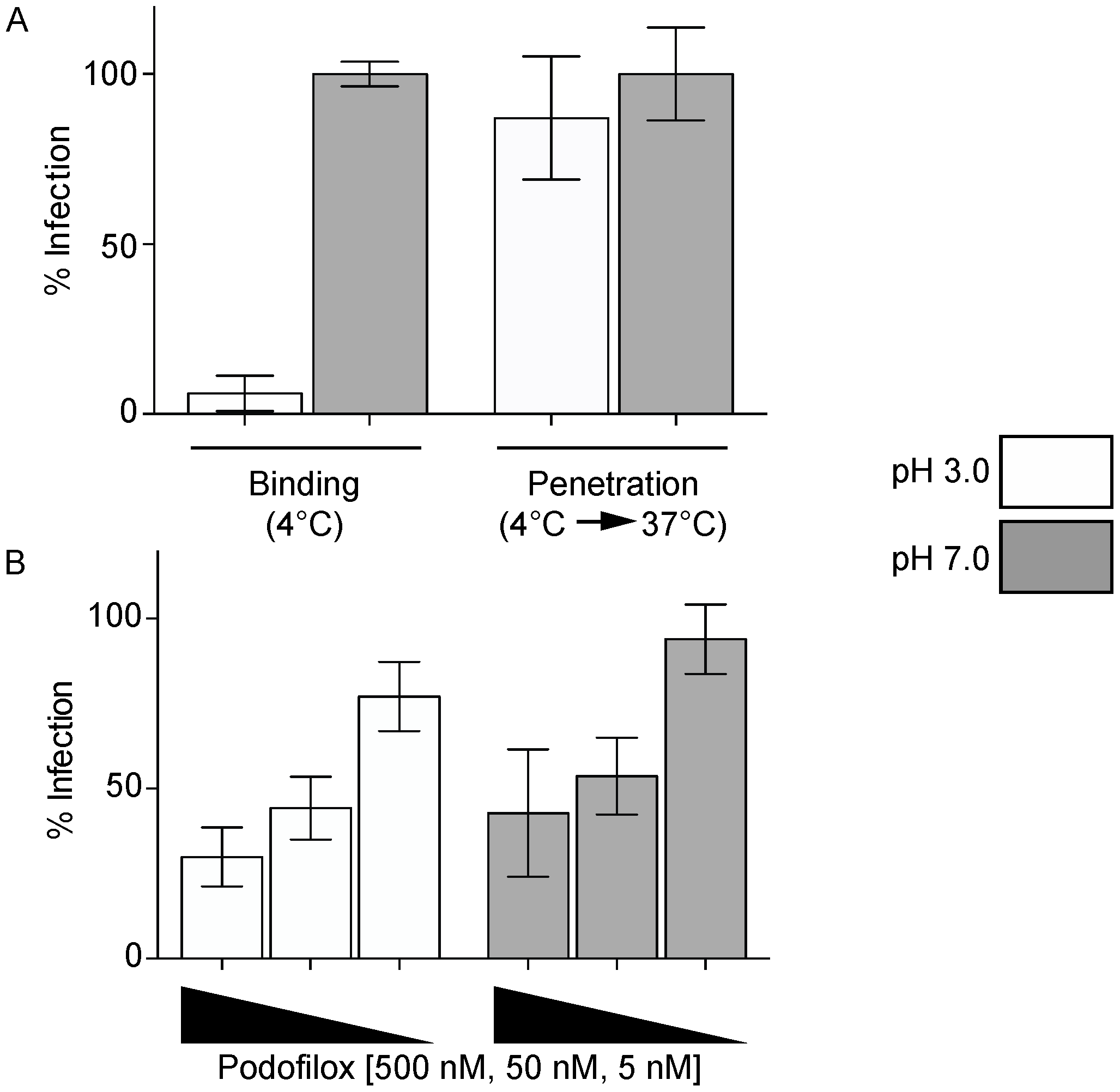
2.3. Virus Entry Assays
2.4. Penetration Assay
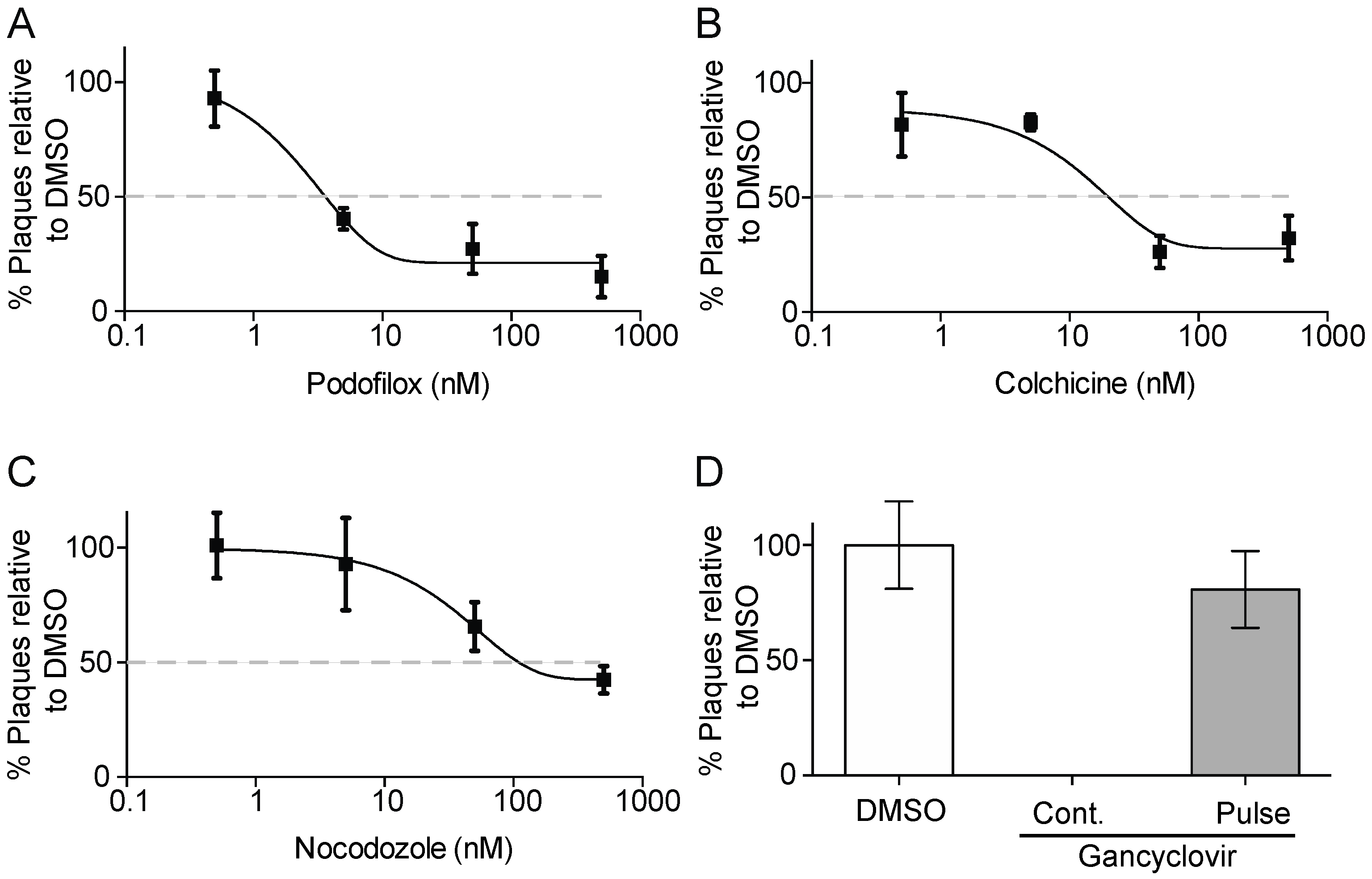
2.5. Plaque Reduction Assay
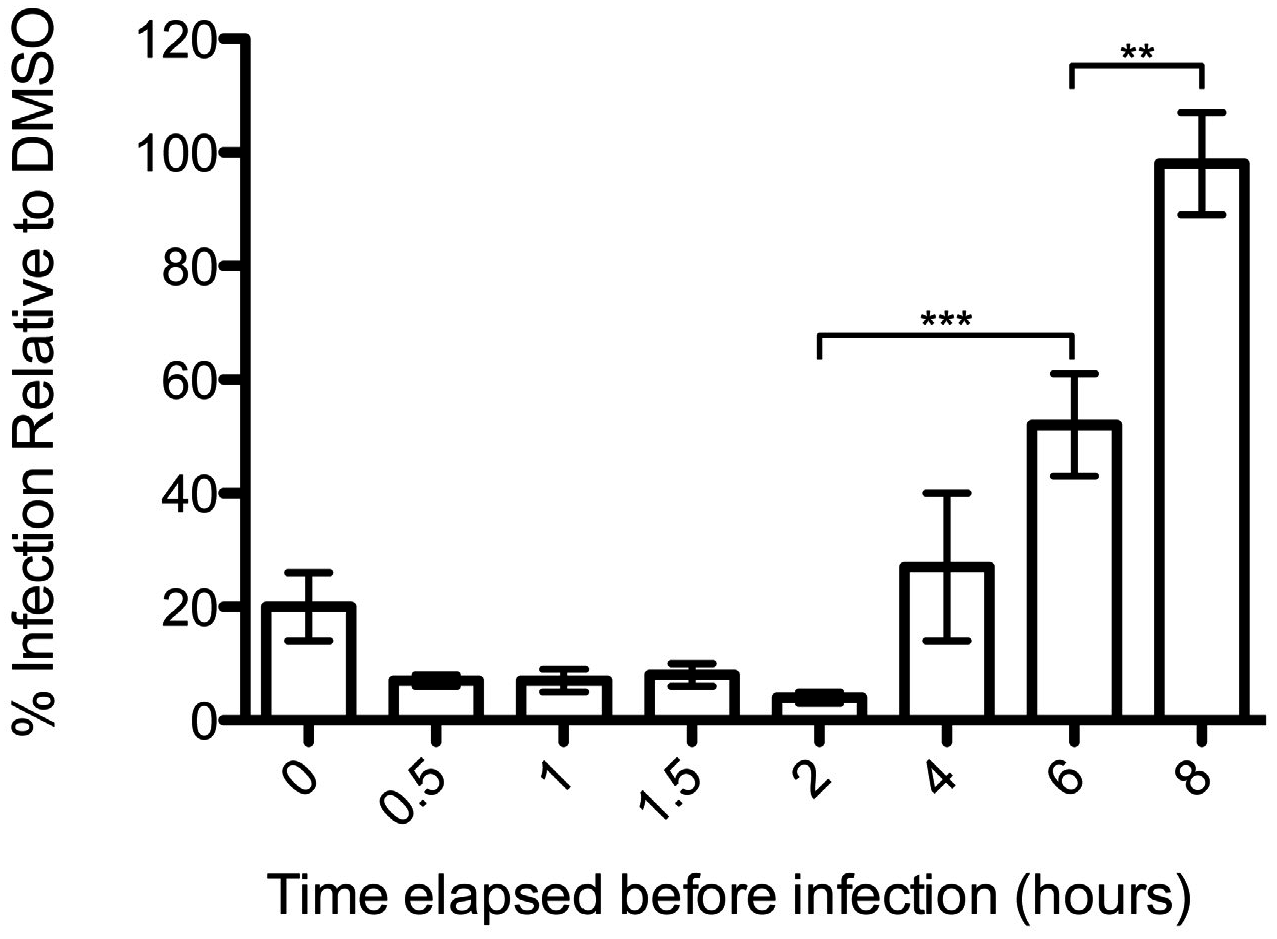
2.6. Reversibility Assay
2.7. Determining Half Maximal Effective Concentration (EC50) of Podofilox on the Expression of IE Genes by the Clinical Strain TB40/E
2.8. Analysis of Podofilox to Inhibit Diverse Viruses
2.9. Analysis of Cells Treated with Microtubule Inhibitors by Microscopy
2.10. Statistical Analysis
3. Results
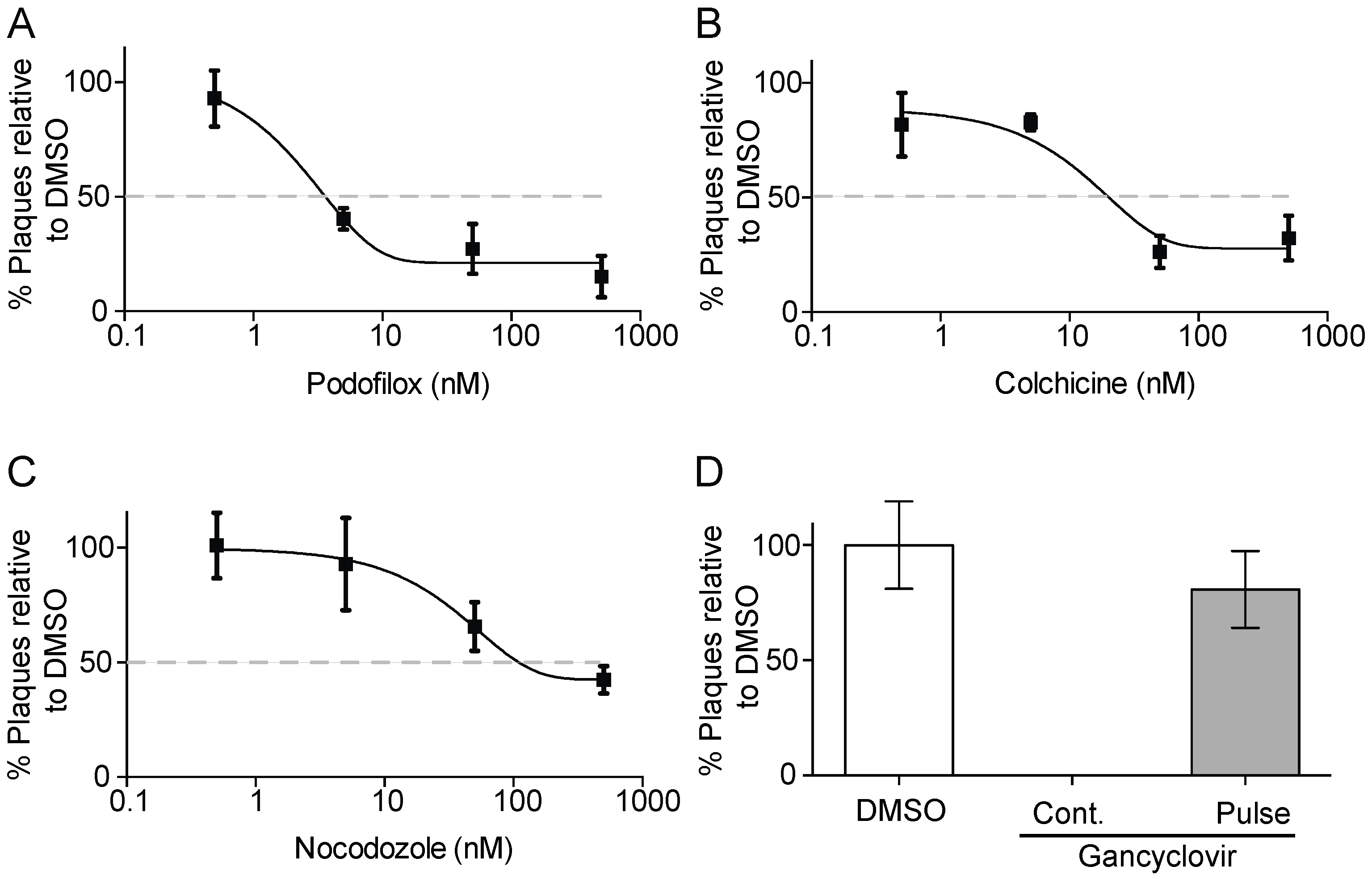
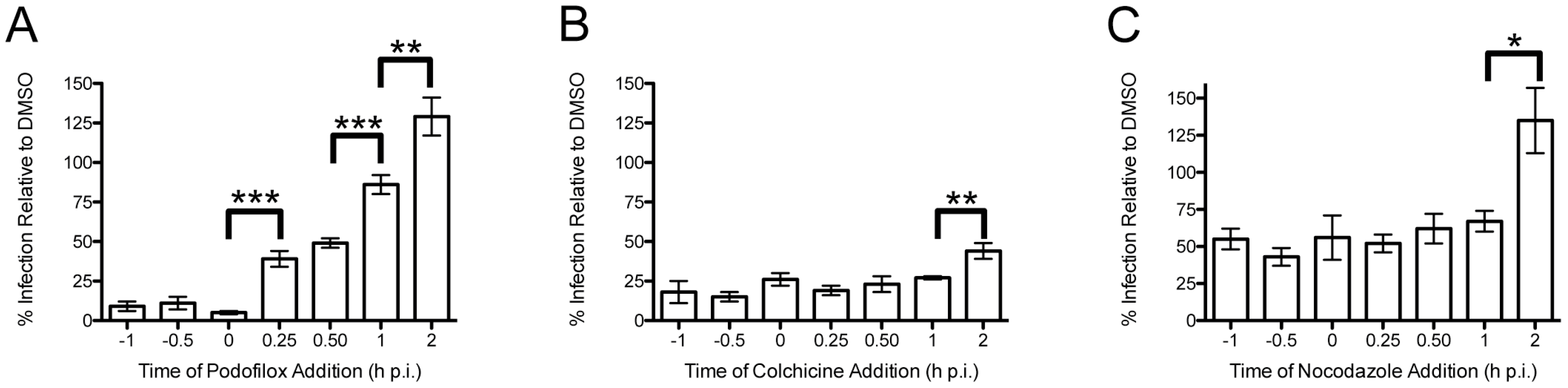
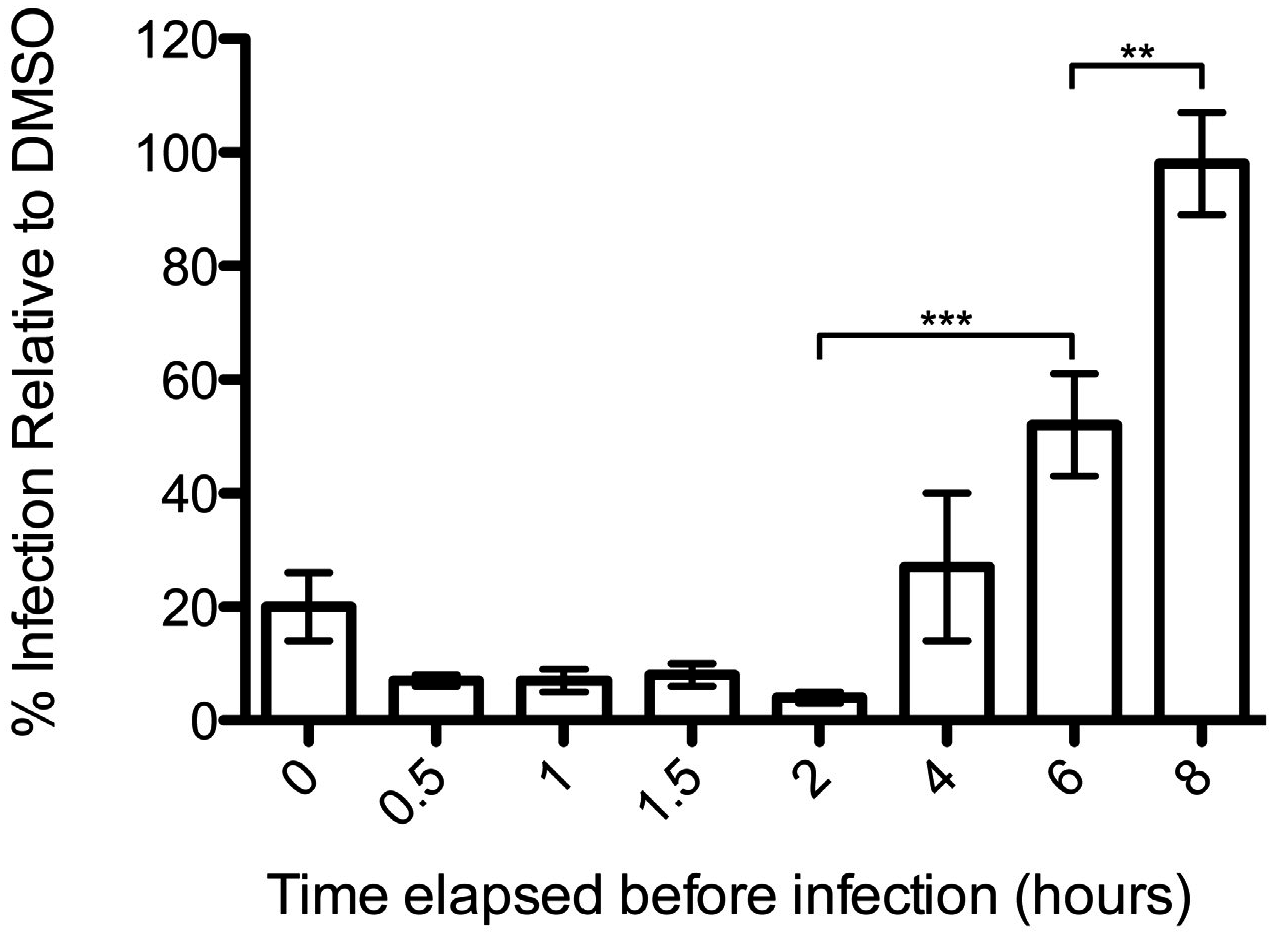
3.1. Podofilox Blocks an Early Step of CMV Entry
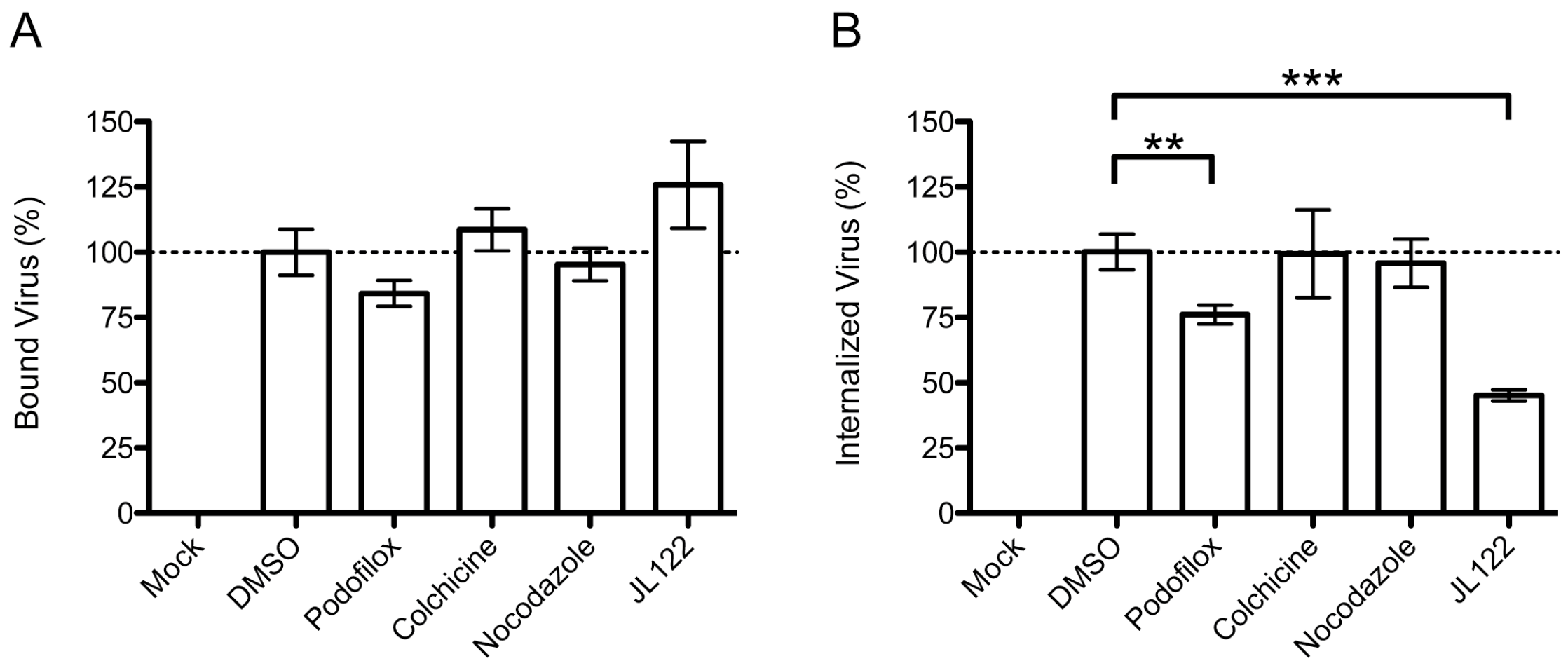
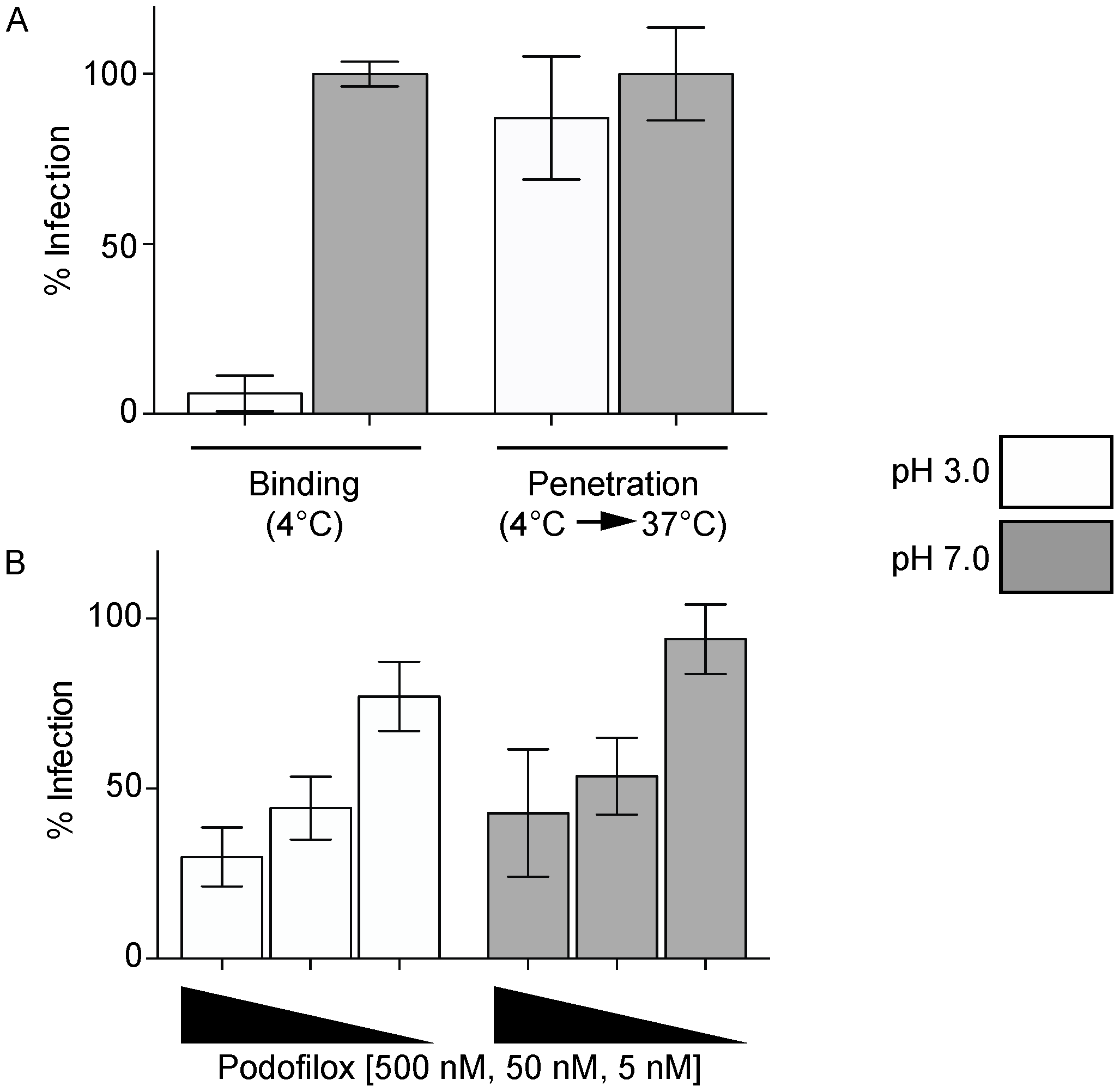
3.2. Podofiox Inhibits a Post-Binding CMV Entry Step
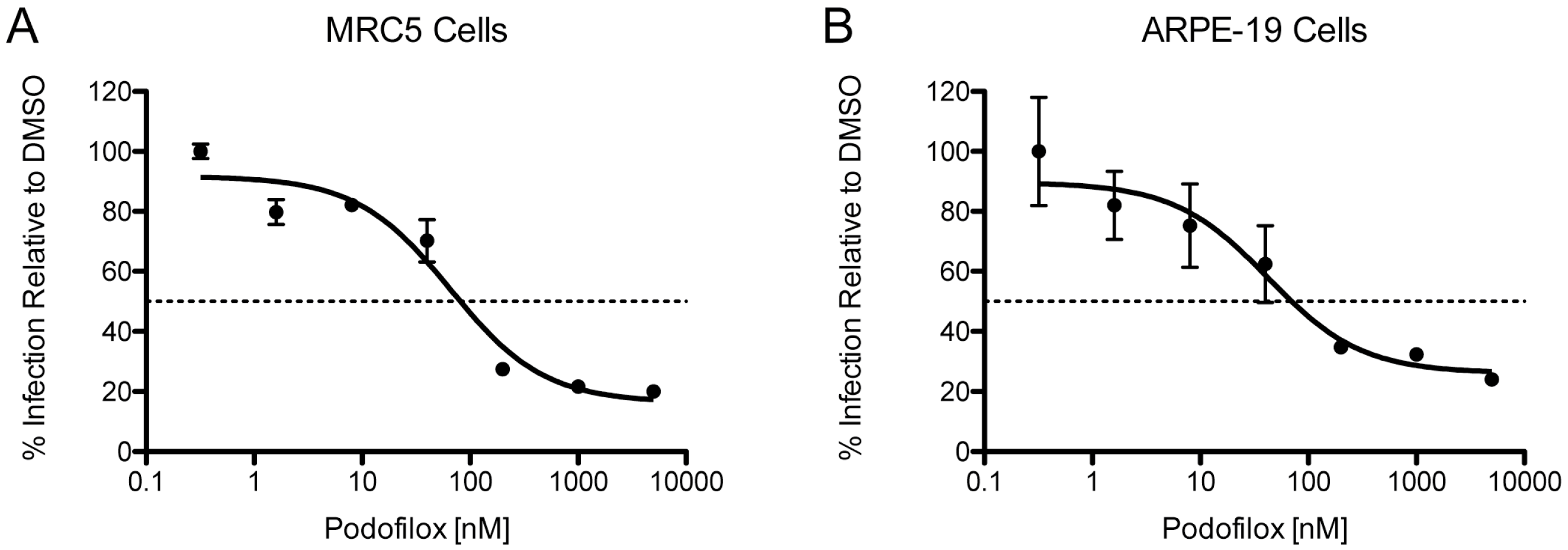
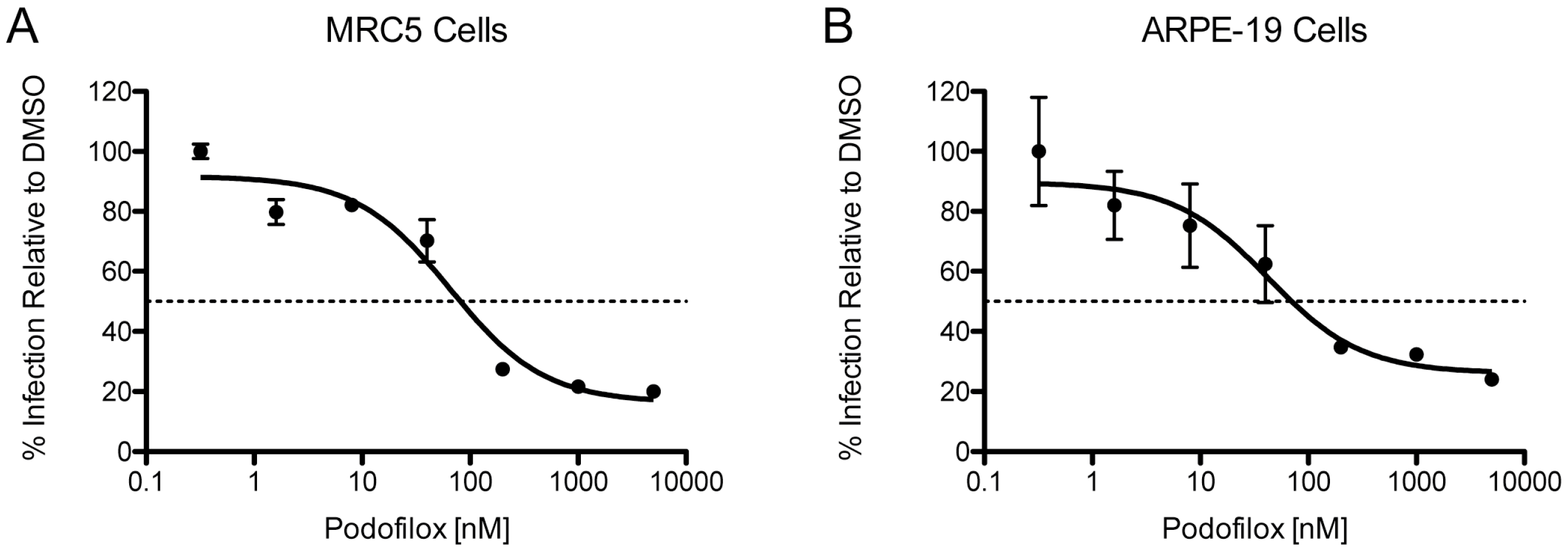
3.3. Efficacy of Podofilox to Inhibit a Clinical-Like CMV Strain
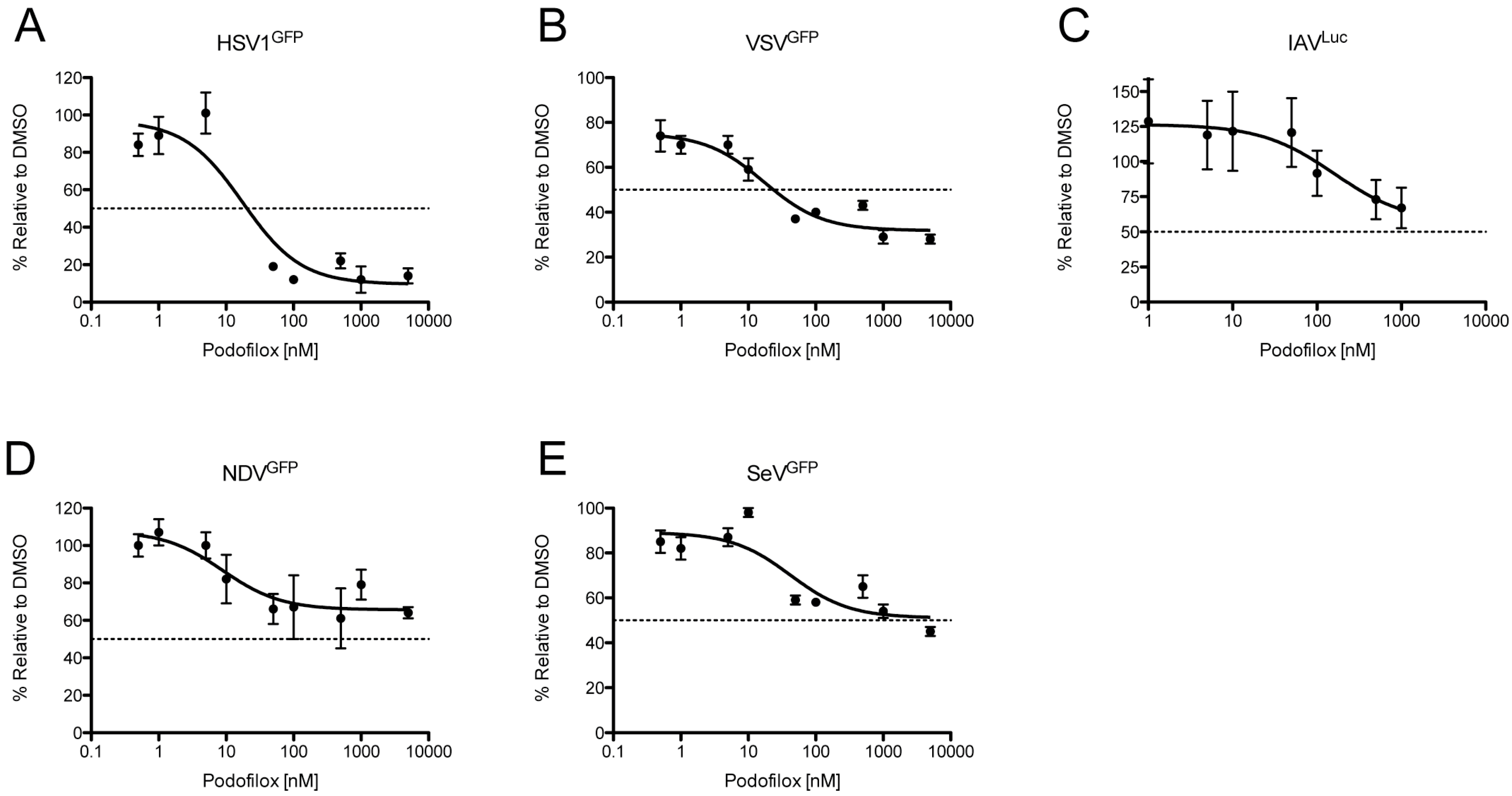
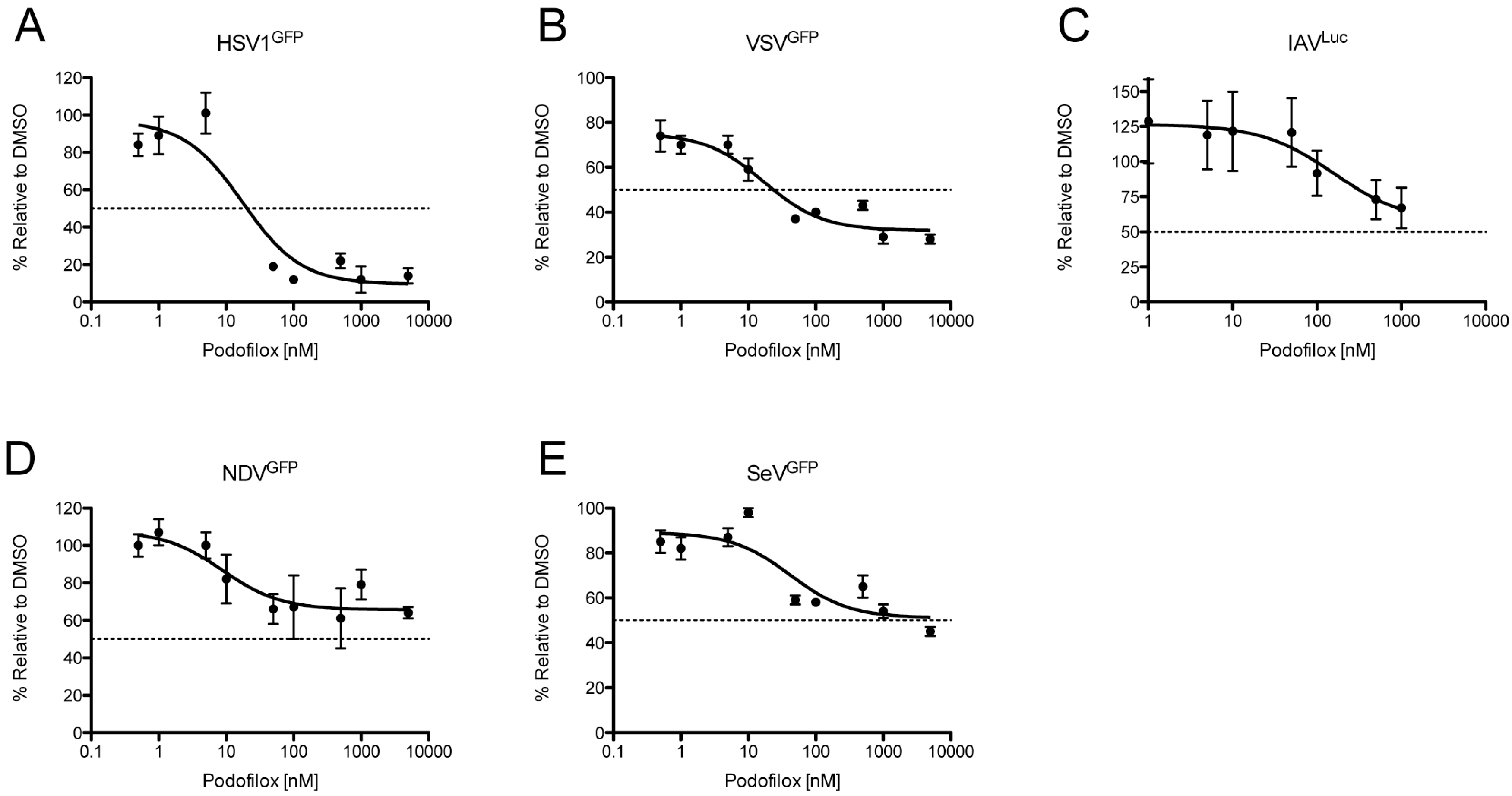
3.4. Podofilox Inhibits Other Enveloped Viruses
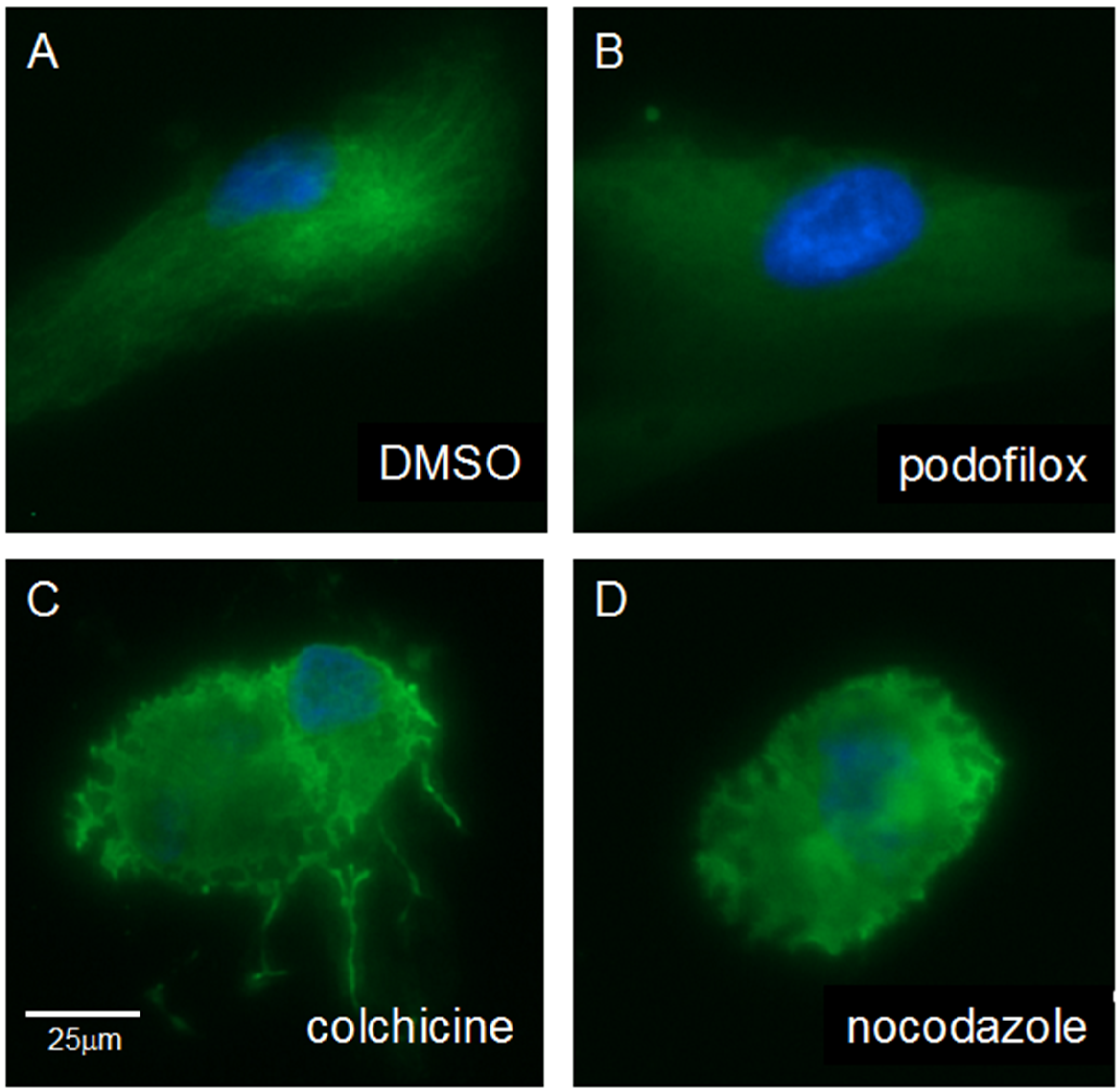
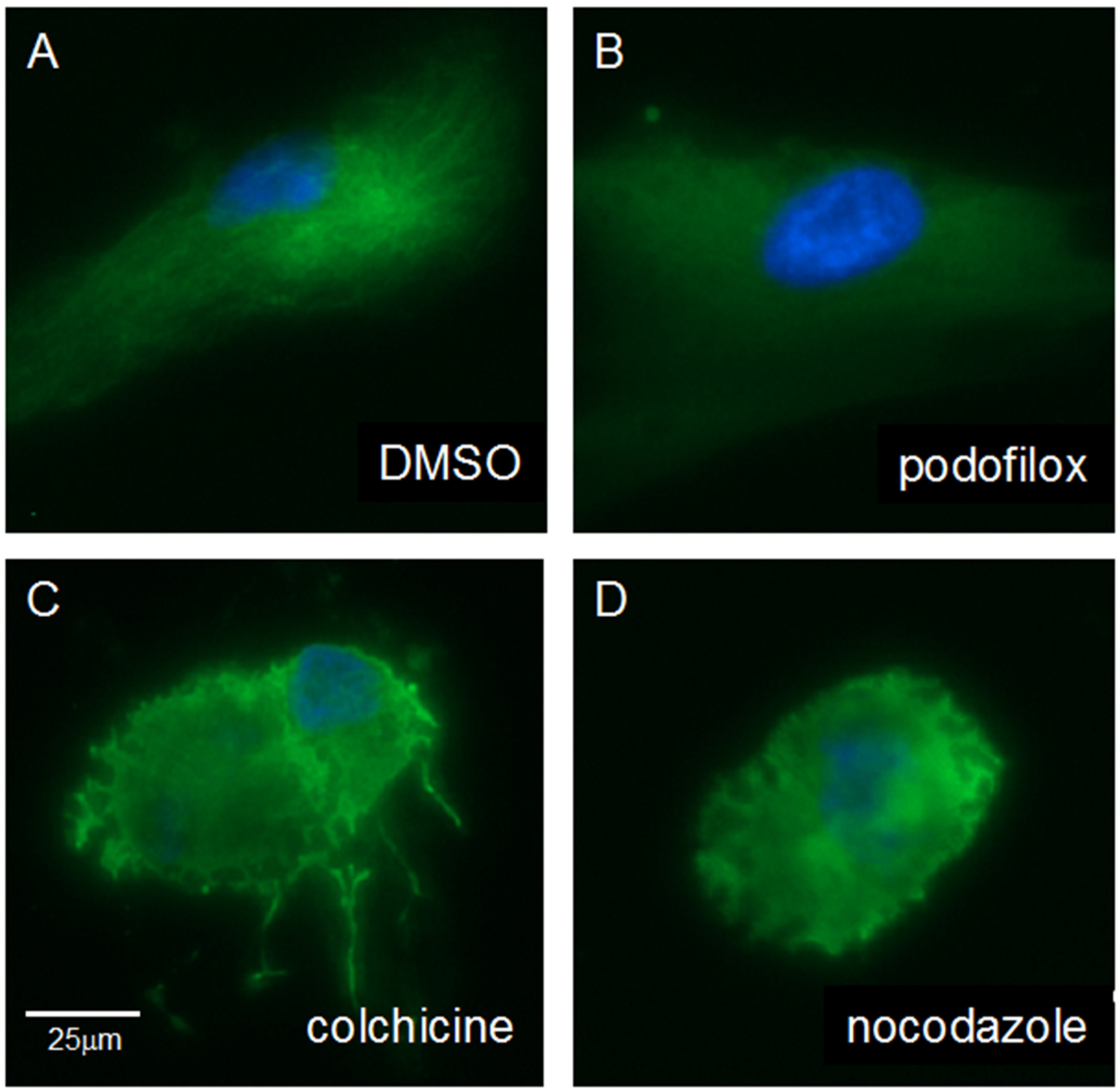
3.5. Podofilox Does Not Disrupt Cellular Morphology
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CMV | Cytomegalovirus |
| EC50 | 50% effective concentration |
| FL | Fusion loops |
| GFP | Green fluorescent protein |
| HSV1 | Herpes simplex virus 1 |
| IAV | Influenza A Virus |
| IE | Immediate early |
| LUC | Luciferase |
| MPI | Maximal plateau inhibition |
| MPR | Membrane proximal region |
| NDV | Newcastle disease virus |
| SeV | Sendai virus |
| UL | Unique long |
| US | Unique short |
| VSV | Vesicular stomatitis virus |
| YFP | Yellow fluorescent protein |
References
- Cohen, J.I.; Corey, G.R. Cytomegalovirus infection in the normal host. Medicine 1985, 64, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Ho, M. Epidemiology of cytomegalovirus infections. Rev. Infect. Dis. 1990, 12, S701–S710. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Davis, K.F. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health 2005. [Google Scholar] [CrossRef] [PubMed]
- Couzi, L.; Pitard, V.; Moreau, J.F.; Merville, P.; Dechanet-Merville, J. Direct and indirect effects of cytomegalovirus-induced gammadelta t cells after kidney transplantation. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.F.; Reyne, A.G.; de Dios, S.; Lopez-Medrano, F.; Jurado, A.; Juan, R.S.; Ruiz-Cano, M.J.; Dolores Folgueira, M.; Gomez-Sanchez, M.A.; Aguado, J.M.; et al. Influence of cytomegalovirus infection in the development of cardiac allograft vasculopathy after heart transplantation. J. Heart Lung Transplant. 2015, 34, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.C.; Westall, G.P.; Widjaja, J.M.; Mifsud, N.A.; Nguyen, T.H.; Meehan, A.C.; Kotsimbos, T.C.; Brooks, A.G. The presence of HLA-E-restricted, CMV-specific CD8+ T cells in the blood of lung transplant recipients correlates with chronic allograft rejection. PLoS ONE 2015, 10, e0135972. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Marousek, G.; Guentzel, S.; Follansbee, S.E.; Poscher, M.E.; Lalezari, J.P.; Miner, R.C.; Drew, W.L. Evolution of mutations conferring multidrug resistance during prophylaxis and therapy for cytomegalovirus disease. J. Infect. Dis. 1997, 176, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Jabs, D.A.; Enger, C.; Dunn, J.P.; Forman, M. Cytomegalovirus retinitis and viral resistance: Ganciclovir resistance. CMV retinitis and viral resistance study group. J. Infect. Dis. 1998, 177, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Faulds, D. Ganciclovir. An update of its therapeutic use in cytomegalovirus infection. Drugs 1994, 48, 455–484. [Google Scholar] [CrossRef] [PubMed]
- Vittecoq, D.; Dumitrescu, L.; Beaufils, H.; Deray, G. Fanconi syndrome associated with cidofovir therapy. Antimicrob. Agents Chemother. 1997, 41, 1846, PMCID: PMC164022. [Google Scholar] [PubMed]
- Wagstaff, A.J.; Bryson, H.M. Foscarnet. A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs 1994, 48, 199–226. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.; Jabs, D.A.; Chou, S.; Martin, B.K.; Lurain, N.S.; Forman, M.S.; Cytomegalovirus Retinitis and Viral Resistance Study Group; The Adult AIDS Clinical Trials Group Cytomegalovirus Laboratories. Mutations conferring foscarnet resistance in a cohort of patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis. J. Infect. Dis. 2003, 187, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Nowlin, D.M.; Cooper, N.R. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 1993, 193, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Mercorelli, B.; Lembo, D.; Palu, G.; Loregian, A. Early inhibitors of human cytomegalovirus: State-of-art and therapeutic perspectives. Pharmacol. Ther. 2011, 131, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Compton, T. Receptor-binding properties of a soluble form of human cytomegalovirus glycoprotein B. J. Virol. 1998, 72, 1826–1833. [Google Scholar] [PubMed]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, Q.C.; Schroer, J.; Murphy, E.; Shenk, T. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 20037–20042. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Carrell, R. Implications of the three-dimensional structure of α 1-antitrypsin for structure and function of serpins. Biochemistry 1989, 28, 8951–8966. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, M.K.; Juckem, L.K.; Compton, T. Virus entry and innate immune activation. Curr. Top. Microbiol. Immunol. 2008, 325, 85–100. [Google Scholar] [PubMed]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed]
- Kalejta, R.F. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr. Top. Microbiol. Immunol. 2008, 325, 101–115. [Google Scholar] [PubMed]
- Ogawa-Goto, K.; Tanaka, K.; Gibson, W.; Moriishi, E.; Miura, Y.; Kurata, T.; Irie, S.; Sata, T. Microtubule network facilitates nuclear targeting of human cytomegalovirus capsid. J. Virol. 2003, 77, 8541–8547. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Chaudhuri, A.R.; Curcio, M.; Tomita, I.; Mizuhashi, F.; Murata, K.; Luduena, R.F. Podophyllotoxin and nocodazole counter the effect of IKP104 on tubulin decay. J. Protein Chem. 1998, 17, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Sabo, Y.; Walsh, D.; Barry, D.S.; Tinaztepe, S.; de Los Santos, K.; Goff, S.P.; Gundersen, G.G.; Naghavi, M.H. HIV-1 induces the formation of stable microtubules to enhance early infection. Cell Host Microbe 2013, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.J.; Cohen, T.; Redmann, V.; Lau, Z.; Felsenfeld, D.; Tortorella, D. Development of a high-content screen for the identification of inhibitors directed against the early steps of the cytomegalovirus infectious cycle. Antiviral Res. 2015, 113, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.J.; Bolovan-Fritts, C.; Teng, M.W.; Redmann, V.; Kraus, T.A.; Sperling, R.; Moran, T.; Britt, W.; Weinberger, L.S.; Tortorella, D. Development of a high-throughput assay to measure the neutralization capability of anti-cytomegalovirus antibodies. Clin. Vaccine Immunol. 2013, 20, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Noriega, V.M.; Gardner, T.J.; Redmann, V.; Bongers, G.; Lira, S.A.; Tortorella, D. Human cytomegalovirus US28 facilitates cell-to-cell viral dissemination. Viruses 2014, 6, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Bolovan-Fritts, C.; Dar, R.D.; Womack, A.; Simpson, M.L.; Shenk, T.; Weinberger, L.S. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell 2012, 151, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Smith, G.A.; Enquist, L.W.; Shenk, T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 2002, 76, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Hahn, G.; Digel, M.; Katona, R.; Sampaio, K.L.; Messerle, M.; Hengel, H.; Koszinowski, U.; Brune, W.; Adler, B. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 2008, 89, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Leyva-Grado, V.H.; Tan, G.S.; Eggink, D.; Hai, R.; Palese, P. In vivo bioluminescent imaging of influenza A virus infection and characterization of novel cross-protective monoclonal antibodies. J. Virol. 2013, 87, 8272–8281. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Hollmann, A.; Lee, J.; Santos, N.C.; Jung, M.E.; Lee, B. The rigid amphipathic fusion inhibitor dUY11 acts through photosensitization of viruses. J. Virol. 2014, 88, 1849–1853. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.C.; Freiberg, A.N.; Zhang, T.; Akyol-Ataman, Z.; Grock, A.; Hong, P.W.; Li, J.; Watson, N.F.; Fang, A.Q.; Aguilar, H.C.; et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 3157–3162. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Chan, G.C.; Yurochko, A.D. Analysis of cytomegalovirus binding/entry-mediated events. Methods Mol. Biol. 2014, 1119, 113–121. [Google Scholar] [PubMed]
- Paeschke, R.; Woskobojnik, I.; Makarov, V.; Schmidtke, M.; Bogner, E. DSTP-27 prevents entry of human cytomegalovirus. Antimicrob. Agents Chemother. 2014, 58, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Castanho, M.A.; Lee, B.; Santos, N.C. Singlet oxygen effects on lipid membranes: Implications for the mechanism of action of broad-spectrum viral fusion inhibitors. Biochem. J. 2014, 459, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Lee, J.; Hollmann, A.; Tanner, L.B.; Akyol Ataman, Z.; Yun, T.; Shui, G.; Aguilar, H.C.; Zhang, D.; Meriwether, D.; et al. A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion. PLoS Pathog. 2013, 9, e1003297. [Google Scholar] [CrossRef] [PubMed]
- Caron, J.M.; Jones, A.L.; Kirschner, M.W. Autoregulation of tubulin synthesis in hepatocytes and fibroblasts. J. Cell Biol. 1985, 101, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Hertel, L. Onset of human cytomegalovirus replication in fibroblasts requires the presence of an intact vimentin cytoskeleton. J. Virol. 2009, 83, 7015–7028. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Smith-Burchnell, C.; Mori, J.; Lewis, M.; Mosley, M.; Stockdale, M.; Dorr, P.; Ciaramella, G.; Perros, M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 2007, 81, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [PubMed]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegalovirus. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Williams: Philadelphia, PA, USA, 2007; pp. 2702–2772. [Google Scholar]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Cantin, C.; Holguera, J.; Ferreira, L.; Villar, E.; Munoz-Barroso, I. Newcastle disease virus may enter cells by caveolae-mediated endocytosis. J. Gen. Virol. 2007, 88, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.G. Structure and function of a paramyxovirus fusion protein. Biochim. Biophys. Acta 2003, 1614, 73–84. [Google Scholar] [CrossRef]
- Regan, A.D.; Whittaker, G.R. Entry of rhabdoviruses into animal cells. Adv. Exp. Med. Biol. 2013, 790, 167–177. [Google Scholar] [PubMed]
- Baquero, E.; Albertini, A.A.; Gaudin, Y. Recent mechanistic and structural insights on class iii viral fusion glycoproteins. Curr. Opin. Struct. Biol. 2015, 33, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.E.; Zeev-Ben-Mordehai, T.; Pandurangan, A.P.; Cairns, T.M.; Hannah, B.P.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Topf, M.; Huiskonen, J.T.; et al. The structure of herpesvirus fusion glycoprotein B-bilayer complex reveals the protein-membrane and lateral protein-protein interaction. Structure 2013, 21, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Bentz, G.L.; Yurochko, A.D. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and β1 and β3 integrins. Proc. Natl. Acad. Sci. USA 2008, 105, 5531–5536. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wilkie, A.R.; Weller, M.; Liu, X.; Cohen, J.I. THY-1 cell surface antigen (CD90) has an important role in the initial stage of human cytomegalovirus infection. PLoS Pathog. 2015, 11, e1004999. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, R.L.; Compton, T. Direct interaction between human cytomegalovirus glycoprotein B and cellular annexin II. J. Virol. 1997, 71, 9803–9807. [Google Scholar] [PubMed]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huong, S.M.; Chiu, M.L.; Raab-Traub, N.; Huang, E.S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Wisner, T.W.; Lei, H.; Kazlauskas, A.; Johnson, D.C. PDGF receptor-α does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog. 2012, 8, e1002905. [Google Scholar] [CrossRef] [PubMed]
- Andreu, J.M.; Timasheff, S.N. Conformational states of tubulin liganded to colchicine, tropolone methyl ether, and podophyllotoxin. Biochemistry 1982, 21, 6465–6476. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.M.; Hamel, E. Effects of inhibitors of tubulin polymerization on GTP hydrolysis. J. Biol. Chem. 1981, 256, 9242–9245. [Google Scholar] [PubMed]
- Head, J.; Lee, L.L.; Field, D.J.; Lee, J.C. Equilibrium and rapid kinetic studies on nocodazole-tubulin interaction. J. Biol. Chem. 1985, 260, 11060–11066. [Google Scholar] [PubMed]
- Luduena, R.F.; Roach, M.C. Interaction of tubulin with drugs and alkylating agents. 2. Effects of colchicine, podophyllotoxin, and vinblastine on the alkylation of tubulin. Biochemistry 1981, 20, 4444–4450. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.R.; Luduena, R.F.; Horowitz, P.M. Bis(8-anilinonaphthalene-1-sulfonate) as a probe for tubulin decay. Biochemistry 1986, 25, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Roth, D.M.; Murray, F.; Swaney, J.S.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 2006, 281, 26391–26399. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Berking, A.; Agerer, F.; Buntru, A.; Neske, F.; Chhatwal, G.S.; Ohlsen, K.; Hauck, C.R. Caveolin limits membrane microdomain mobility and integrin-mediated uptake of fibronectin-binding pathogens. J. Cell Sci. 2010, 123, 4280–4291. [Google Scholar] [CrossRef] [PubMed]
- Grove, L.M.; Southern, B.D.; Jin, T.H.; White, K.E.; Paruchuri, S.; Harel, E.; Wei, Y.; Rahaman, S.O.; Gladson, C.L.; Ding, Q.; et al. Urokinase-type plasminogen activator receptor (upar) ligation induces a raft-localized integrin signaling switch that mediates the hypermotile phenotype of fibrotic fibroblasts. J. Biol. Chem. 2014, 289, 12791–12804. [Google Scholar] [CrossRef] [PubMed]
- Jovasevic, V.; Naghavi, M.H.; Walsh, D. Microtubule plus end-associated clip-170 initiates HSV-1 retrograde transport in primary human cells. J. Cell Biol. 2015, 211, 323–337. [Google Scholar] [CrossRef] [PubMed]
- De Hoop, M.J.; Dotti, C.G. Membrane traffic in polarized neurons in culture. J. Cell Sci. Suppl. 1993, 17, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Samji, H.; Cescon, A.; Hogg, R.S.; Modur, S.P.; Althoff, K.N.; Buchacz, K.; Burchell, A.N.; Cohen, M.; Gebo, K.A.; Gill, M.J.; et al. Closing the gap: Increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS ONE 2013, 8, e81355. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Cell | EC50 (nM) | MPI (%) |
|---|---|---|---|
| TB40/EWT | MRC5 | 81 | 84 |
| TB40/EWT | ARPE-19 | 70 | 74 |
| Virus | EC50 (nM) | MPI (%) |
|---|---|---|
| HSV1 | 20 | 90 |
| IAV | N/A | 44 |
| NDV | N/A | 34 |
| SeV | N/A | 49 |
| VSV | 24 | 68 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, T.; Schwarz, T.M.; Vigant, F.; Gardner, T.J.; Hernandez, R.E.; Lee, B.; Tortorella, D. The Microtubule Inhibitor Podofilox Inhibits an Early Entry Step of Human Cytomegalovirus. Viruses 2016, 8, 295. https://doi.org/10.3390/v8100295
Cohen T, Schwarz TM, Vigant F, Gardner TJ, Hernandez RE, Lee B, Tortorella D. The Microtubule Inhibitor Podofilox Inhibits an Early Entry Step of Human Cytomegalovirus. Viruses. 2016; 8(10):295. https://doi.org/10.3390/v8100295
Chicago/Turabian StyleCohen, Tobias, Toni M. Schwarz, Frederic Vigant, Thomas J. Gardner, Rosmel E. Hernandez, Benhur Lee, and Domenico Tortorella. 2016. "The Microtubule Inhibitor Podofilox Inhibits an Early Entry Step of Human Cytomegalovirus" Viruses 8, no. 10: 295. https://doi.org/10.3390/v8100295
APA StyleCohen, T., Schwarz, T. M., Vigant, F., Gardner, T. J., Hernandez, R. E., Lee, B., & Tortorella, D. (2016). The Microtubule Inhibitor Podofilox Inhibits an Early Entry Step of Human Cytomegalovirus. Viruses, 8(10), 295. https://doi.org/10.3390/v8100295