1. Introduction
The vaccinia virus (Poxviridae family) is among the most “famous” viruses; it is primarily known for its successful use in vaccination and its role in smallpox eradication [
1]. Vaccinia virus (VACV) possesses many unique properties that place this virus at a leading position in molecular biology and genetic engineering. Its ability to kill cancer cells is one of the fundamental biological properties of VACV, and was first reported by Levaditi C. and Nicolau S. in 1923 in the Annals of the Pasteur Institute [
2]. Subsequent studies confirmed the oncolytic activity of VACV [
3]; however, over the next few decades researchers failed to achieve stable results in treatment of cancer patients using VACV, and most importantly, to avoid complications caused by introducing the infectious virus into human organism. The interest to studies of VACV application in oncology seemed to wane. However, progress in genetic engineering now enables researchers to change the biological properties of VACV over a wide range, and this has led to a surge of interest to oncolytic abilities of genetically modified VACV. Modern studies exploit many properties of VACV in order to reach its full oncolytic potential. These properties include the VACV’s ability to infect a wide range of eukaryotic cells [
4], and to actively replicate in the cytoplasm and thus spread among neighboring cells to infect and kill them. Most importantly for genetic engineering are the absence of VACV integration with the host genome and the possibility to insert up to 25,000 base pairs into the viral genome without loss of infectivity [
5,
6,
7]. To enhance VACV oncolytic properties, various genes are inserted: cytokines [
8,
9,
10,
11], inhibitors of angiogenesis [
12,
13], enzymes that convert non-toxic precursors within tumors in their cytotoxic derivatives [
14,
15], and genes of apoptosis-inducing proteins [
16,
17]. Detailed reviews and analysis of studies with genetically modified oncolytic VACV, including some which are in clinical trials, recently were published in several comprehensive reviews [
7,
18,
19].
Advances in development of genetically modified VACV variants overshadowed the question: how original parental VACV implements oncolytic (antitumor) effect? We think that it is necessary to know “native” antitumor properties of the parental VACV in order to understand the mechanisms of recombinant viruses’ oncolytic activities and to evaluate the real effects of the genetic modifications. The aim of this work was to study antitumor properties of the VACV strain L-IVP (Lister-Institute of Vaccine Preparations, Moscow, Russia, GenBank accession number: Bank It1780508 LIVP KP233807). It is believed that Lister strain of VACV was isolated in Cologne (Germany) at the Institute of Vaccines in 1870 from a soldier suffering from smallpox, and most probably the strain appeared after the additional infection of a soldier with the laboratory vaccine strain [
1].
The Lister strain was used in the program to fight smallpox in the United Kingdom since 1892 and in the Lister Institute (France) since 1916. After transfer to other laboratories, this strain was also known as Liverpool, Merieux 37 and Nigeria. The Lister strain also was transferred to the Institute for Viral Preparations (Moscow, USSR) and used under the acronym L-IVP for vaccination against smallpox and for scientific research [
17,
20,
21]. Many studies have shown the advantages of genetically modified Lister strain as an oncolytic agent in comparison with other VACV strains and other viruses [
22].
Our study of apoptin-producing recombinant obtained using L-IVP strain in comparison with the parental strain revealed rapid destruction of human carcinoma A431 xenografts in
nude mice after intratumoral injection of both viruses [
20]. The L-IVP strain clearly demonstrated oncolytic effects via direct destruction of tumor cells (signs of inflammatory reactions and leukocyte accumulation in tumor tissue, and viral destruction of blood vessels were not observed). The question arose: what are other mechanisms may contribute to the antitumor effects of the VACV?
In this study, we examined antitumor effect of the L-IVP strain using murine Ehrlich carcinoma in C57Bl mice and compared that with oncolytic effect of this virus in human A431 carcinoma xenografts in nude mice. In contrast with human cells, murine cells are not naturally susceptible to VACV, so it was interesting to compare viral antitumor effects in these two models. Our study showed that the L-IVP strain of VACV possesses antitumor activity towards murine tumor, which is mainly related with mitotic arrest in murine tumor cells.
2. Materials and Methods
2.1. Virus and Cells
The L-IVP strain of VACV was obtained from the State Collection of Viral and Rickettsial Disease Agents of the State Research Center of Virology and Biotechnology “Vector” (SRC VB “Vector”, Koltsovo, Russia). The strain was cloned and has been passed 6 times in CV-1 cells and purified by centrifugation in sucrose density gradient (25%–45%). The viral preparation was sonicated and titrated using the plaque formation assay in CV-1 cell monolayers. Virus titers were expressed as plaque forming units (PFU) per mL. The viral stock represented 109 PFU/mL in sterile saline and aliquots were stored at −80 °C. Human cancer cell lines (A549, A431, C33A, U87MG, RD, DU145, MCF7, Mel8, SW480, HeLa) of different origin were grown in DMEM (Invitrogen, Waltham, MA, USA) supplemented with 10% fetal calf serum (FCS, HyClone, Logan, UT, USA). Diploid human embryonic LECH-240 cells were grown in F-12 medium (Invitrogen) supplemented with 10% fetal calf serum (FCS, HyClone). MCF10A cells were grown in a specialized culture medium for mammary epithelial cells MEGM Bullet Kit (Lonza, Allendale, NJ, USA).
2.2. Cytotoxic Activity of VACV Strain L-IVP toward Human Tumor Cell Lines
Cytotoxic activity of VACV strain L-IVP toward human tumor cell lines was evaluated by XTT microassay (using 2,3_bis_(2-methoxy-4_nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, Sigma-Aldrich, St. Louis, MO, USA) in 96-well plates (Greiner, Pleidelsheim, Germany) [
8]. This method employs the fact that mitochondrial dehydrogenases can convert soluble XTT into formazan, which crystallizes within the cell. Formazan can be solubilized by phenazine methosulfate (PMS) treatment, and the optical density of the solution determined by spectrophotometry accurately reflects the changes of formazan quantities in viable cells. The specific rate of cell death in infected cultures was assessed in relation to uninfected control cells (100% viabilityπ). Cytolytic activity was evaluated as the 50% cytotoxic dose (CD
50), that is, the virus concentration causing death of 50% of cells. To determine CD
50, cells growing in a 50% monolayer were infected with sequential tenfold dilutions of viral suspension in 100 μL of 199 medium supplemented with 2% FCS (0.001 to 10 PFU/cell). Following 72 h incubation at 37 °C;, in an atmosphere of 5% CO
2 and 85% humidity, 50 μL of XTT/PMS mixture were added to each well (the mixture was prepared with 20 μL of 1.25 mM PMS (Fluka, St. Louis, MO, USA) per 1 mL of 1 mg/mL XTT working solution). Plates were incubated for additional 4 h, and optical densities (
ОD490/620) were determined using the SpectraCount Plate Reader photometer (Packard, Missouri, TX, USA). To obtain a curve that represents the dependence between
OD and multiplicity of infection, data obtained from five replicates were used to calculate the mean and SD values for each point corresponding to different virus concentrations. This curve was used to determine CD
50 as the virus concentration at which the
ОD490/620 value of infected wells was 50% of
ОD490/620 measured in wells with uninfected culture. CD
50 values were compared across cell cultures; higher CD
50 indicated the lower oncolytic activity of the given virus strain in a particular cell culture. All computations were performed using LabView software.
2.3. Animal Studies
Studies with mice were performed under protocols approved by the SRC VB VECTOR Institutional Animal Care and Use Committee (NIH Office 85 of Laboratory Animal Welfare, Number A5505-02).
A431 human carcinoma model. Female
Nu/Nu mice (Nursery for Laboratory Animals, Institute of Bioorganic Chemistry, Moscow, Russia) were used for the xenograft model of human A431 carcinoma. Tumors were generated in 8–10 week old
Nu/Nu mice (20–26 g) by subcutaneous injections of 5 × 10
6 A431 cells (in 100 mL PBS) in the left hind region. Tumors were measured in two dimensions using a digital caliper, and their volume was calculated by the following formula: [length × width
2 × 0.5] [
20]. When tumors reached 200–250 mm
3 in volume (10 days after tumor cells injection), the mice received a single injection of the LIVP strain (1 × 10
7 PFU/mouse in 100 mL of saline) into tumor. The control group of mice received 100 mL of saline.
To assess the dynamics of virus accumulation in tissues and organs, mice were sacrificed every 48 h in the period from 2 to 20 days after injection; and every 96 h in the period from 20th to 55th days (two mice at each time-point from each experimental group). Specimens of xenografts, spleen, liver, lungs, kidneys and blood were collected from two mice from experimental and control groups at each time point. To evaluate viral content, homogenates of organs and tissues (10%, v/v) were prepared in saline, sonicated, centrifuged and then titrated by plaque formation assay on a monolayer of CV-1 cells. For microscopic studies xenografts were collected from mice of each control and infected groups at 2, 4 and 8 days after VACV injection. Xenografts of infected mice also were sampled on 36th and 55th days after virus injection. The tumors and adjacent tissues were dissected and fixed in 4% paraformaldehyde solution.
Murine Ehrlich carcinoma model. Male mice C57Bl were inoculated subcutaneously with murine Ehrlich ascites carcinoma cells (13 × 106 in 0.4 mL of sterile saline) in the thigh right hind paw. After 7 days, when tumors reached 200–220 mm3 in volume, mice received a single injection of the L-IVP stain (1 × 107 PFU/mouse in 100 mL of sterile saline) into tumor. Mice of the control group received 100 mL of saline. Mice were sacrificed on days 2, 4, 6, 8, 12 and 14 after virus injection (three mice at each time-point from each group); volume of their tumors was measured as described above. Tumors were dissected and fixed in 4% paraformaldehyde solution.
Female C57/B1 mice were injected intraperitoneally with 3–3.5 × 106 cells of Ehrlich carcinoma cells in 0.2 mL of saline. Four days after cell injection 15 mice received intraperitoneally 1.3 × 107 PFU of L-IVP strain in 100 mL of saline, and 10 mice were injected with 100 mL of saline (control). Mice were sacrificed on days 3, 6, 9 and 10 after VACV injection (three mice from at each time-point from each group), abdominal cavity was opened and ascitic fluid was collected. Some fluid (0.3–0.4 mL) was used for VACV titration by plaque formation assay on a monolayer of CV-1 cells. The remaining ascitic fluid was cleaned by three repeats of dilution with saline and centrifugation at 2000 RPM for 5 min. Final pellets were fixed in 4% paraformaldehyde solution.
We attempted to propagate cells of Ehrlich carcinoma in vitro without success; in agreement with published studies, the tumor was only able to be grown in vivo.
2.4. Microscopic Studies
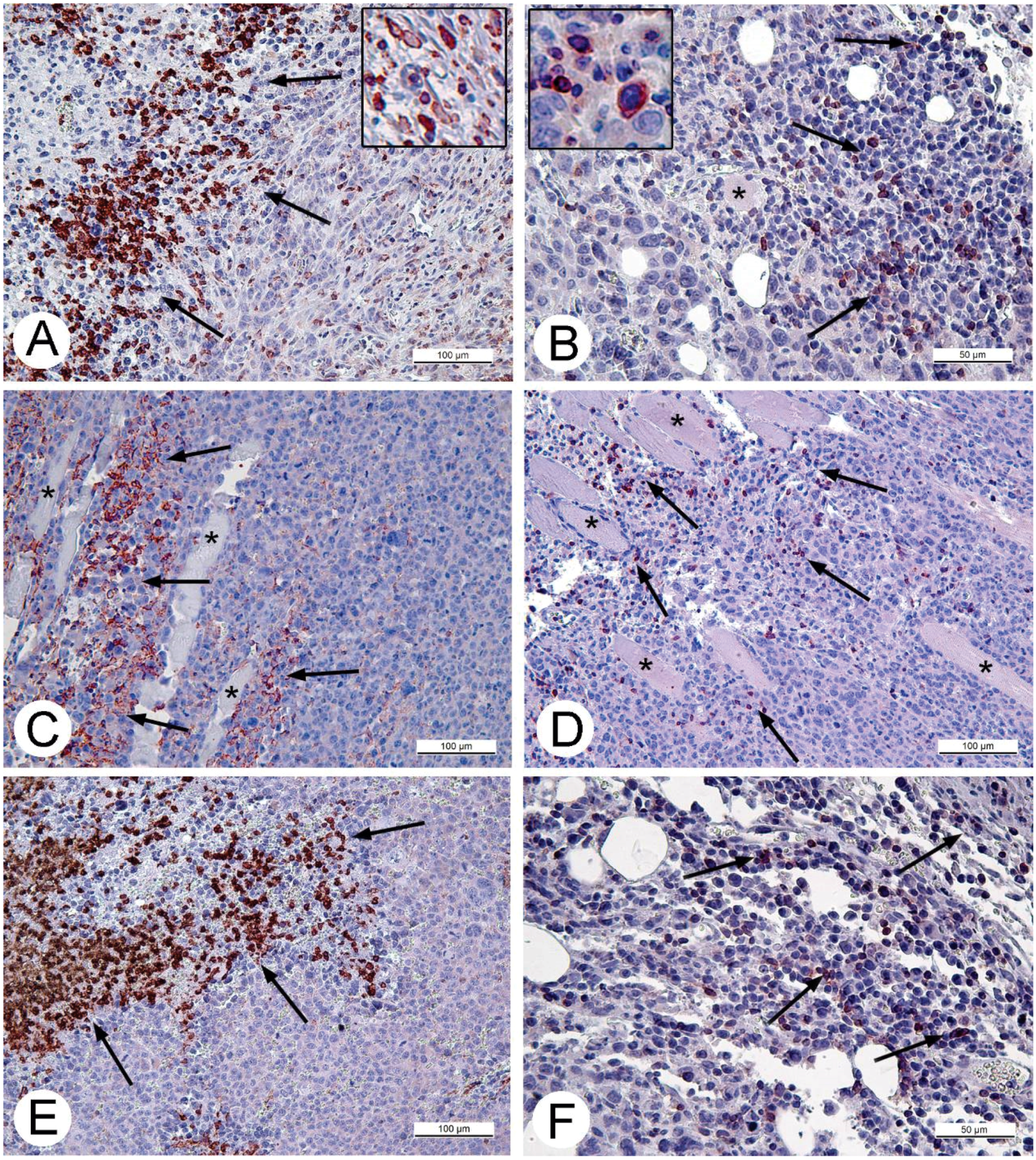
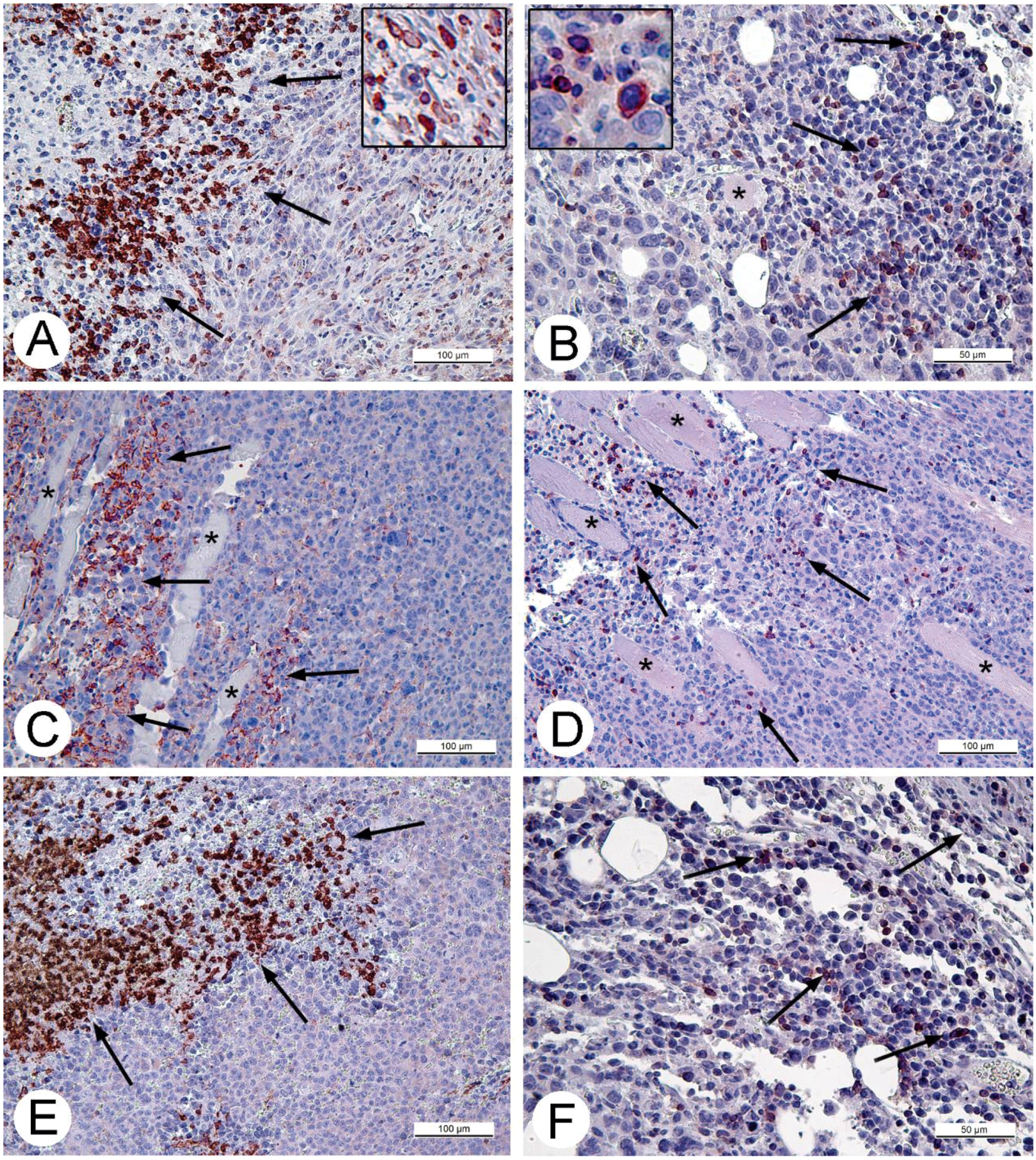
For histologic studies, specimens of tumors and ascitic cells were routinely processed using Sakura Tissue-TEK II device (Sakura Finetek, Tokyo, Japan) and embedded in Histomix. Paraffin sections were stained with hematoxylin and eosin. The sections for immunohistochemistry were mounted on polylysine-coated slides (Thermo Scientific, Waltham, MA, USA). Rabbit polyclonal antibodies (Abcam, Cambridge, UK) in concentrations 2.5 and 5 mg/mL were applied to detect proteins Apaf-1 (a marker of apoptosis) and Ki-67 (proliferation marker). The cell cycle S-phase marker, PCNA, was detected using corresponding mouse antibodies in concentration 5 mg/mL (BioLegend, San Diego, CA, USA). Rabbit polyclonal antibodies against CD3 and CD11b (Abcam) were used to detect T-lymphocytes or monocytes and granulocytes on paraffin sections of solid Ehrlich carcinoma correspondingly. The antibodies were diluted in accordance with manufacturer’s recommendations. The antigens were visualized using AEC Single Solution detection kit (Abcam) in accordance with the manufacturer's recommendations. Paraffin sections were examined using Leica DM 2500 (Leica, Wetzlar, Germany) microscope supplied with digital camera Leica DFC420 C (Leica).
For electron microscopy, fixed in 4% paraform samples of xenografts and ascitic cells were postfixed in 1% osmium tetroxide solution, then routinely processed and embedded into a mixture of epon-araldite (EMS, Hatfield, PA, USA). Semithin sections were prepared from hard blocks, stained with Azur II and examined in light microscope for selection of the areas for ultrathin sectioning and counting mitotic cells. Ultrathin sections were cut using Leica EM UC7 ultramicrotome (Leica), routinely contrasted by uranylacetate and lead citrate (SPI, West Chester, PA, USA) and examined in transmission electron microscope JEM 1400 (Jeol, Tokyo, Japan). Images were collected using the side-mounted digital camera Veleta (Olympus Corporation, Tokyo, Japan).
Tumor structures on paraffin sections were measured using Axio Vision SE64 Rel. 4.9.1 software. The number of mitotic cells was calculated at ×400 magnification in 10 randomly selected fields in peripheral zone of A431 carcinoma. In paraffin sections of Ehrlich ascitic carcinoma the amount of mitotic and immunohistochemically positive cells were counted in randomly selected fields, not less than 2000 cells were used for each calculation.
Statistical analysis was performed using STATISTICA 8.0.360.0 [
23]. The significance of differences was assessed by Mann-Whitney-Wilcoxon method (U-Mann-Whitney test).
4. Discussion
The antitumor effect of various recombinants of VACV is a subject for hundreds of studies and tens of reviews, but information about antitumor properties of initial, genetically unmodified VACV or “natural” is scarce. We became interested in the features of antitumor effect of genetically unmodified VACV when we studied the antitumor effects of apoptin-expressing recombinant toward A431 human adenocarcinoma xenografts in comparison with parental the L-IVP strain. We found that extensive replication of the L-IVP strain in tumor cells was main factor providing antitumor effect [
20]. The present study was conducted to understand better what kind of mechanisms could exploit properties of genetically unmodified VACV in the destruction of the tumors.
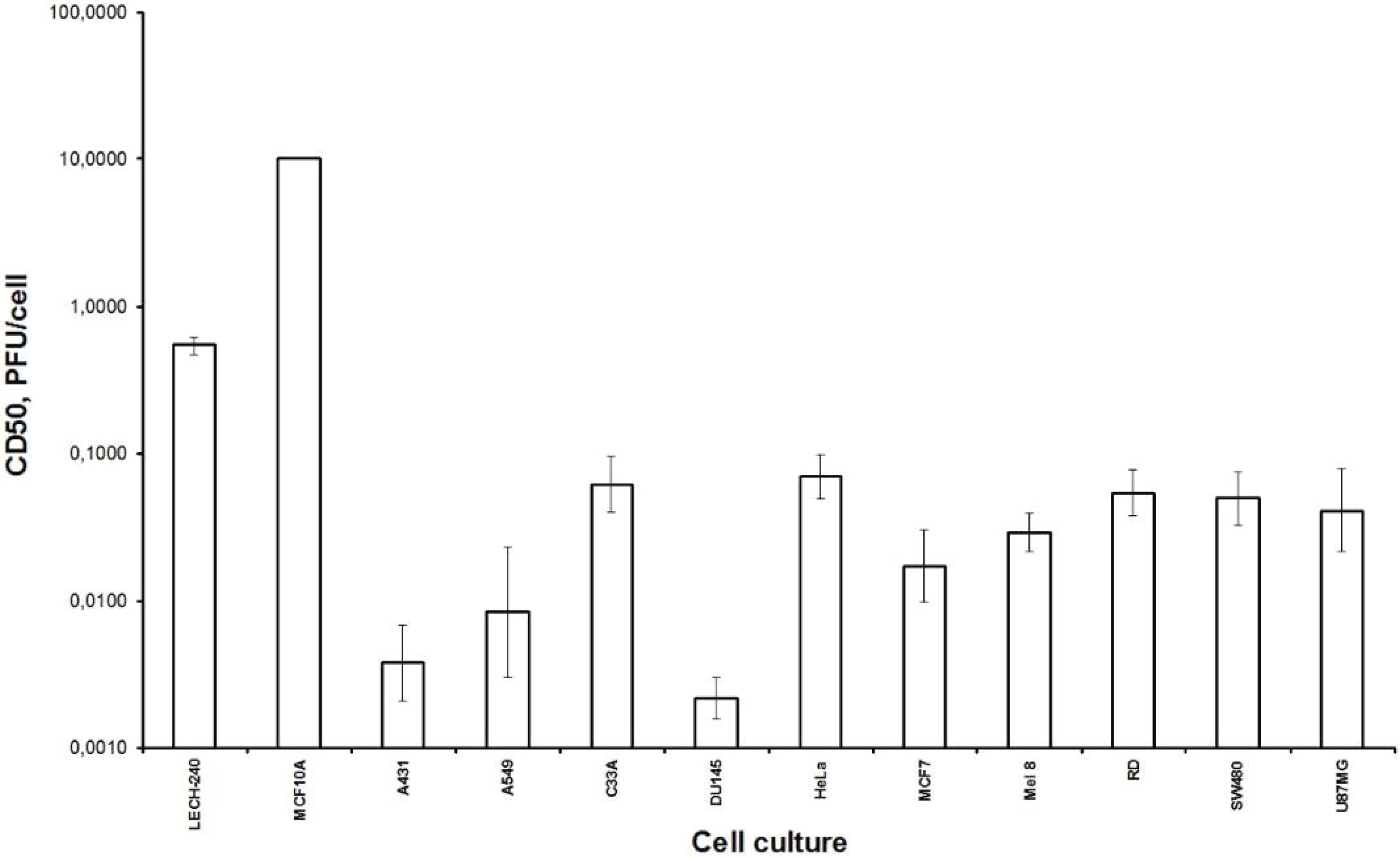
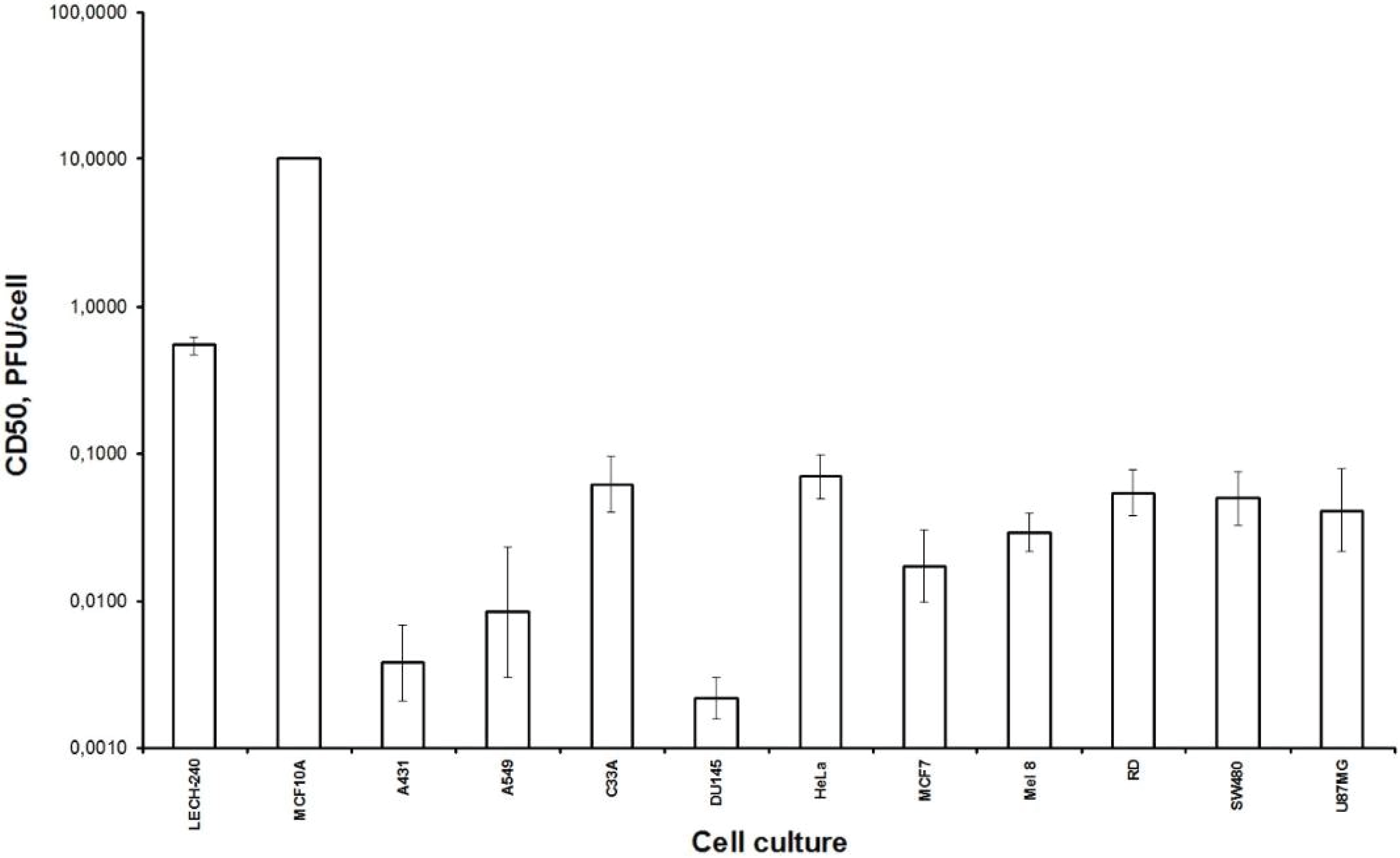
Firstly, we examined the lytic effect of the L-IVP strain replication in the cells originating from various types of human tumors in comparison with diploid human cells (
Figure 1), and we found high selectivity of the virus strain toward cancer cells
in vitro. This phenomenon was described earlier [
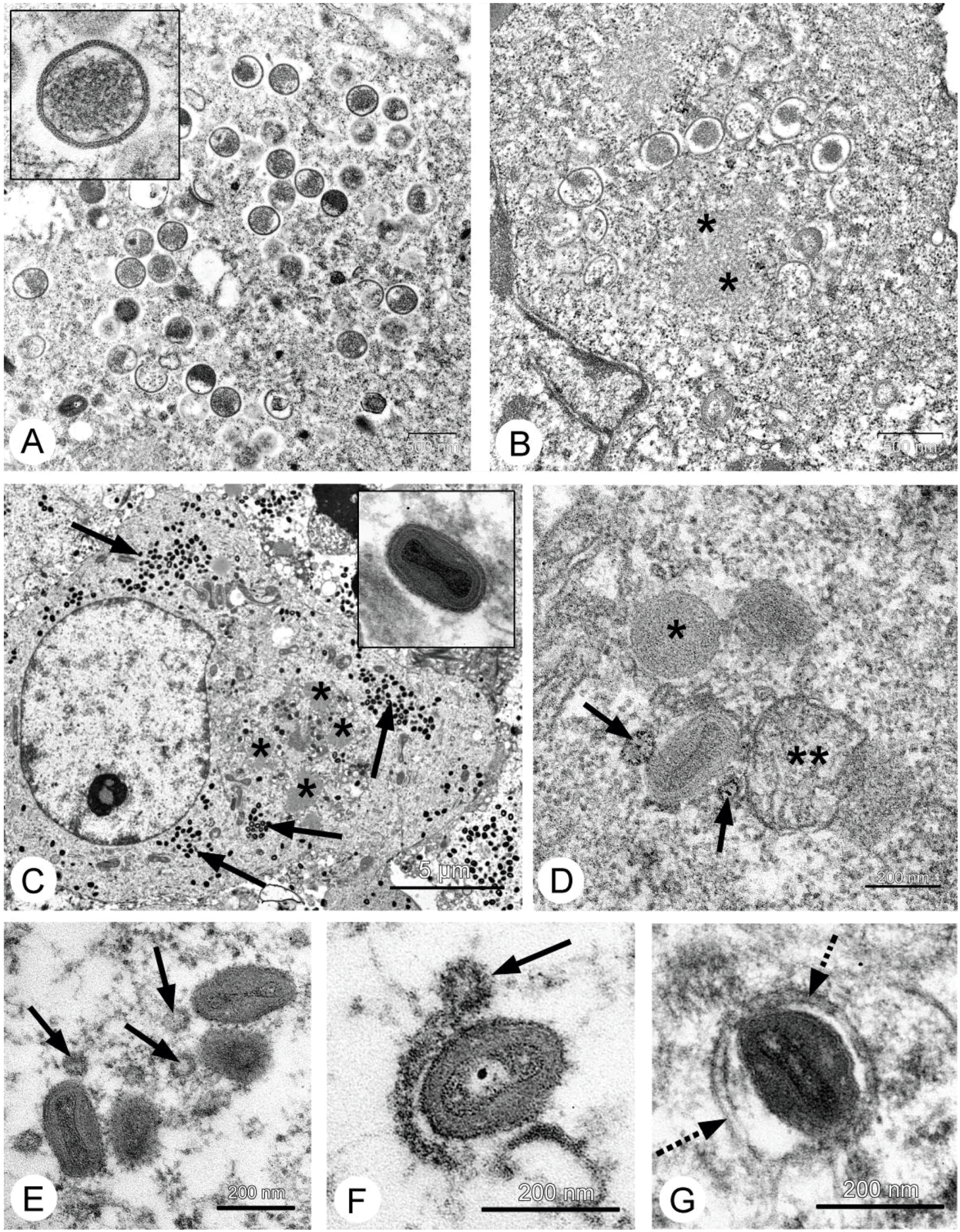
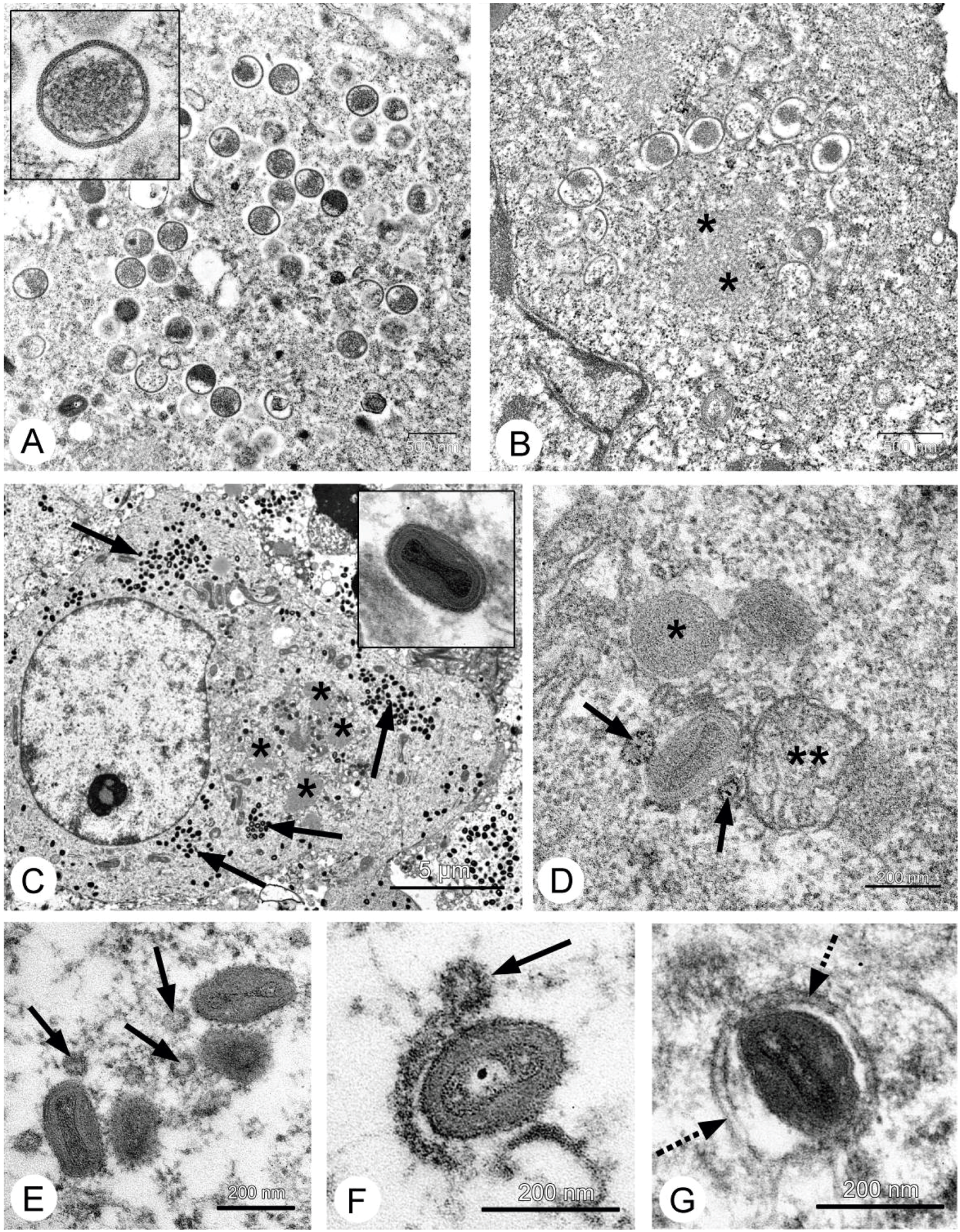
26], and our data fully confirmed this. Localization of the VACV in tumor nodes of experimental animals was shown using GFP-expressing recombinants [
13,
27,
28], and we directly showed selective infection of the tumor cells with the L-IVP strain by electron microscopy: the virus did not replicate in cells of blood vessels and connective tissue [
20].
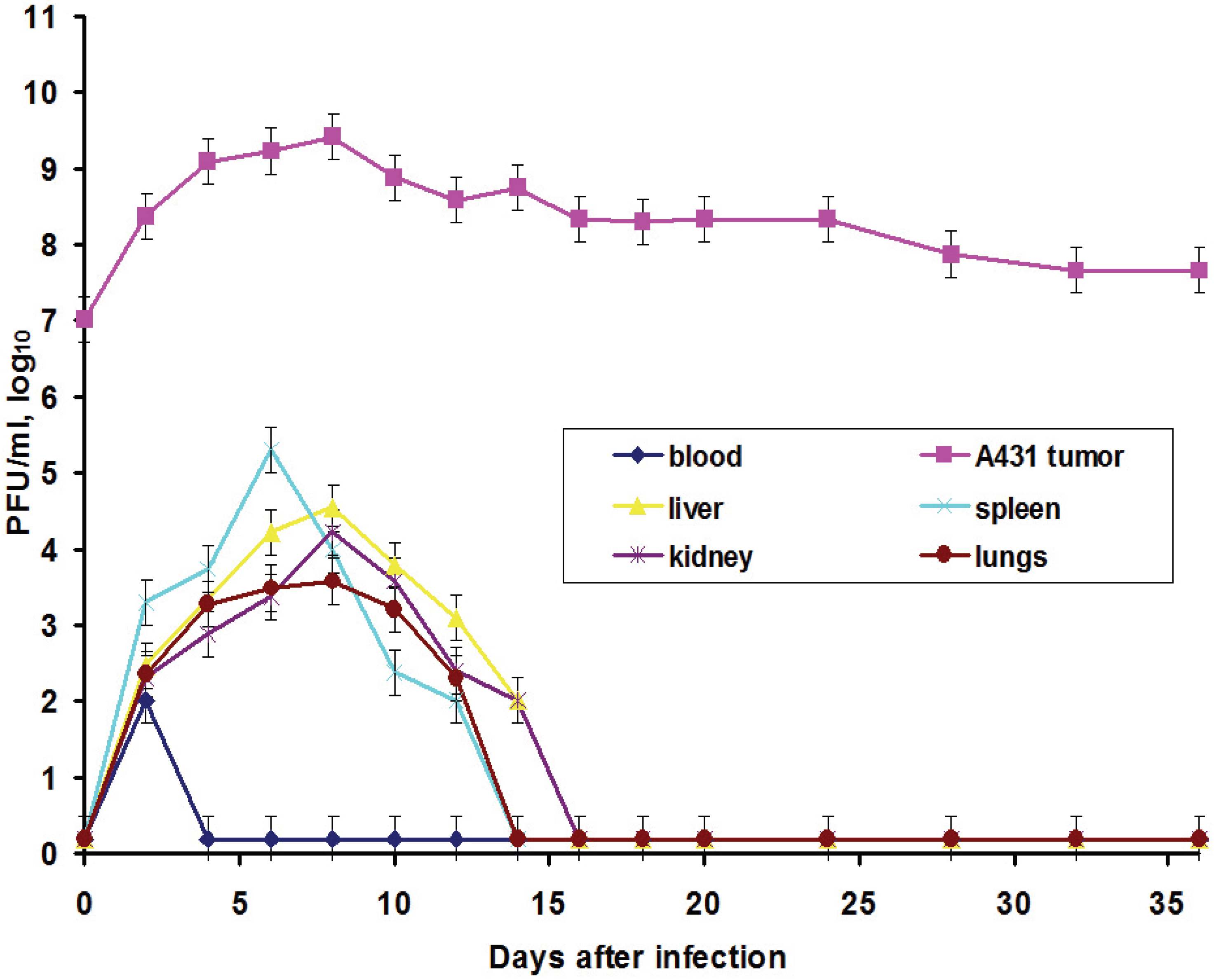
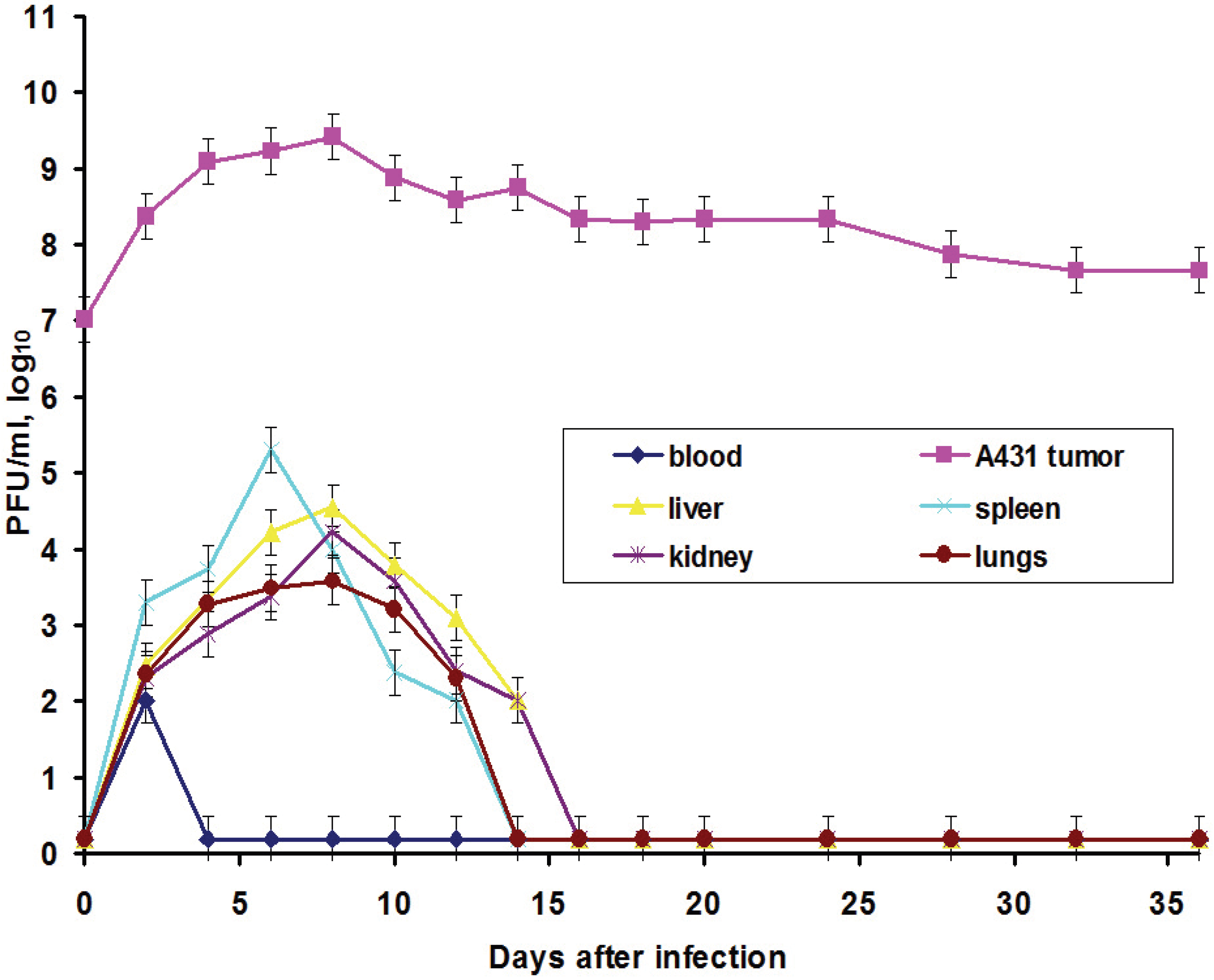
In this work, we also elucidated another important characteristic of the VACV with respect to tumors: the ability of the virus to spread in organism and to be present in visceral organs. Titration of the L-IVP virus using the PFU method revealed the presence of the infectious (living) virus in liver, spleen, lungs and kidneys for two weeks, while the virus rapidly disappeared from blood (
Figure 3).
We suppose that virus entered the blood flow in the moment of injection, because the data do not give support or describe the migration of the virus outside the tumor. The titers in the visceral organs increase for some time and their values are higher than maximal value in blood, evidencing for the replication of the L-IVP strain in cells of the examined organs. Release of viral progeny from infected cells and infection of distant cells was considered as advantageous feature for the realization of the VACV oncolytic effect [
7,
18,
29]. Replication of the L-IVP strain in mouse visceral organs illustrates ability of virus to reach distant targets and demonstrates the possibility of the virus to infect tumor cells distant from primary virus injection site, for example in metastases.
The main goal of this work was to study the VACV antitumor effect in mice with non-compromised immune system using a murine tumor, because the human carcinoma A431 xenografts in
nude mice represents an artificial experimental system. We used murine Ehrlich carcinoma, which can grow in solid and ascitic forms in different lines of mice [
30,
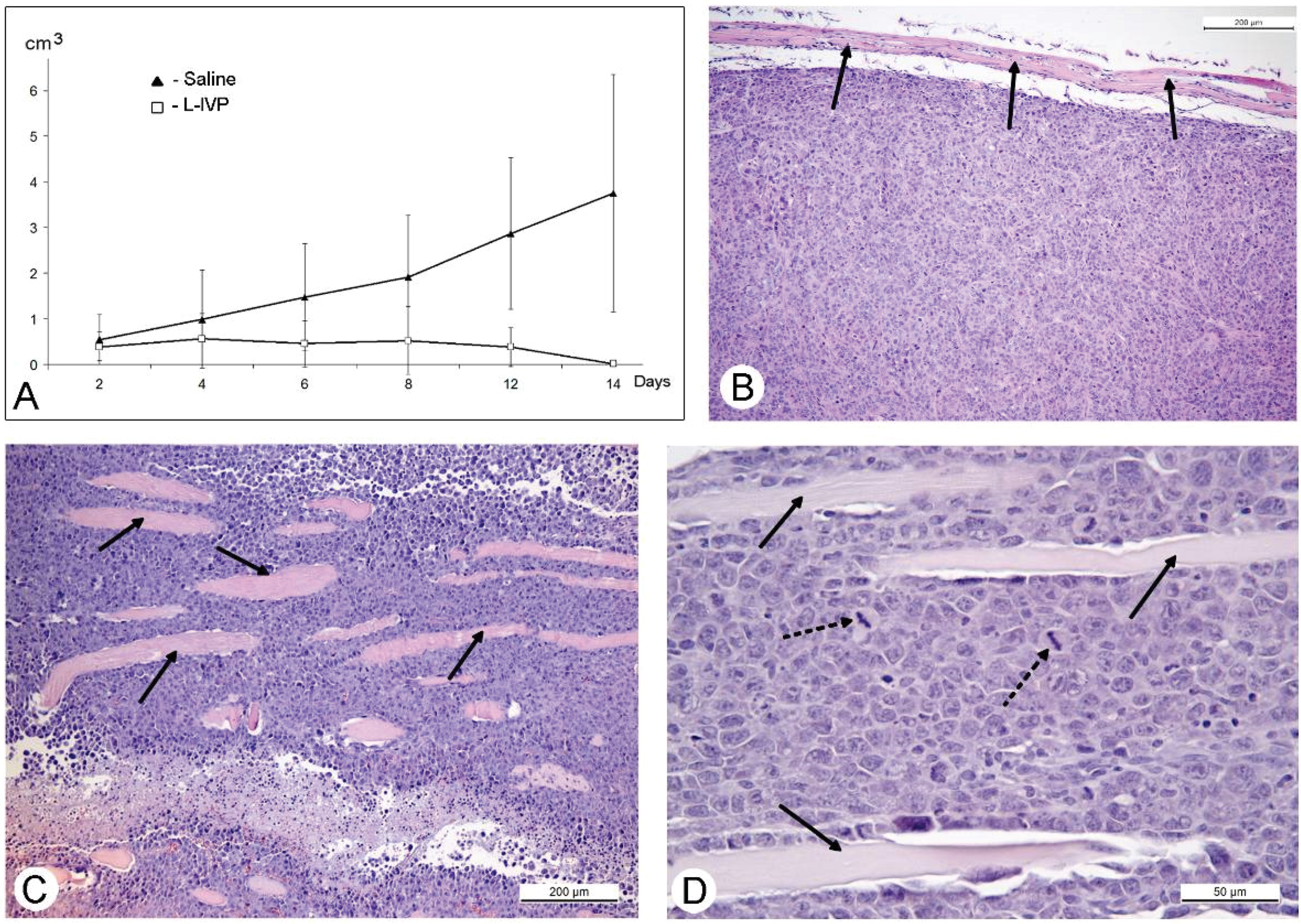
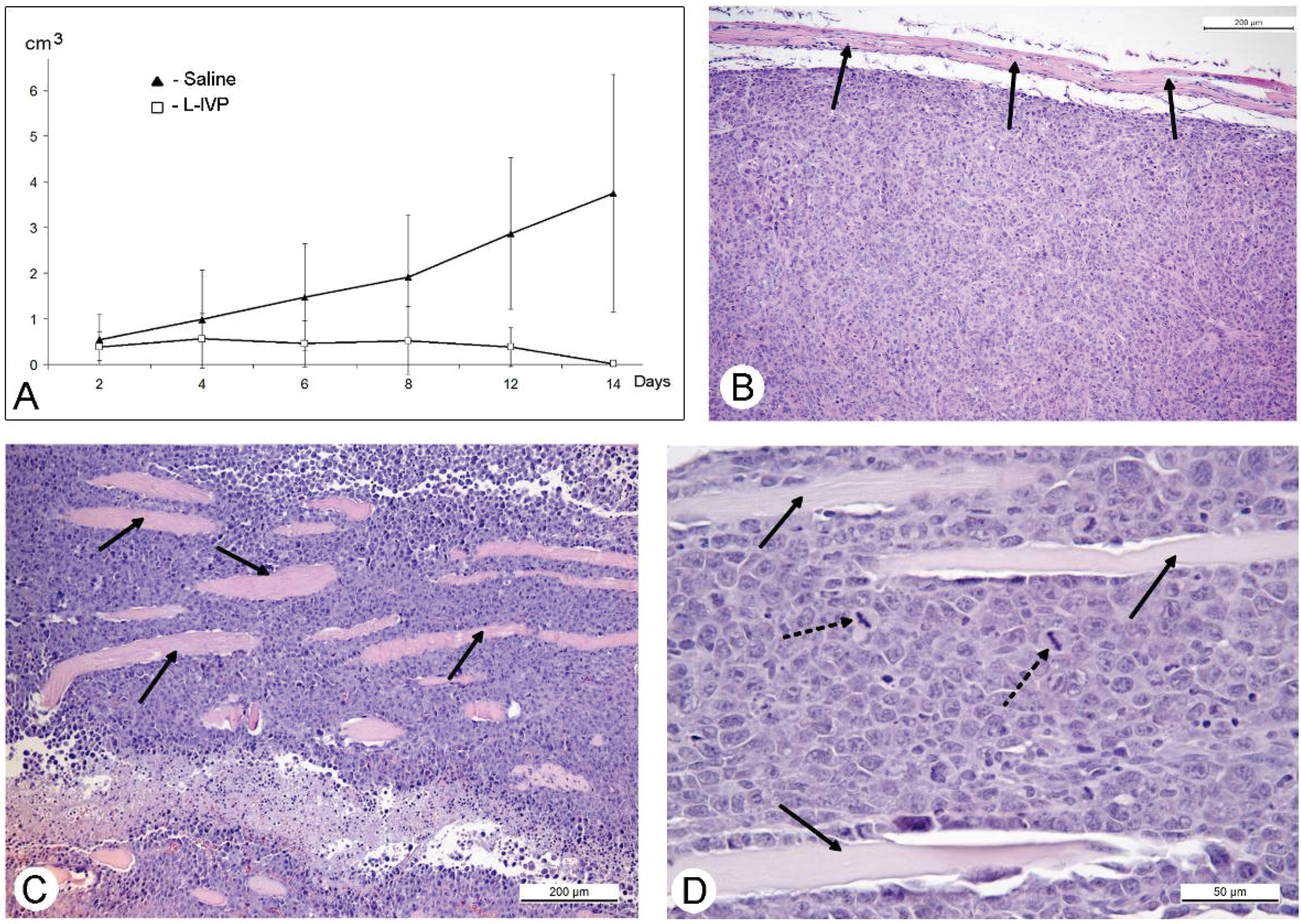
31]. The protocol of this study was similar to those of A431 carcinoma, and similar to those, we observed a decrease in volume of the solid tumor in comparison to saline-treated control (
Figure 4). However, in contrast to A431 carcinoma, we failed to find signs of virus replication in the tumor using electron microscopy, although we examined numerous sections in all virus-injected animals. This could be explained by a small amount of infected cells, which is below the detection capabilities of the method. Examination of paraffin sections of Ehrlich solid carcinoma revealed no signs of selective destruction and apoptosis in VACV-injected tumors compared with saline-injected during the whole period of observations.
Solid tumors, growing in living organism, reach the stage when their supply with oxygen and nutrients does not keep pace with the growth of a tumor, leading to formation of necroses. This process promotes entry of tumor tissue antigens in blood and development of immune response to tumor [
32]. Obviously, CD11b- and CD3-positive cells in solid Ehrlich carcinoma found after 9 days post tumor transplantation (2 days after injection of saline), represent development of mouse immune response to the carcinoma. However, the differences in localization and numbers of CD11b- and CD3-positive cells between saline- and VACV-injected mice were not detected within 14 days of the experiment. We suppose that the duration of the experiment (14 days, time of the death of mice in control group) was too short for the development of detectable signs of cell immune response to the VACV in the Ehrlich carcinoma. The immune response to oncolytic viruses is a complicated event, unfolding at the background of tumor evading the immune system using various mechanisms including damage of antigen presentation and induction of immune tolerance [
33,
34]. In addition, the environment in the tissue of a tumor is immunosuppressive [
35]. All these circumstances could contribute to a delay in the development of virus-induced cell immune response in the Ehrlich carcinoma.
Therefore, we could not understand how the L-IVP strain causes the decrease in volume of Ehrlich solid carcinoma. We proposed that the virus replicates in small number of tumor cells and releases some substances altering the tumor growth. To test this, we examined the effect of L-IVP strain on ascitic form of Ehrlich carcinoma, which allows one to obtain homogeneous preparations of tumor cells. As we expected, replication of the L-IVP strain in carcinoma cells was weak (the titers did not exceed 105 PFU/mL), and electron microscopy revealed few infected cells.
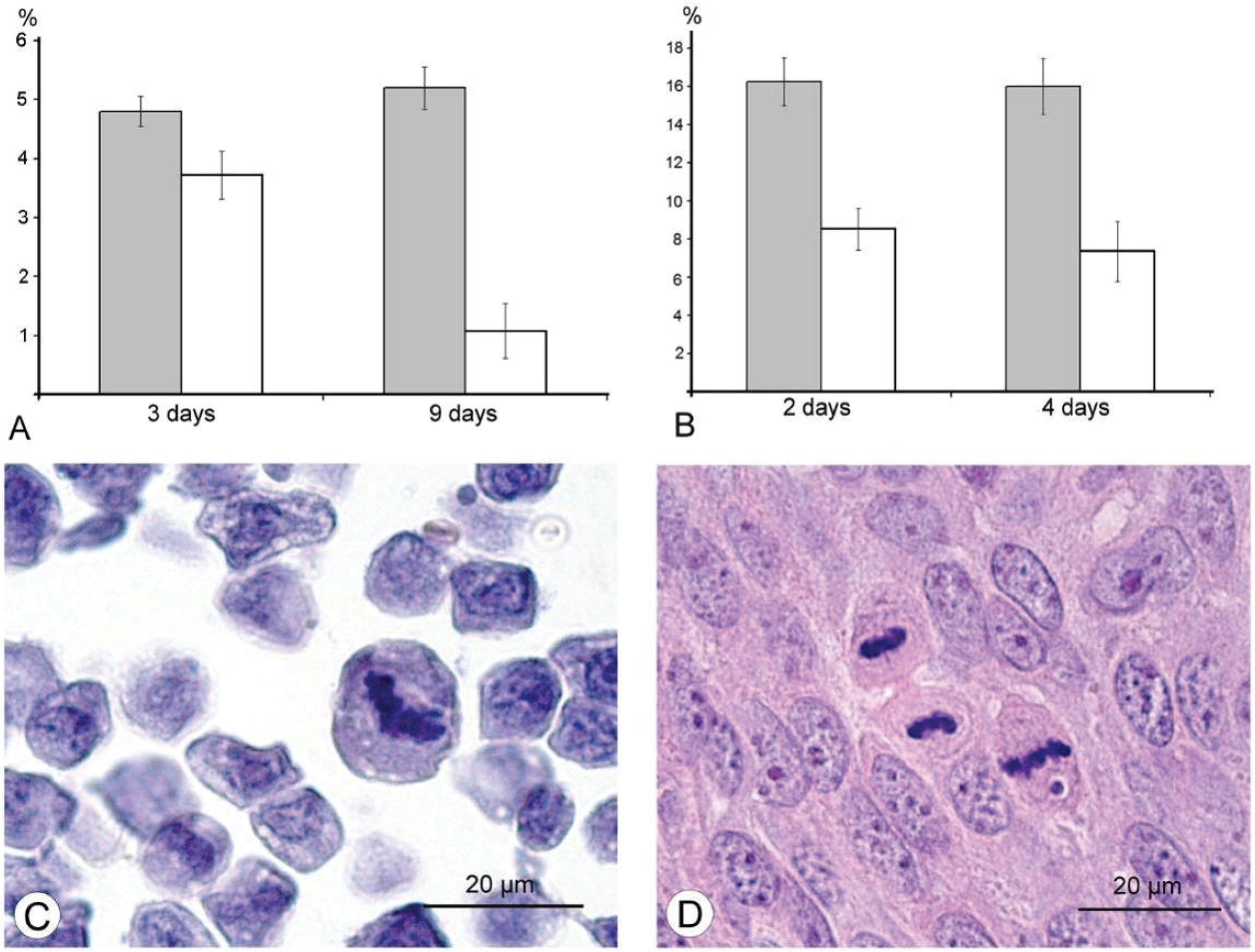
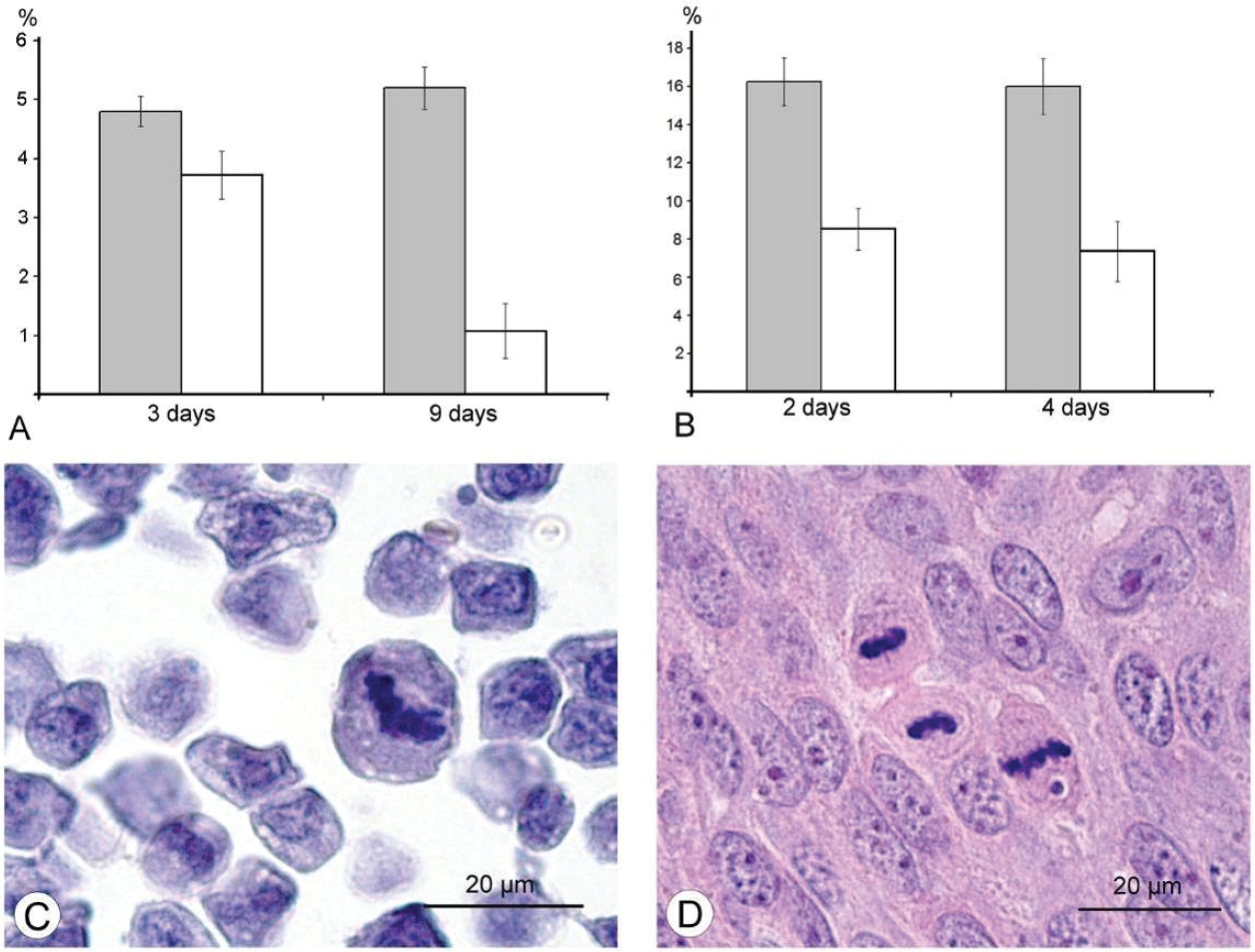
The results of our study excluded direct destruction of tumor cells by VACV replication and immune response as possible factors for the decreasing sizes of Ehrlich carcinoma. The ability of the VACV to reduce amount of mitotic cells in a tumor was noted more than 60 years ago [
36], and we supposed that this could play a role in decrease of tumor size after injection of VACV in our experiments. Indeed, a comparison of number of mitoses counted in the L-IVP- and saline-treated carcinomas revealed that the decrease occurs after treatment with the virus (
Figure 6). The same effect of the L-IVP strain was observed in A431 carcinoma xenografts on days 2 and 4 after the L-IVP strain injection, however their rapid destruction by VACV do not permit to observe this effect longer than 4 days (
Figure 6A).
It would have been interesting to follow this antimitotic effect of VACV on the same tumor cells
in vitro, however, Ehrlich carcinoma is unable grow
in vitro. The cells of A431 carcinoma are highly sensitive to VACV and are destroyed too quickly to observe this effect. The effect of VACV on mitoses of tumor cells was observed about 50 years ago; authors used different viral strains and cell cultures, and various experimental designs and methods, these showed that antimitotic effect of the virus could present in both tumor and non-tumor cells [
37,
38,
39,
40]. Recent studies of VACV influence on cell cycle are devoted to molecular mechanisms of VACV interaction with a cell, and showed that the virus shifts the cell to S-phase and thereby provides more efficient viral replication. This phenomenon was shown to be related with hyperphosphorylation of p53 by viral early B1R kinase, leading to p53 downregulation, which, in turn, could promote synthesis of DNA [
40,
41].
Na-Kyung Yoo and co-workers, 2008 [
42] examined the shifting of VACV infected cells to S-phase using recombinant vTF7-3 VACV and rapidly proliferating human osteosarcoma 143B and HeLa cells. The results showed that VACV possesses a unique cell-cycle control mechanism, involving inactivation of p53 and Rb, which are associated with the RNA polymerase III transcription factor B (TFIIIB) subunits, TBP and Brf1, respectively. All the examined events unfold inside infected cells
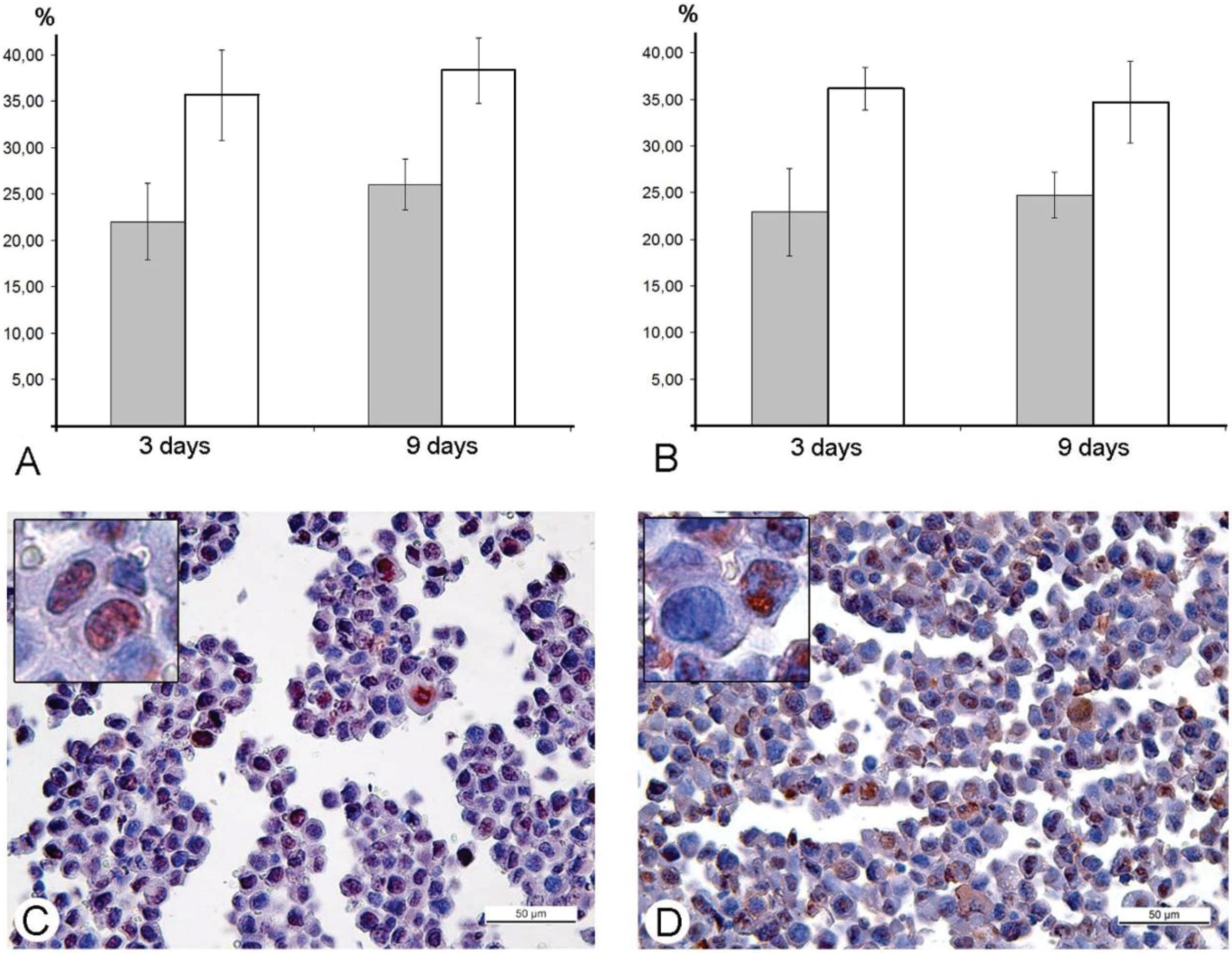
in vitro. Our study was performed in animal models, and immunohistochemical reaction for the PCNA-antigen (marker of S-phase) showed that the amount of the cells in the S-phase is incomparably larger, than the amount of infected cells in Ehrlich ascitic carcinoma. One of the possible explanations of damage of cell cycle is blockage of VACV replication in the early stages, when viral early B1R kinase is expressed, but visual signs of the virus replication are not seen. In such case, we will not see infected cells, and viral yield will be low, just as we observed. It is interesting that the ability of the virus to promote entry of infected cells into S-phase is considered as advantageous mechanism, providing more efficient replication of VACV and many other DNA- and RNA-viruses [
43]. Our study showed another facet of this ability—the stopping of mitotic division of tumor cells resulting in delays of tumor growth. Usage of mouse Ehrlich carcinoma in mice demonstrated this effect clearly, but pointed new questions about its mechanisms. One of these questions is what is possible influence of immune system mediators induced by VACV on mitotic division of tumor cells. However, decrease in amount of mitoses was evident from day 2, while specific immune response to a virus develops later.
Our study showed that VACV achieves its antitumor abilities in both A431 and Ehrlich carcinomas; however, this antitumor activity is achieved in different ways, presumably depending on the tumor origination. Humans are a natural host for VACV, and cells of human A431 carcinoma support active replication of the virus, which rapidly destroys tumor cells. In this case, the influence of the VACV infection on cell cycle recedes into the background, and is overwhelmed by the complete destruction of the tumor. A mouse is not naturally susceptible to VACV, so it was difficult to expect high destructive action of the virus on the tumor, and our research showed this. Despite of poor replication, VACV demonstrated an antitumor effect, and our study revealed that this effect is associated with accumulation of tumor cells in S-phase, which reduces number of dividing cells and, correspondingly, delays tumor growth. The susceptibility of the tumor cells to the virus determines predominance of first or second mechanism. The VACV is “a perfect parasite” utilizing all cellular mechanisms for successful survival, and a clear understanding of how the virus can alter undesired cells could be useful for development of effective anticancer therapeutics.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}