
Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade



Abstract
:1. Introduction
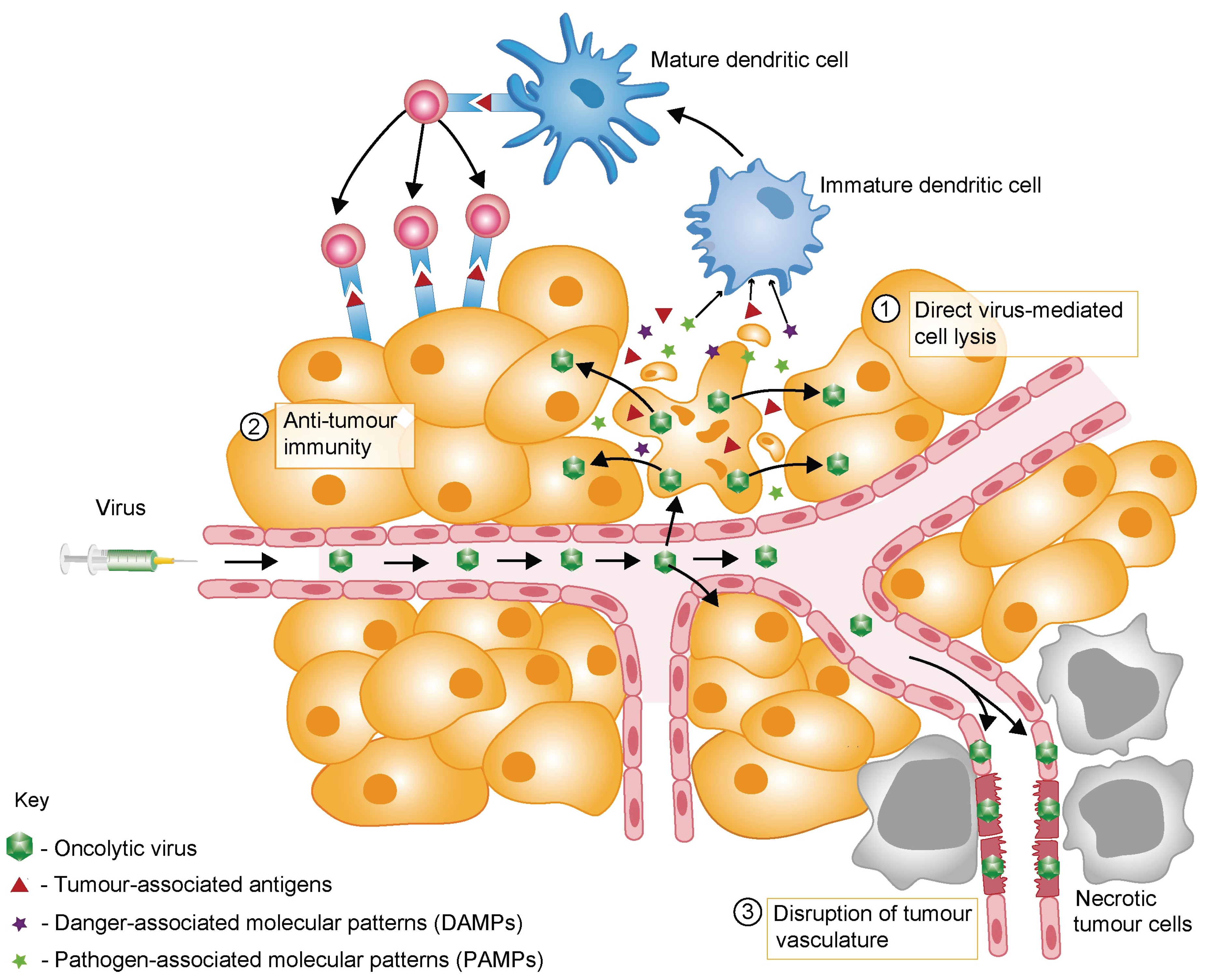
2. Mechanisms of OV-Mediated Tumour Destruction
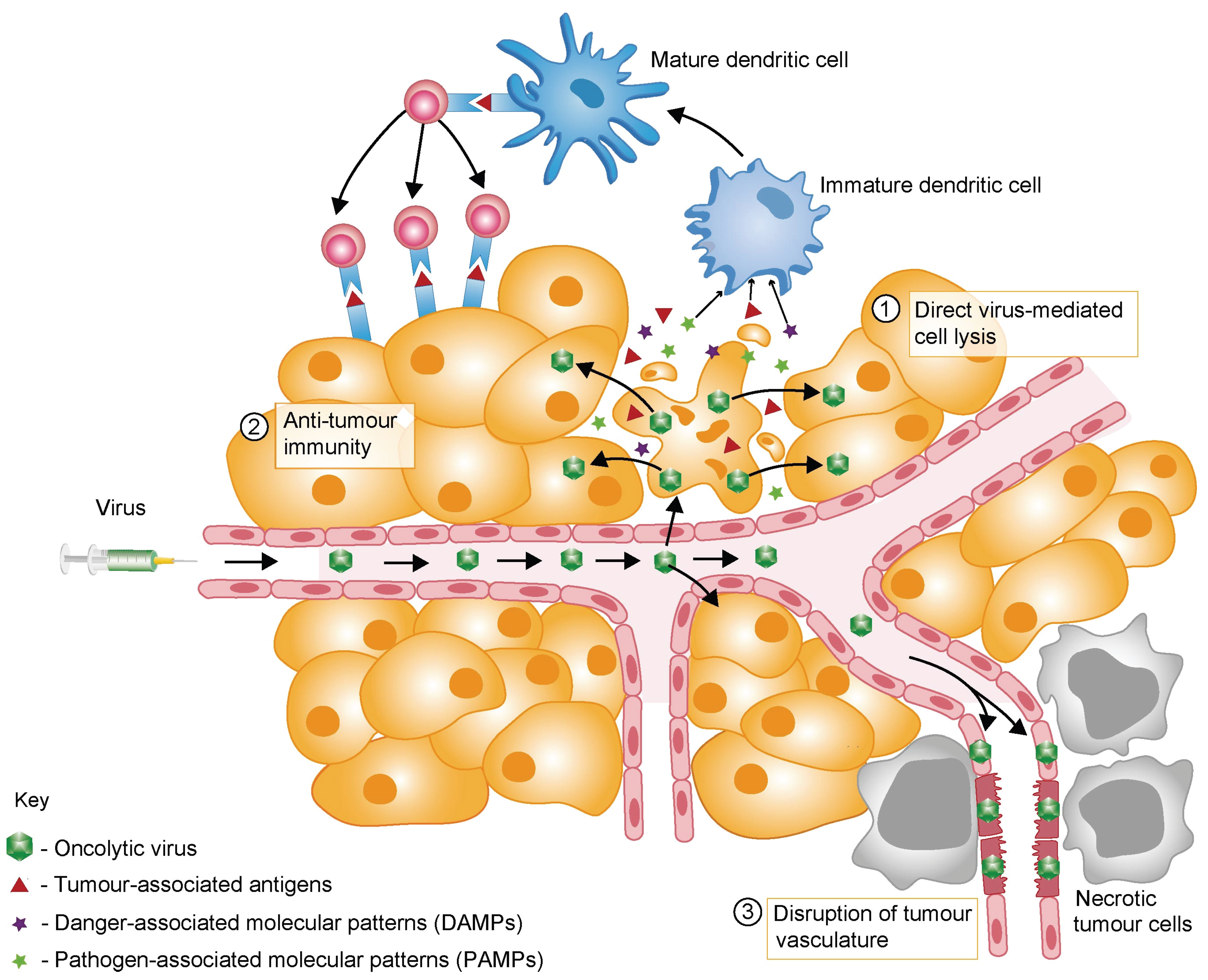
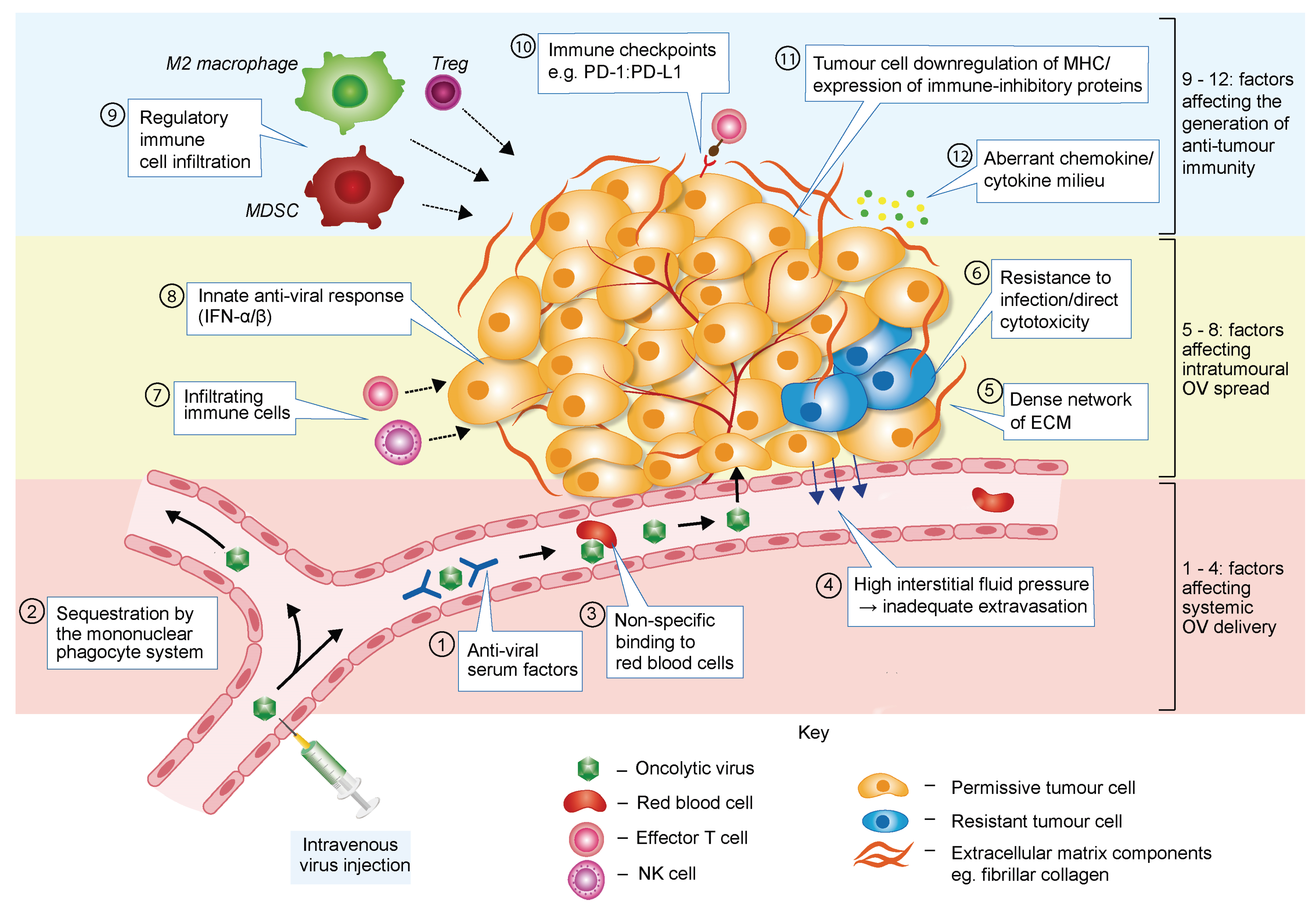
3. Barriers to Successful Oncolytic Virotherapy
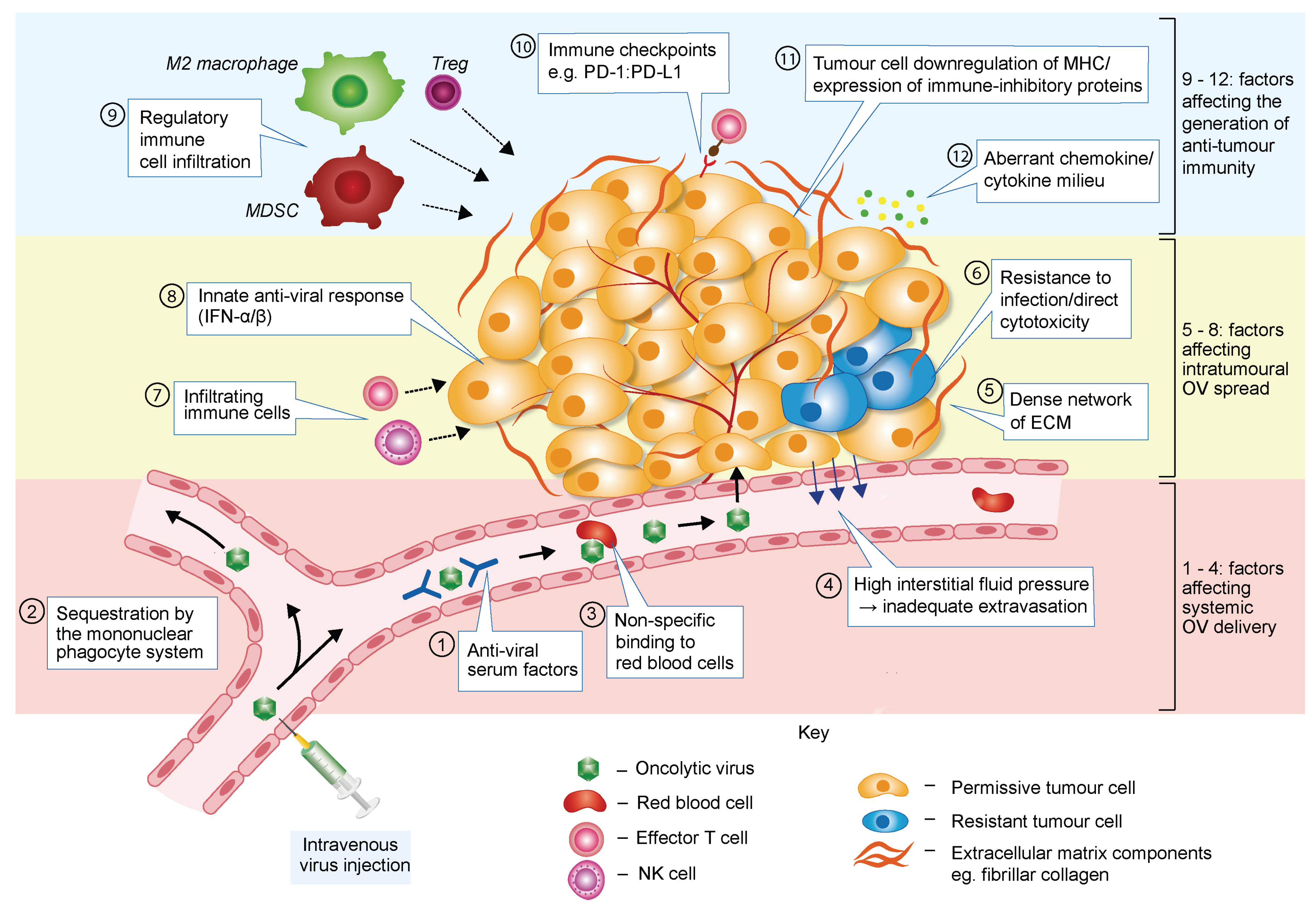
3.1. Barriers Limiting Systemic OV Delivery
3.2. Barriers Affecting Intratumoural Virus Infection and Spread
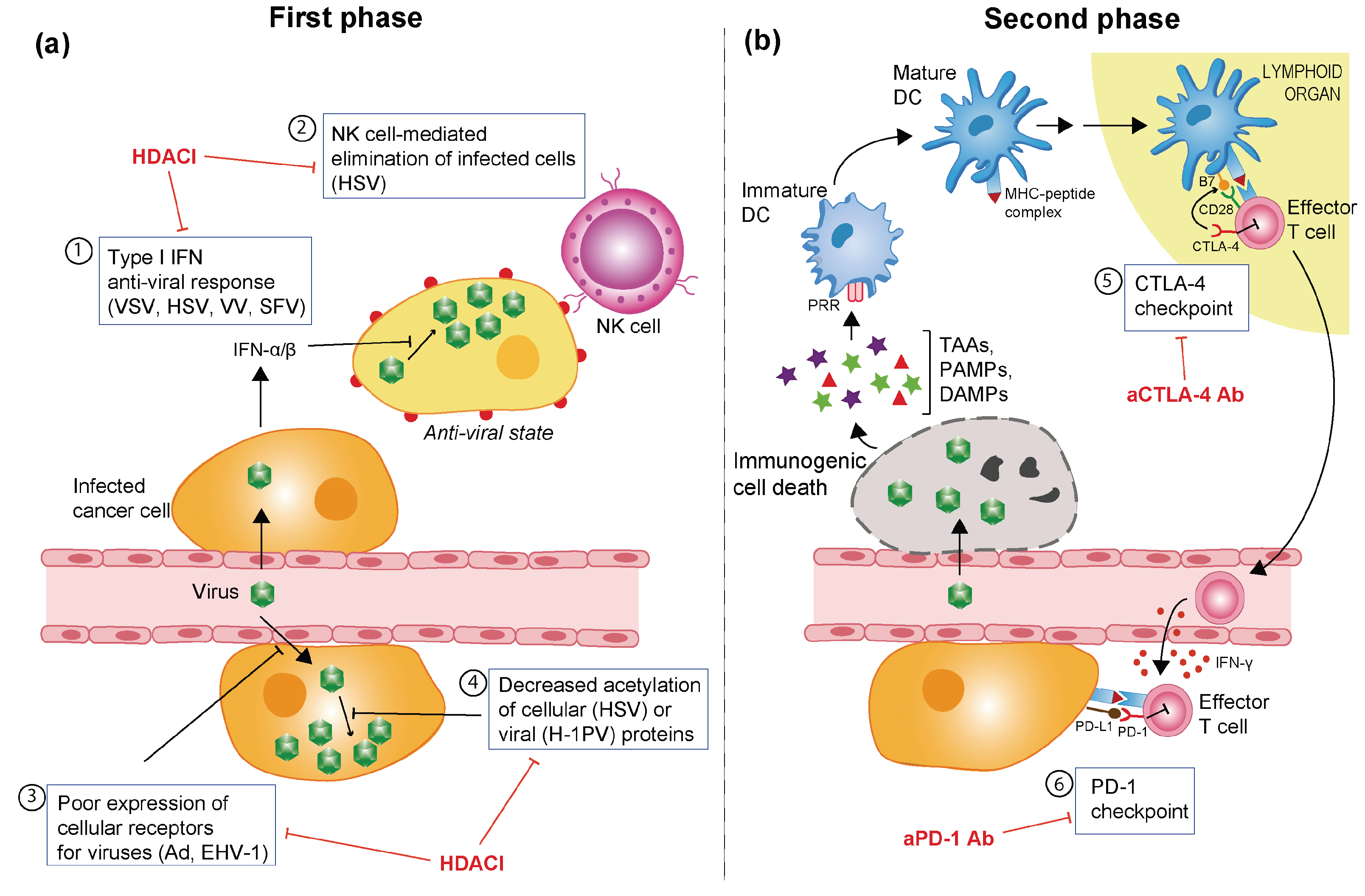
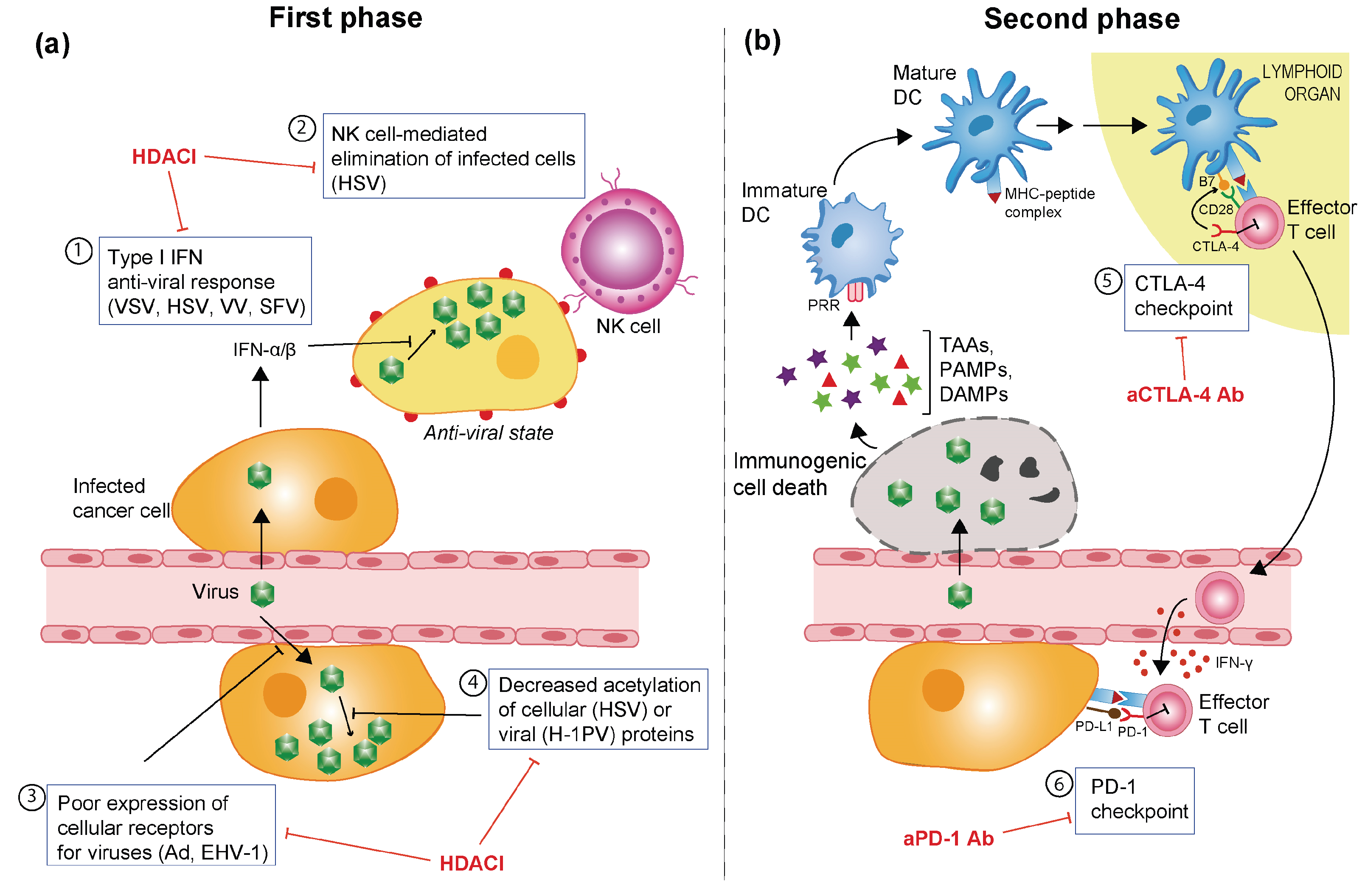
3.3. Barriers Hampering OV-Induced Anti-Tumour Immune Response
4. OVs in Combination Therapy
5. OVs in Combination with Histone Deacetylase Inhibitors

{kind=link}
{kind=link}
{kind=link}
| Virus | Viral Variant | HDACI(s) | HDAC Selectivity | Cancer Type(s) | In vivo Model (Route of OV Delivery) | Mode of Action | Ref. |
|---|---|---|---|---|---|---|---|
| VSV | VSVΔ51 | Vorinostat, MS-275 | Classes I and II (Vorinostat) | Various solid tumours | Athymic nude mice (IT or IP) | ↓ IFN and IFN-responsive gene expression; ↑ virus multiplication; ↑ intrinsic apoptosis | [71] |
| Class I (MS-275) | |||||||
| VSVΔ51 | Vorinostat | Classes I and II | Prostate cancer | - | ↑ NF-κB activity; ↑ autophagy; ↓ IFN-mediated response; ↑ viral replication and apoptosis | [72] | |
| HSV-1 | G47Δ | TSA | Classes I and II | Glioma and colorectal cancer | Athymic nude mice (IT) | ↓ VEGF secretion; ↓ angiogenesis; ↓ cyclin D1 | [73] |
| rQNestin34.5 | VPA (pre-treatment) | Classes I and IIa | Glioma | Athymic nude mice (IT) | ↓ IFN-inducible gene expression; ↑ viral replication | [74] | |
| R849 | TSA | Classes I and II | Oral squamous cell carcinoma | - | ↑ NF-κB activity; ↑ viral replication; ↑p21→G1 cell cycle arrest | [75] | |
| rQNestin34.5 | VPA | Classes I and IIa | Glioma | Athymic nude mice (IT) | ↓ Innate immune responses; ↓ NK cell activity, through inhibition of STAT5/T-BET signalling | [76] | |
| ΔICP34.5 | Various | - | Breast cancer | - | ↑ Viral replication | [77] | |
| EHV-1 | Wild type (WT) | VPA (pre-treatment) | Classes I and IIa | Glioma | - | ↑ Viral entry | [78] |
| Ad | Ad5.CMV-LacZ | Romidepsin | Class I | Various solid tumours | - | ↑ Viral entry receptors | [79] |
| OBP-301 | Romidepsin | Class I | Non-small cell lung cancer | - | ↑ Viral entry receptors | [80] | |
| Ad5.CMV-GFP | Romidepsin | Class I | Melanoma | Athymic nude mice (IT) | ↑ Viral entry receptors | [81] | |
| Delta24-RGD | Scriptaid, LBH589 | Class I (Scriptaid) | Glioma-initiating stem-like cells | - | ↑ Cell death pathways | [82] | |
| Classes I and II (LBH589) | |||||||
| VV | VVdd | TSA | Classes I and II | Various solid tumours | Immunocompetent C57BL/6 mice (IV) | ↓ IFN-response; ↑ viral replication and spread | [83] |
| Western Reserve | TSA | Classes I and II | Various solid tumours | - | ↑ Viral replication | [83] | |
| Western Reserve B18R-TK-Luc+ | TSA | Classes I and II | Various solid tumours | Athymic nude mice (IV) | ↑ Viral replication | [83] | |
| H-1PV | WT | VPA, sodium butyrate | Classes I and IIa (VPA) | Cervical and pancreatic carcinomas | Athymic nude rats and NOD/SCID mice (IT) | ↑ Acetylation and activity of viral effector protein; ↑ virus multiplication; ↑ oxidative stress | [84] |
| Classes I and IIa (sodium butyrate) | |||||||
| SFV | WT | Vorinostat, MS-275 | Classes I and II (Vorinostat) | Breast cancer | - | ↑ Viral replication and spread | [71] |
5.1. Vesicular Stomatitis Virus
5.2. Herpesvirus
5.3. Adenovirus
5.4. Vaccinia Virus
5.5. H-1 Parvovirus
5.6. Semliki Forest Virus
6. Potentiating OV-Elicited Anti-Tumour Immune Responses with Immune Checkpoint Inhibitors
6.1. Pre-Clinical Studies
6.2. Clinical Studies
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Bines, S.D. OPTIM trial: A Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010, 6, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Cancer-fighting viruses win approval. Nature 2015, 526, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Kyula, J.N.; Roulstone, V.; Karapanagiotou, E.M.; Melcher, A.A.; Harrington, K.J. Oncolytic reovirus type 3 (Dearing) as a novel therapy in head and neck cancer. Expert Opin. Biol. Ther. 2012, 12, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Eggermont, A.; Sautes-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Yaacov, B.; Shiloach, T.; Eliahoo, E.; Kadouri, L.; Lotem, M.; Perlman, R.; Zakay-Rones, Z.; Panet, A.; Ben-Yehuda, D. The oncolytic activity of Newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein Livin. J. Virol. 2010, 84, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Roth, J.C.; Friedman, G.K.; Gillespie, G.Y. Oncolytic viral therapy: Targeting cancer stem cells. Oncol.Virother. 2014, 2014, 21–33. [Google Scholar]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; de Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J., Jr.; Michaelis, M.; Driever, P.H.; Cinatl, J.; Hrabeta, J.; Suhan, T.; Doerr, H.W.; Vogel, J.U. Multimutated herpes simplex virus g207 is a potent inhibitor of angiogenesis. Neoplasia 2004, 6, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.; Nieva, J.; Hwang, T.H.; Moon, A.; Patt, R.; et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Joensuu, T.; Jager, E.; Karbach, J.; Wahle, C.; Kairemo, K.; Alanko, T.; Partanen, K.; Turkki, R.; Linder, N.; et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8 T-cell response, prominent infiltration of CD8 lymphocytes and Th1 type polarization. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.; Ranki, T.; Joensuu, T.; Jager, E.; Karbach, J.; Wahle, C.; Partanen, K.; Kairemo, K.; Alanko, T.; Turkki, R.; et al. Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8 T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Kratzke, R.A. Oncolytic virus therapy for cancer: The first wave of translational clinical trials. Transl. Res. 2013, 161, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Wakimoto, H.; Ichikawa, T.; Jhung, S.; Hochberg, F.H.; Louis, D.N.; Chiocca, E.A. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J. Virol. 2000, 74, 4765–4775. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Fulci, G.; Tyminski, E.; Chiocca, E.A. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysis. Gene Ther. 2004, 11, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Tian, J.; Smith, J.S.; Byrnes, A.P. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J. Virol. 2008, 82, 11705–11713. [Google Scholar] [CrossRef] [PubMed]
- Iankov, I.D.; Blechacz, B.; Liu, C.; Schmeckpeper, J.D.; Tarara, J.E.; Federspiel, M.J.; Caplice, N.; Russell, S.J. Infected cell carriers: A new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol. Ther. 2007, 15, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, D.C.; Charlton, D.; Henderson, D.R. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of CN706 in the nude mouse LNCaP xenograft model: Implications and proposals for human therapy. Hum. Gene Ther. 2000, 11, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.W.; Myers, R.; Greenslade, A.; Mader, E.; Greiner, S.; Federspiel, M.J.; Dispenzieri, A.; Russell, S.J. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013, 20, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.; Onion, D.; Green, N.K.; Aslan, K.; Rajaratnam, R.; Bazan-Peregrino, M.; Phipps, S.; Hale, S.; Mautner, V.; Seymour, L.W.; et al. Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol. Ther. 2006, 14, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Breznik, J.; Lichty, B.D. Strategies to enhance viral penetration of solid tumors. Hum. Gene Ther. 2011, 22, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Critchley-Thorne, R.J.; Simons, D.L.; Yan, N.; Miyahira, A.K.; Dirbas, F.M.; Johnson, D.L.; Swetter, S.M.; Carlson, R.W.; Fisher, G.A.; Koong, A.; et al. Impaired interferon signaling is a common immune defect in human cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 9010–9015. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Cramer, S.D.; Lyles, D.S. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 2004, 330, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Haralambieva, I.; Iankov, I.; Hasegawa, K.; Harvey, M.; Russell, S.J.; Peng, K.W. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol. Ther. 2007, 15, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Vaha-Koskela, M.J.; Heikkila, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef] [PubMed]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Calcinotto, A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Joss, A.; Akdis, M.; Faith, A.; Blaser, K. A molecular basis for T cell suppression by IL-10: CD28-associated IL-10 receptor inhibits CD28 tyrosine phosphorylation and phosphatidylinositol 3-kinase binding. FASEB J. 2000, 14, 1666–1668. [Google Scholar] [CrossRef] [PubMed]
- Perillo, N.L.; Pace, K.E.; Seilhamer, J.J.; Baum, L.G. Apoptosis of T cells mediated by galectin-1. Nature 1995, 378, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Zamanakou, M.; Germenis, A.E.; Karanikas, V. Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunol. Lett. 2007, 111, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.T.; Liu, J.; McFadden, G. Bugs and drugs: Oncolytic virotherapy in combination with chemotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1817–1833. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Breckenridge, C.; Kaur, B.; Chiocca, E.A. Pharmacologic and chemical adjuvants in tumor virotherapy. Chem. Rev. 2009, 109, 3125–3140. [Google Scholar] [CrossRef] [PubMed]
- Eager, R.M.; Nemunaitis, J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011, 18, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.E.; Abdelbary, H.; Lupien, M.; Bell, J.C.; Diallo, J.S. Exploiting tumor epigenetics to improve oncolytic virotherapy. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.E.; Krishnan, R.; Diallo, J.S. Pharmacological modulation of anti-tumor immunity induced by oncolytic viruses. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; la Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, A.J.; West, A.; Banks, K.M.; Haynes, N.M.; Teng, M.W.; Smyth, M.J.; Johnstone, R.W. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar] [PubMed]
- Chang, H.M.; Paulson, M.; Holko, M.; Rice, C.M.; Williams, B.R.; Marie, I.; Levy, D.E. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 9578–9583. [Google Scholar] [CrossRef] [PubMed]
- Nusinzon, I.; Horvath, C.M. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. USA 2003, 100, 14742–14747. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Abdelbary, H.; Arguello, M.; Breitbach, C.; Leveille, S.; Diallo, J.S.; Yasmeen, A.; Bismar, T.A.; Kirn, D.; Falls, T.; et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc. Natl. Acad. Sci. USA 2008, 105, 14981–14986. [Google Scholar] [CrossRef] [PubMed]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; van Grevenynghe, J.; Belgnaoui, S.M.; Nguyen, T.L.; di Lenardo, T.; Semmes, O.J.; Lin, R.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Castelo-Branco, P.; Rabkin, S.D.; Martuza, R.L. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol. Ther. 2008, 16, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, A.; Patel, A.; Kasai, K.; Suzuki, M.; Kurozumi, K.; Chiocca, E.A.; Saeki, Y. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 2008, 16, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Katsura, T.; Iwai, S.; Ota, Y.; Shimizu, H.; Ikuta, K.; Yura, Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009, 16, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.; Wei, M.; Wang, Y.; Nowicki, M.O.; Ha, Y.P.; Bergin, S.; Hwang, C.; Fernandez, S.A.; et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 2012, 86, 4566–4577. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.J.; Markert, J.M.; Hurst, D.R. Histone deacetylase inhibitors improve the replication of oncolytic herpes simplex virus in breast cancer cells. PLoS ONE 2014, 9, e92919. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, M.J.; White, M.C.; Stanfield, B.A.; Frampton, A.R., Jr. Equine herpesvirus type 1-mediated oncolysis of human glioblastoma multiforme cells. J. Virol. 2012, 86, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Kitazono, M.; Goldsmith, M.E.; Aikou, T.; Bates, S.; Fojo, T. Enhanced adenovirus transgene expression in malignant cells treated with the histone deacetylase inhibitor FR901228. Cancer Res. 2001, 61, 6328–6330. [Google Scholar] [PubMed]
- Watanabe, T.; Hioki, M.; Fujiwara, T.; Nishizaki, M.; Kagawa, S.; Taki, M.; Kishimoto, H.; Endo, Y.; Urata, Y.; Tanaka, N.; et al. Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase-specific replication-selective adenoviral agent OBP-301 in human lung cancer cells. Exp. Cell Res. 2006, 312, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.E.; Aguila, A.; Steadman, K.; Martinez, A.; Steinberg, S.M.; Alley, M.C.; Waud, W.R.; Bates, S.E.; Fojo, T. The histone deacetylase inhibitor FK228 given prior to adenovirus infection can boost infection in melanoma xenograft model systems. Mol. Cancer Ther. 2007, 6, 496–505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berghauser Pont, L.M.; Kleijn, A.; Kloezeman, J.J.; van den Bossche, W.; Kaufmann, J.K.; de Vrij, J.; Leenstra, S.; Dirven, C.M.; Lamfers, M.L. The HDAC inhibitors scriptaid and LBH589 combined with the oncolytic virus Delta24-RGD exert enhanced anti-tumor efficacy in patient-derived glioblastoma cells. PLoS ONE 2015, 10, e0127058. [Google Scholar] [CrossRef] [PubMed]
- MacTavish, H.; Diallo, J.S.; Huang, B.; Stanford, M.; le Boeuf, F.; de Silva, N.; Cox, J.; Simmons, J.G.; Guimond, T.; Falls, T.; et al. Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors. PLoS ONE 2010, 5, e14462. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnolzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Bridle, B.W.; Chen, L.; Lemay, C.G.; Diallo, J.S.; Pol, J.; Nguyen, A.; Capretta, A.; He, R.; Bramson, J.L.; Bell, J.C.; et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol. Ther. 2013, 21, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Frampton, A.R., Jr. The histone deacetylase inhibitor valproic acid enhances equine herpesvirus type 1 (EHV-1)-mediated oncolysis of human glioma cells. Cancer Gene Ther. 2013, 20, 88–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khan, A.N.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.E.; Kitazono, M.; Fok, P.; Aikou, T.; Bates, S.; Fojo, T. The histone deacetylase inhibitor FK228 preferentially enhances adenovirus transgene expression in malignant cells. Clin. Cancer Res. 2003, 9, 5394–5401. [Google Scholar] [PubMed]
- Kitazono, M.; Rao, V.K.; Robey, R.; Aikou, T.; Bates, S.; Fojo, T.; Goldsmith, M.E. Histone deacetylase inhibitor FR901228 enhances adenovirus infection of hematopoietic cells. Blood 2002, 99, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Maguire, C.A.; Ramirez, S.H.; Bradel-Tretheway, B.; Sapinoro, R.; Sui, Z.; Chakraborty-Sett, S.; Dewhurst, S. Valproic acid enhances gene expression from viral gene transfer vectors. J. Virol. Methods 2005, 125, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Sachs, M.D.; Ramamurthy, M.; Poel, H.; Wickham, T.J.; Lamfers, M.; Gerritsen, W.; Chowdhury, W.; Li, Y.; Schoenberg, M.P.; Rodriguez, R. Histone deacetylase inhibitors upregulate expression of the coxsackie adenovirus receptor (CAR) preferentially in bladder cancer cells. Cancer Gene Ther. 2004, 11, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Segura-Pacheco, B.; Avalos, B.; Rangel, E.; Velazquez, D.; Cabrera, G. HDAC inhibitor valproic acid upregulates CAR in vitro and in vivo. Genet.Vaccines Ther. 2007, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.R.; Park, M.Y.; Lee, C.S.; Shim, S.H.; Yoon, H.I.; Lee, J.H.; Sung, M.W.; Kim, Y.S.; Lee, C.T. Combination of vorinostat and adenovirus-TRAIL exhibits a synergistic antitumor effect by increasing transduction and transcription of TRAIL in lung cancer cells. Cancer Gene Ther. 2011, 18, 467–477. [Google Scholar] [CrossRef] [PubMed]
- VanOosten, R.L.; Earel, J.K., Jr.; Griffith, T.S. Histone deacetylase inhibitors enhance Ad5-TRAIL killing of TRAIL-resistant prostate tumor cells through increased caspase-2 activity. Apoptosis 2007, 12, 561–571. [Google Scholar] [CrossRef] [PubMed]
- VanOosten, R.L.; Earel, J.K., Jr.; Griffith, T.S. Enhancement of Ad5-TRAIL cytotoxicity against renal cell carcinoma with histone deacetylase inhibitors. Cancer Gene Ther. 2006, 13, 628–632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vanoosten, R.L.; Moore, J.M.; Ludwig, A.T.; Griffith, T.S. Depsipeptide (FR901228) enhances the cytotoxic activity of TRAIL by redistributing TRAIL receptor to membrane lipid rafts. Mol. Ther. 2005, 11, 542–552. [Google Scholar] [CrossRef] [PubMed]
- El-Zawahry, A.; Lu, P.; White, S.J.; Voelkel-Johnson, C. In vitro efficacy of AdTRAIL gene therapy of bladder cancer is enhanced by trichostatin A-mediated restoration of CAR expression and downregulation of cFLIP and Bcl-XL. Cancer Gene Ther. 2006, 13, 281–289. [Google Scholar] [CrossRef] [PubMed]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Kirn, D.H.; Wang, Y.; le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007, 4, e353. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Wilson, M.G.; Hiscott, J. Oncolytic viruses and histone deacetylase inhibitors—A multi-pronged strategy to target tumor cells. Cytokine Growth Factor Rev. 2010, 21, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kondo, S.; Tada, K.; Kitano, S. Clinical development of immune checkpoint inhibitors. Biomed. Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase IIItrials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Postow, M.A. Immune checkpoint modulation: Rational design of combination strategies. Pharmacol. Ther. 2015, 150, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Wang, Z.; Donovan, C.; He, H.; Mark, D.; Guan, G.; Wang, Y.; Walunas, T.; Bluestone, J.; Listman, J.; et al. Regulation of CTLA-4 expression during T cell activation. J. Immunol. 1996, 156, 4154–4159. [Google Scholar] [PubMed]
- Rojas, J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Loskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Andtbacka, R.H.I.; Minor, D.R.; Hamid, O.; Li, A.; Chastain, M.; Gorski, K.; Anderson, A.; Vanderwalde, A.M.; et al. Primary analysis of a phase 1b multicenter trial to evaluate safety and efficacy of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB-IV melanoma. J. Clin. Oncol. 2014, 32 (Suppl. S5). [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses 2016, 8, 9. https://doi.org/10.3390/v8010009
Marchini A, Scott EM, Rommelaere J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses. 2016; 8(1):9. https://doi.org/10.3390/v8010009
Chicago/Turabian StyleMarchini, Antonio, Eleanor M. Scott, and Jean Rommelaere. 2016. "Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade" Viruses 8, no. 1: 9. https://doi.org/10.3390/v8010009
APA StyleMarchini, A., Scott, E. M., & Rommelaere, J. (2016). Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses, 8(1), 9. https://doi.org/10.3390/v8010009