
Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry



 and
and
Abstract
:
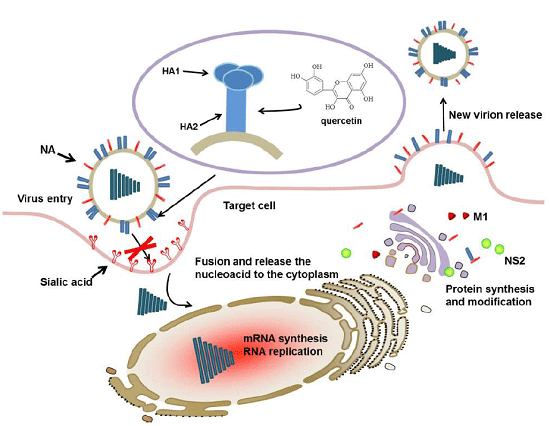
1. Introduction
2. Result
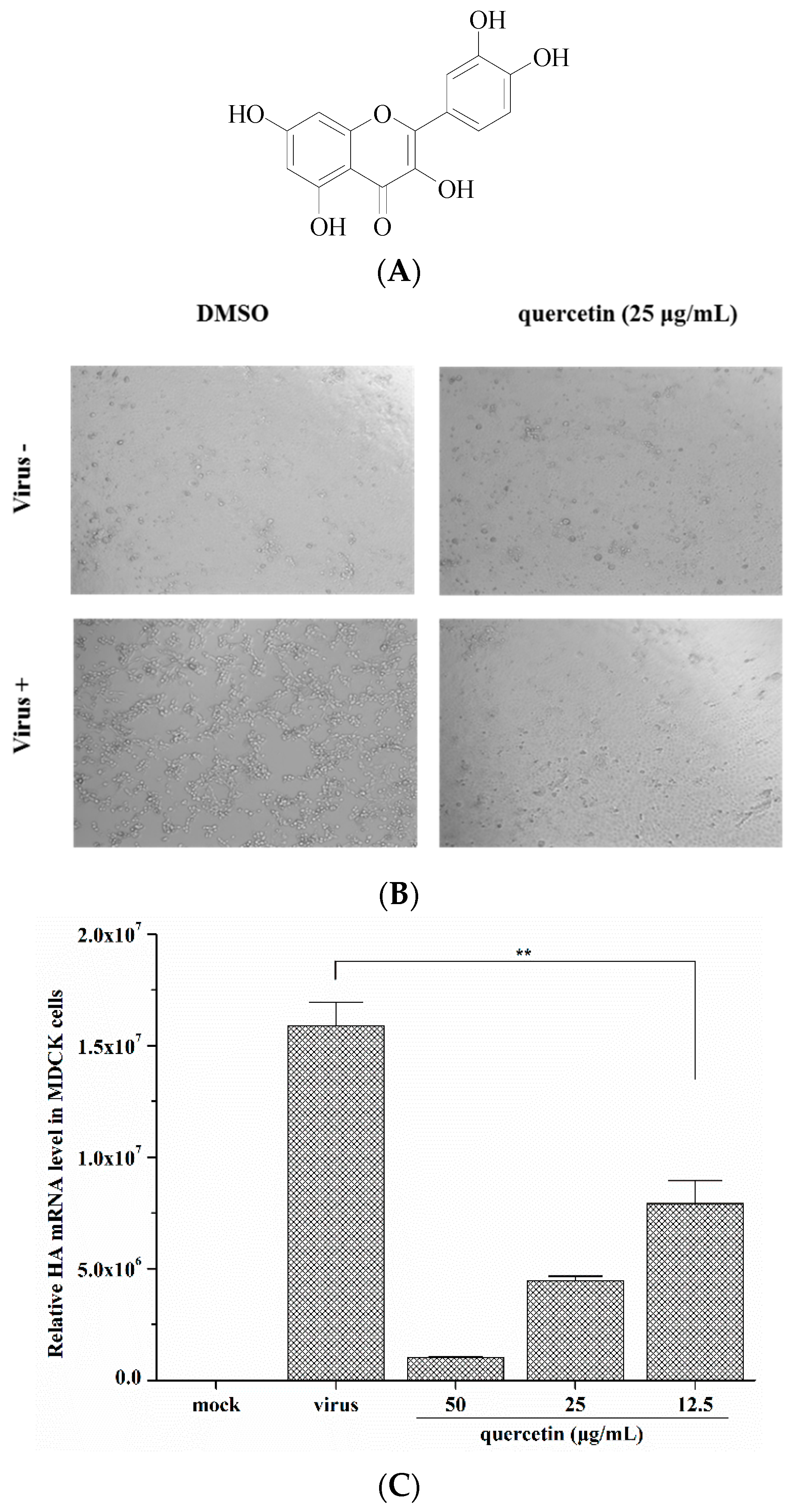
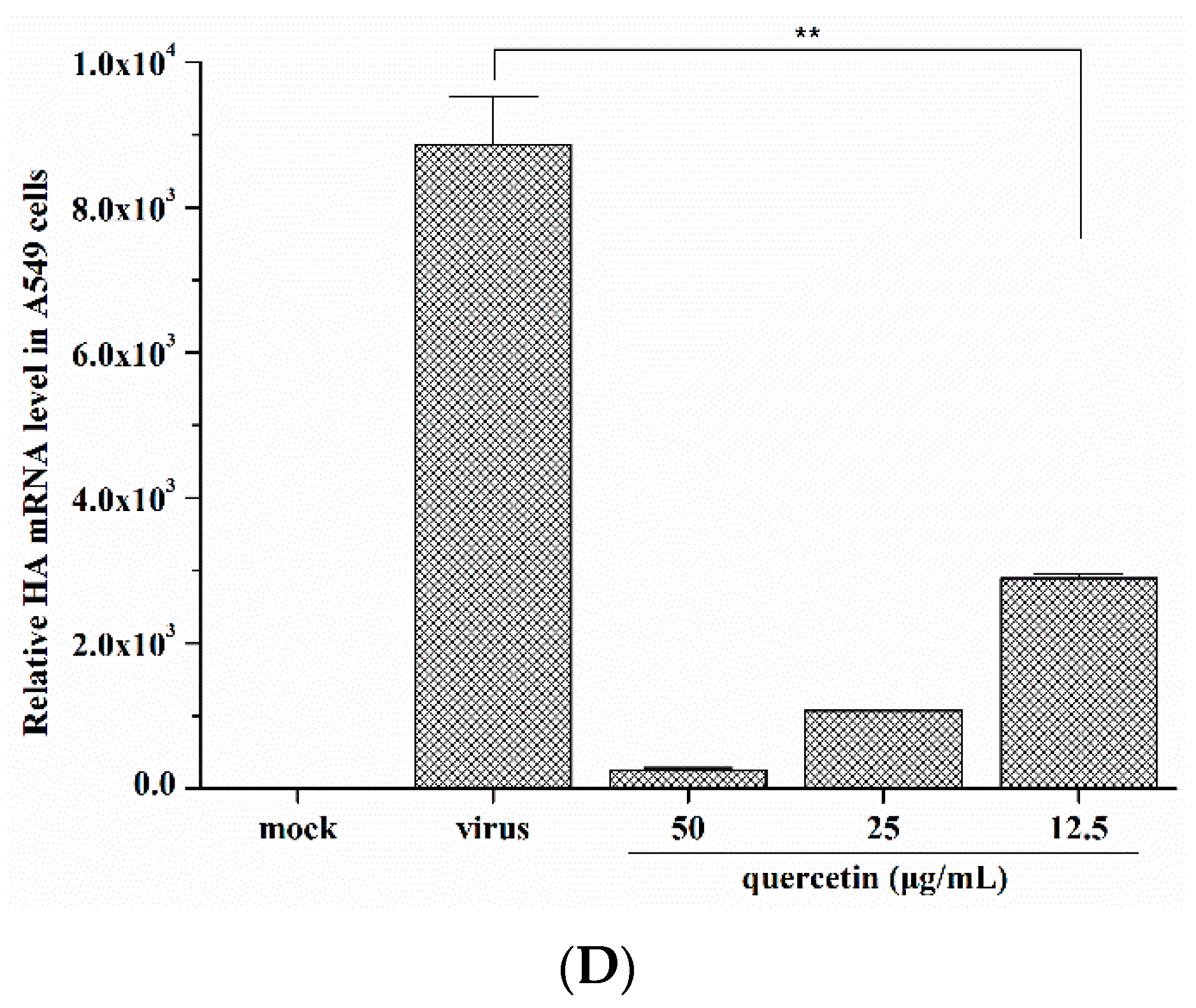
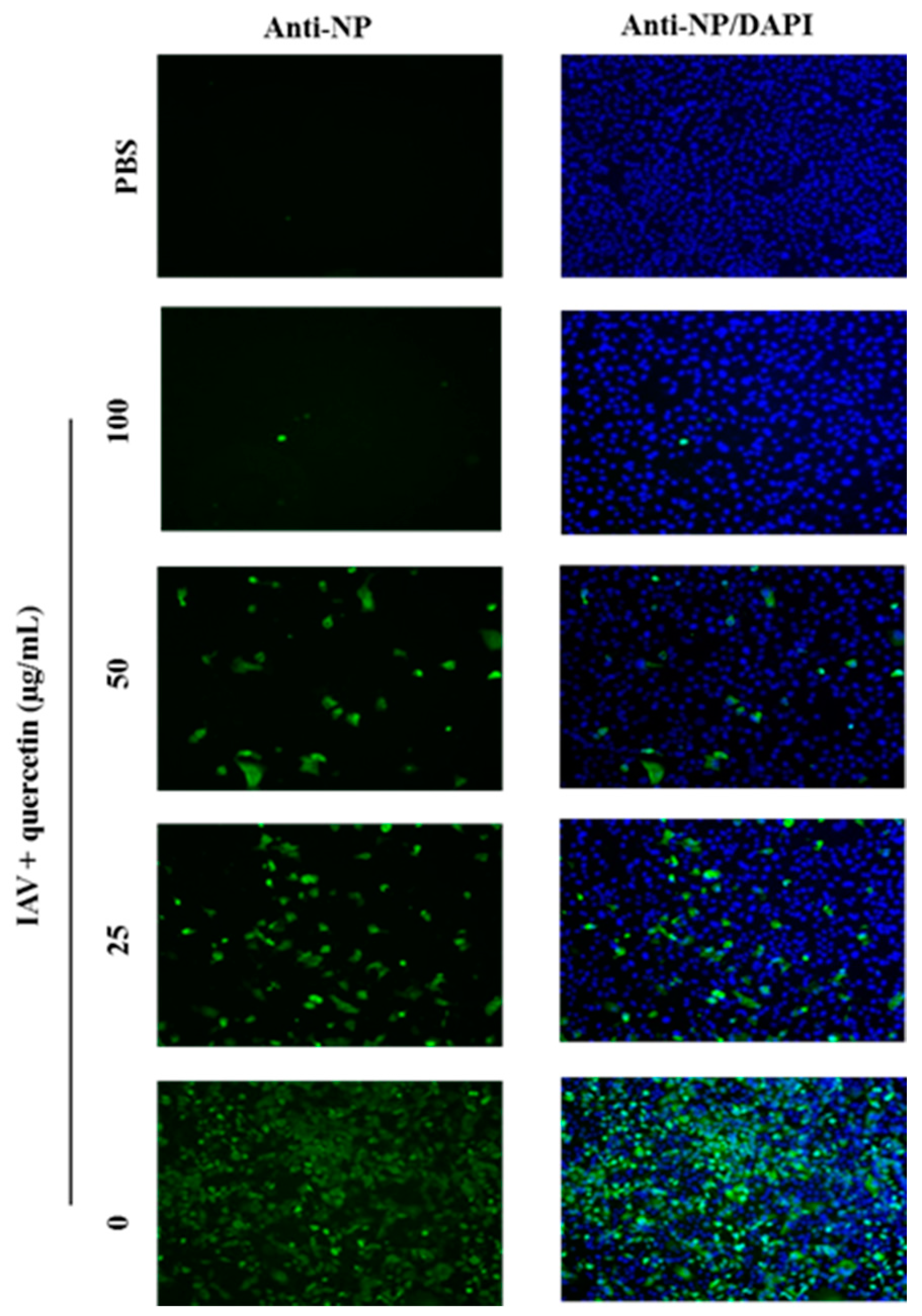
2.1. Quercetin Inhibited Influenza A Virus Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Influenza A Virus Strains | Inhibitory Activity (Mean ± S.D.) a | |
|---|---|---|
| Quercetin | ||
| IC50 (µg/mL) | IC90 (µg/mL) | |
| A/Puerto Rico/8/34 (H1N1) | 7.76 ± 1.10 | 24.58 ± 6.71 |
| A/FM-1/47/1 (H1N1) | 6.23 ± 0.47 | 20.47 ± 3.97 |
| A/Aichi/2/68 (H3N2) | 2.74 ± 1.93 | 8.24 ± 2.84 |
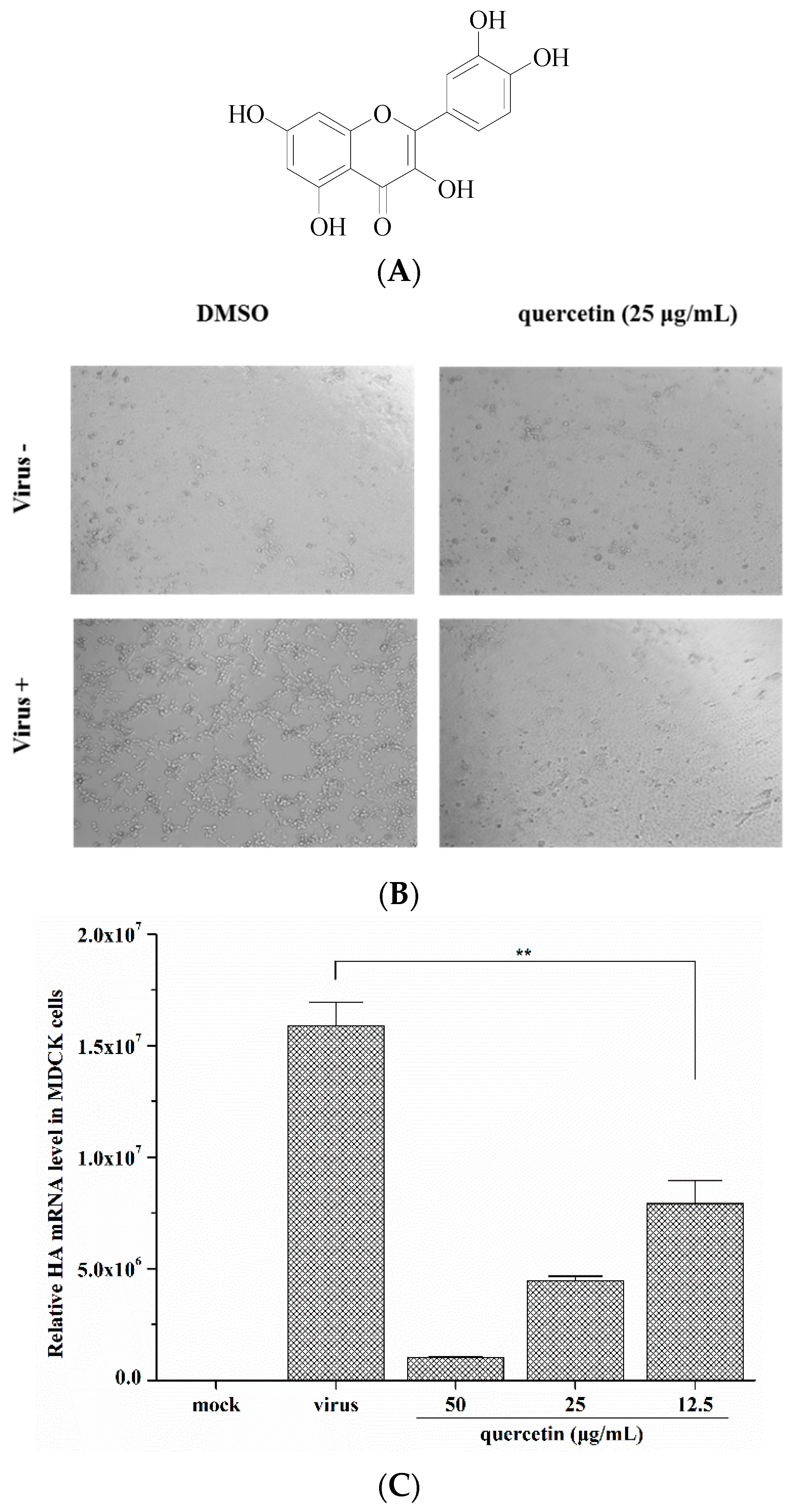
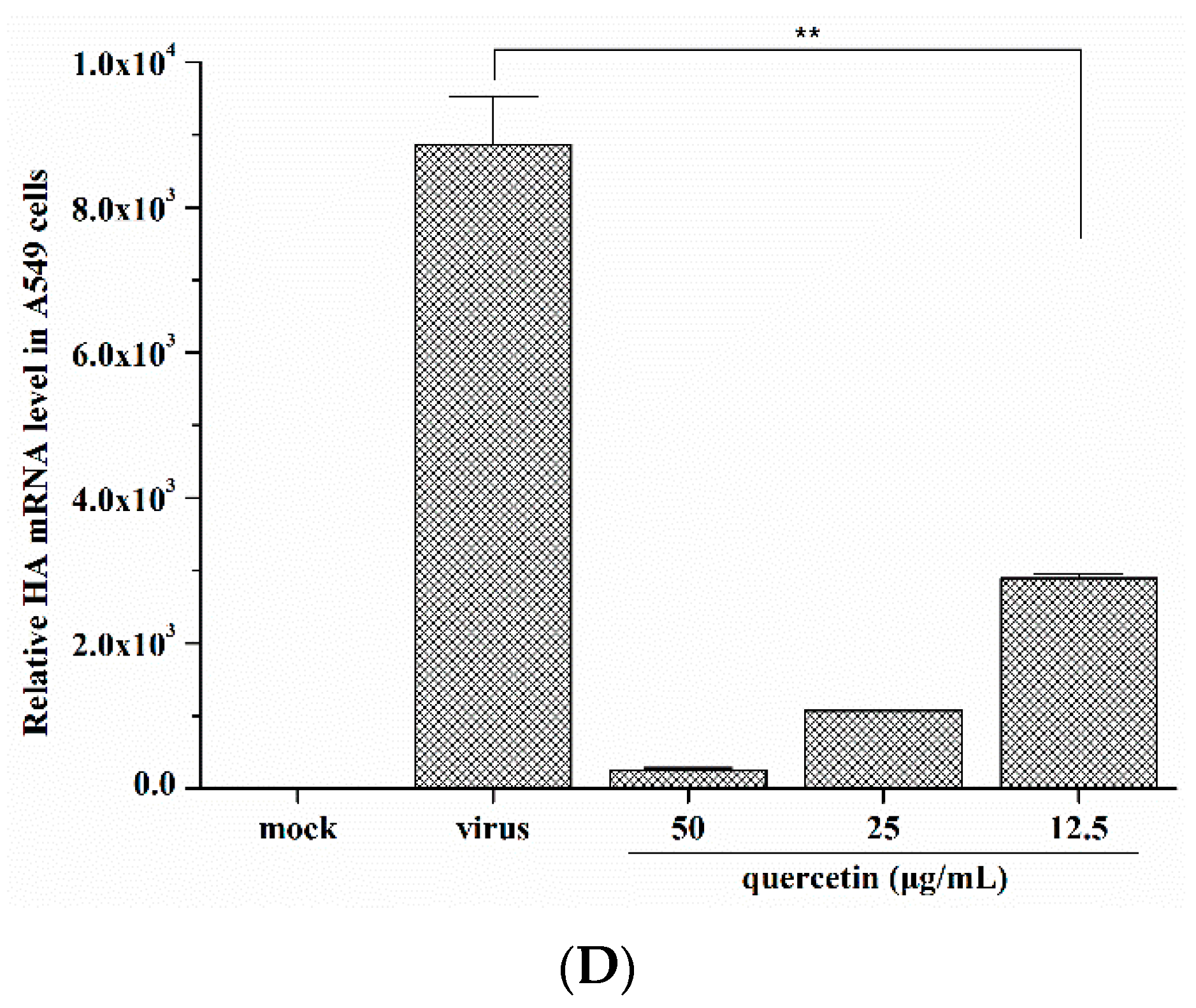
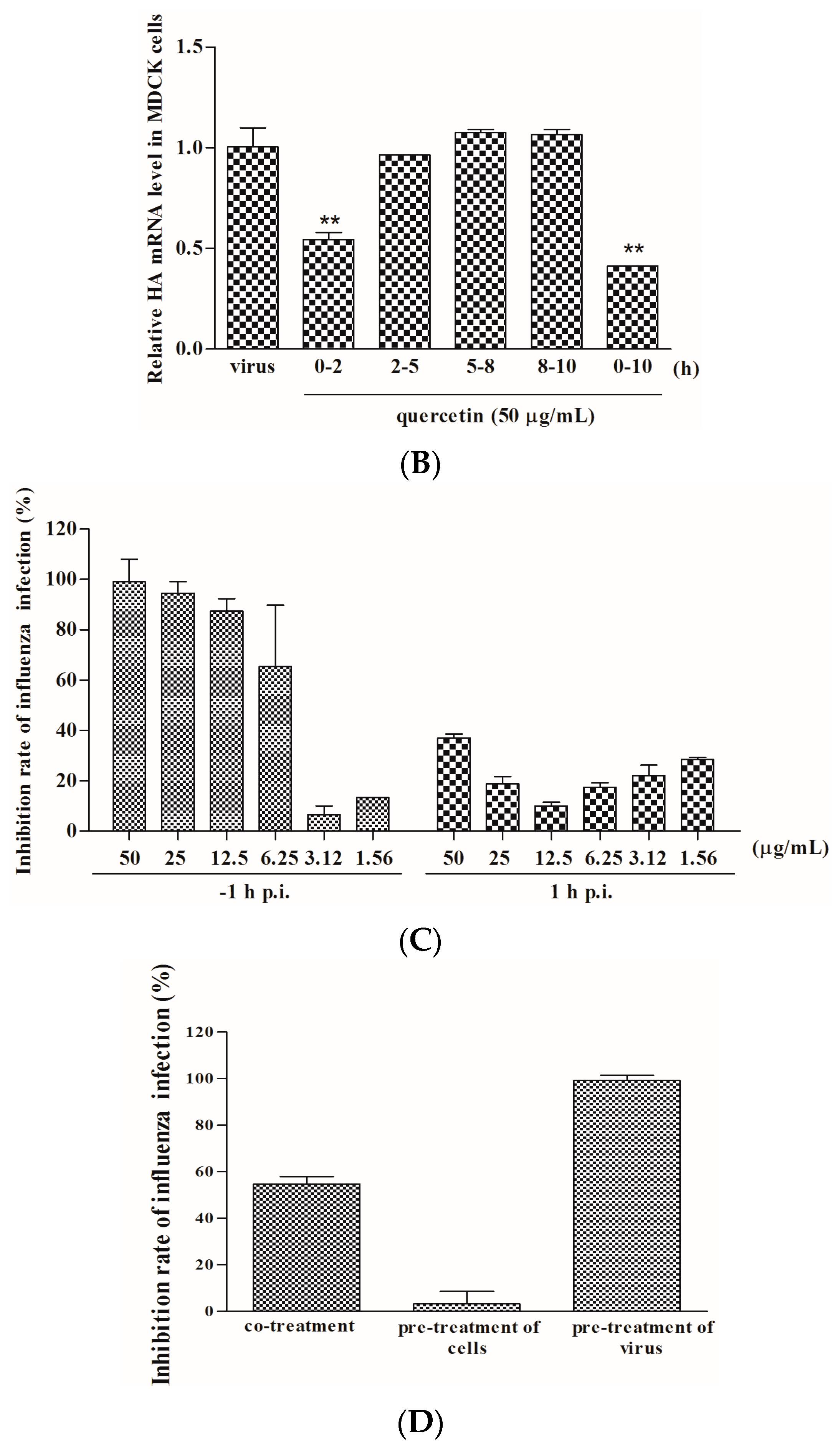
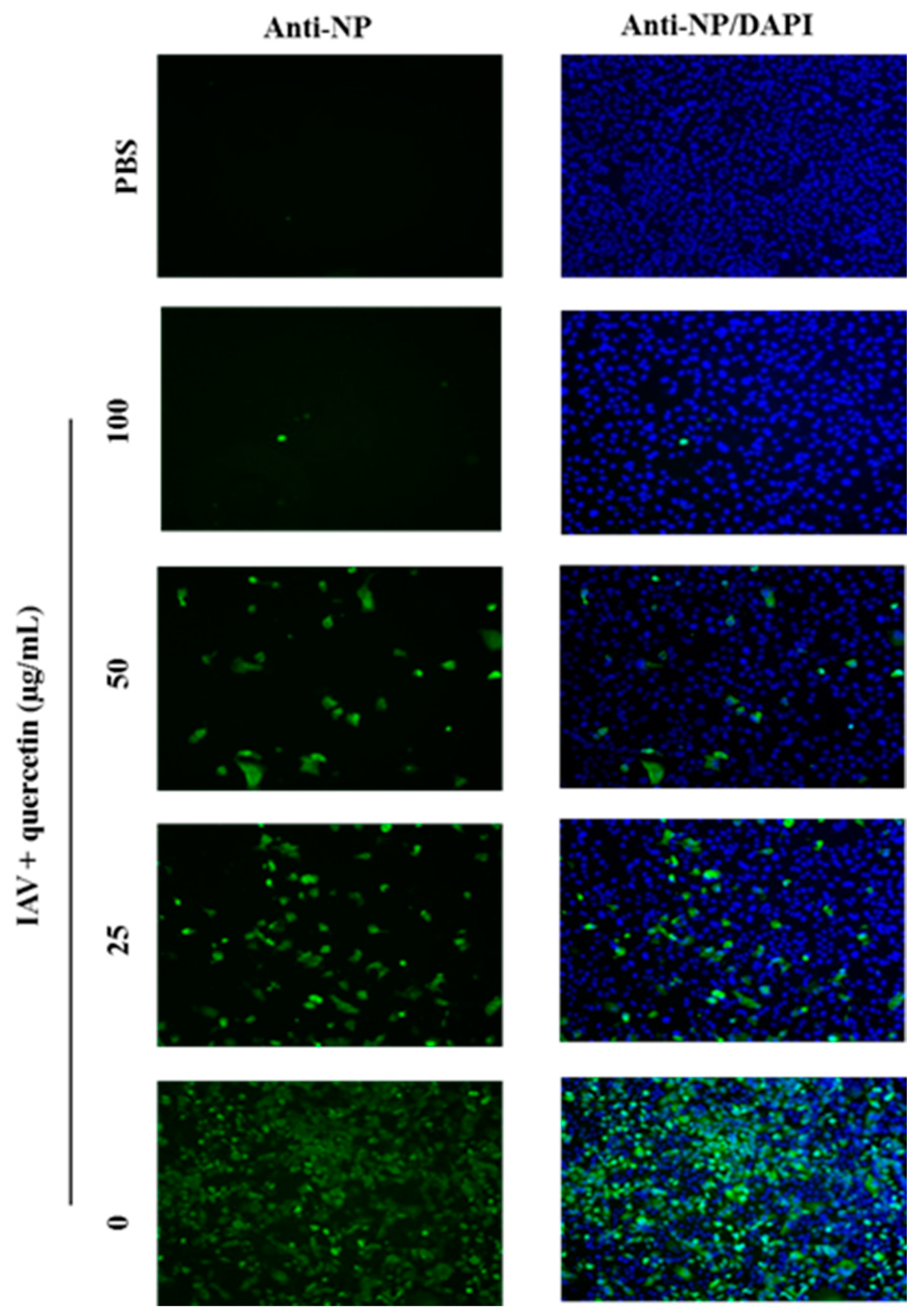
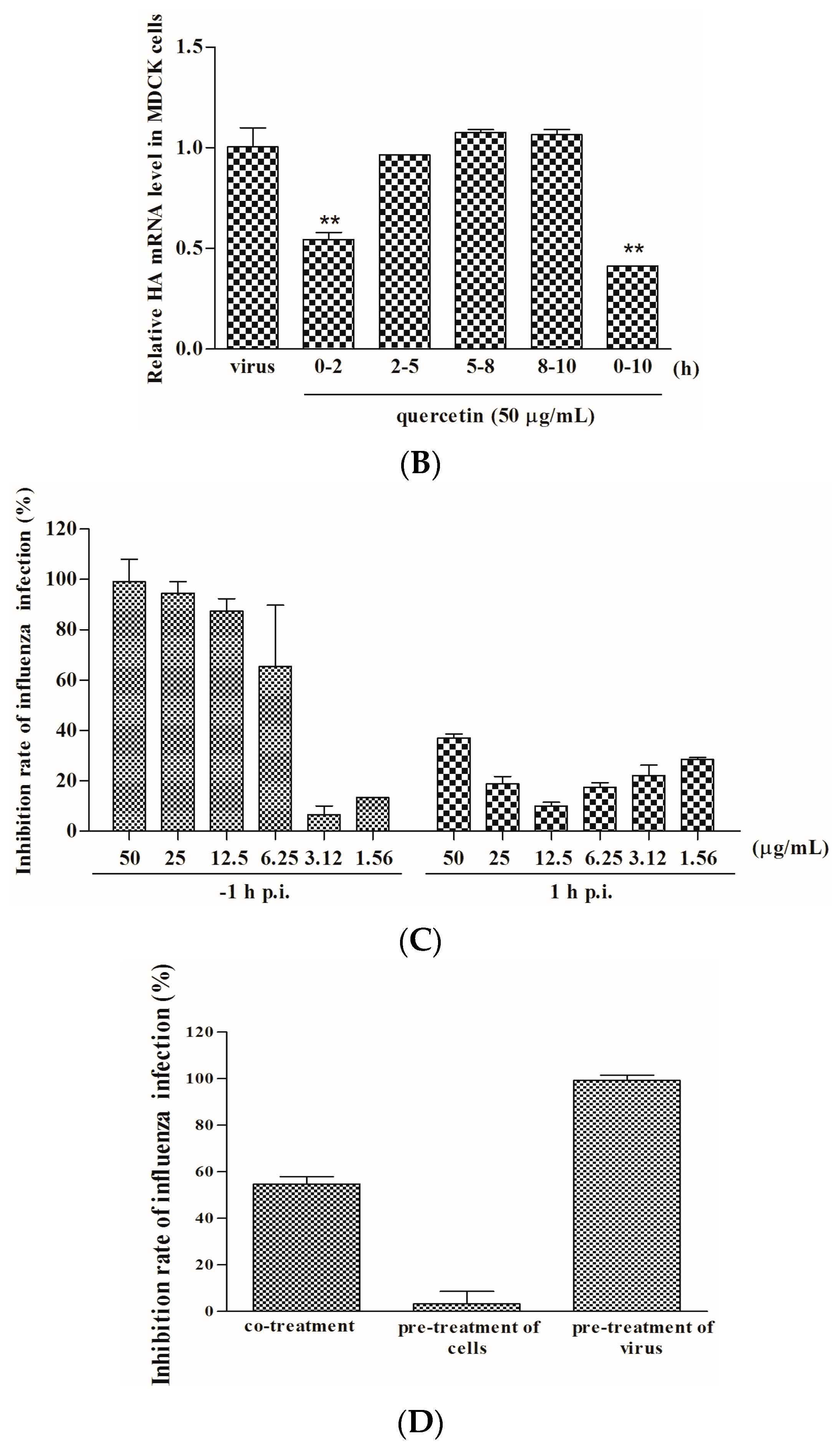
2.2. Quercetin Performed the Inhibitory Activity in the Initial Stage of Influenza Virus Infection

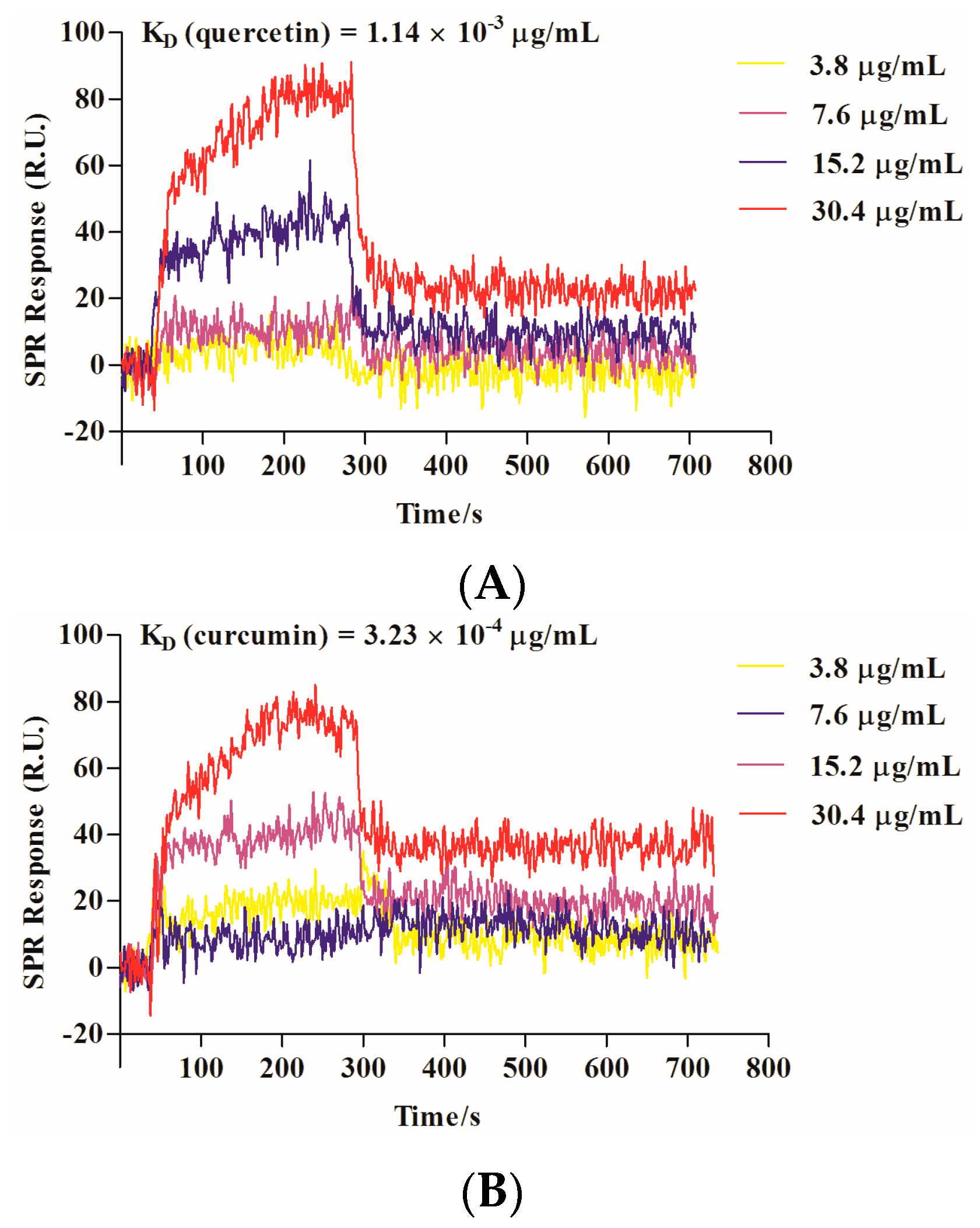
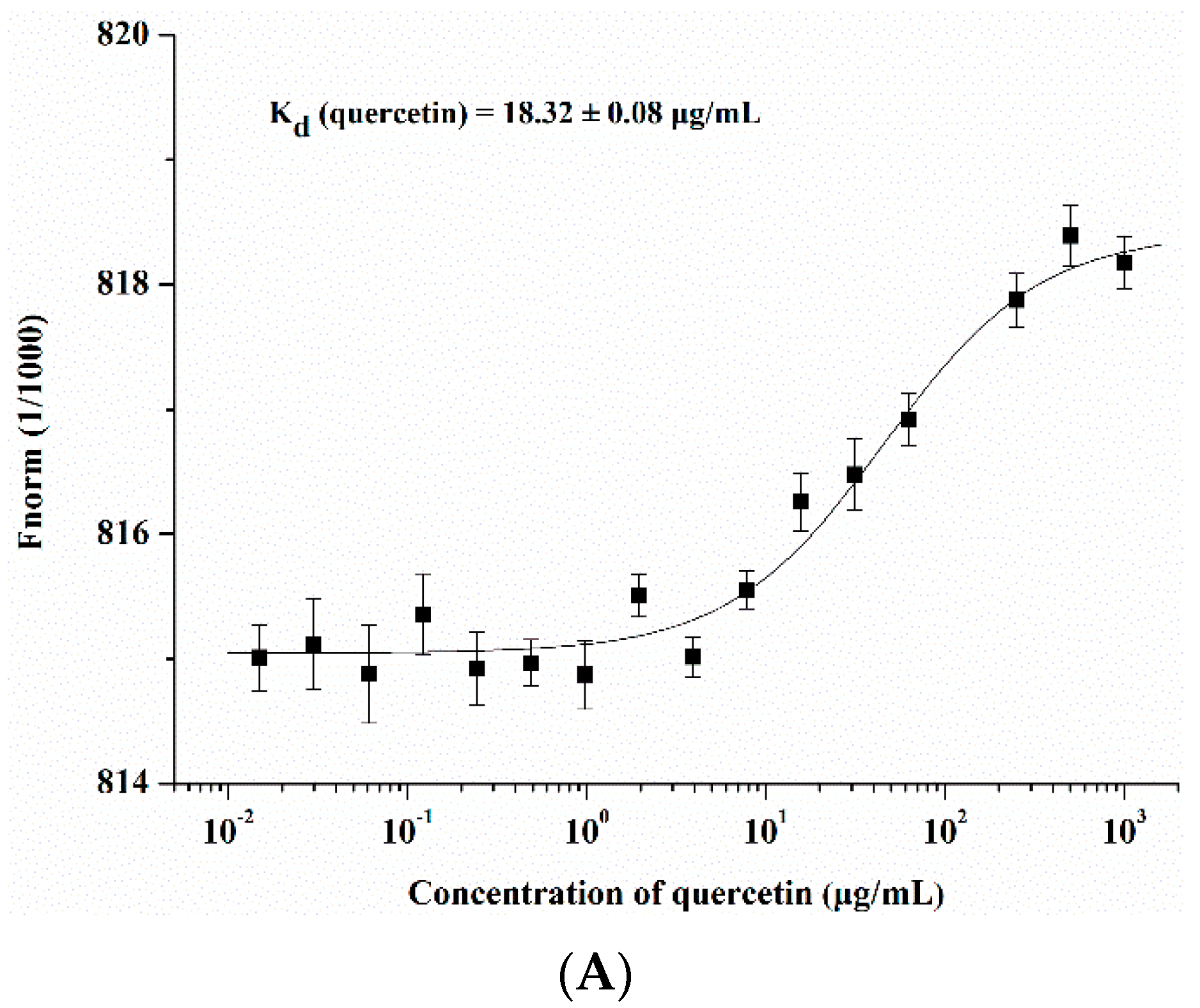
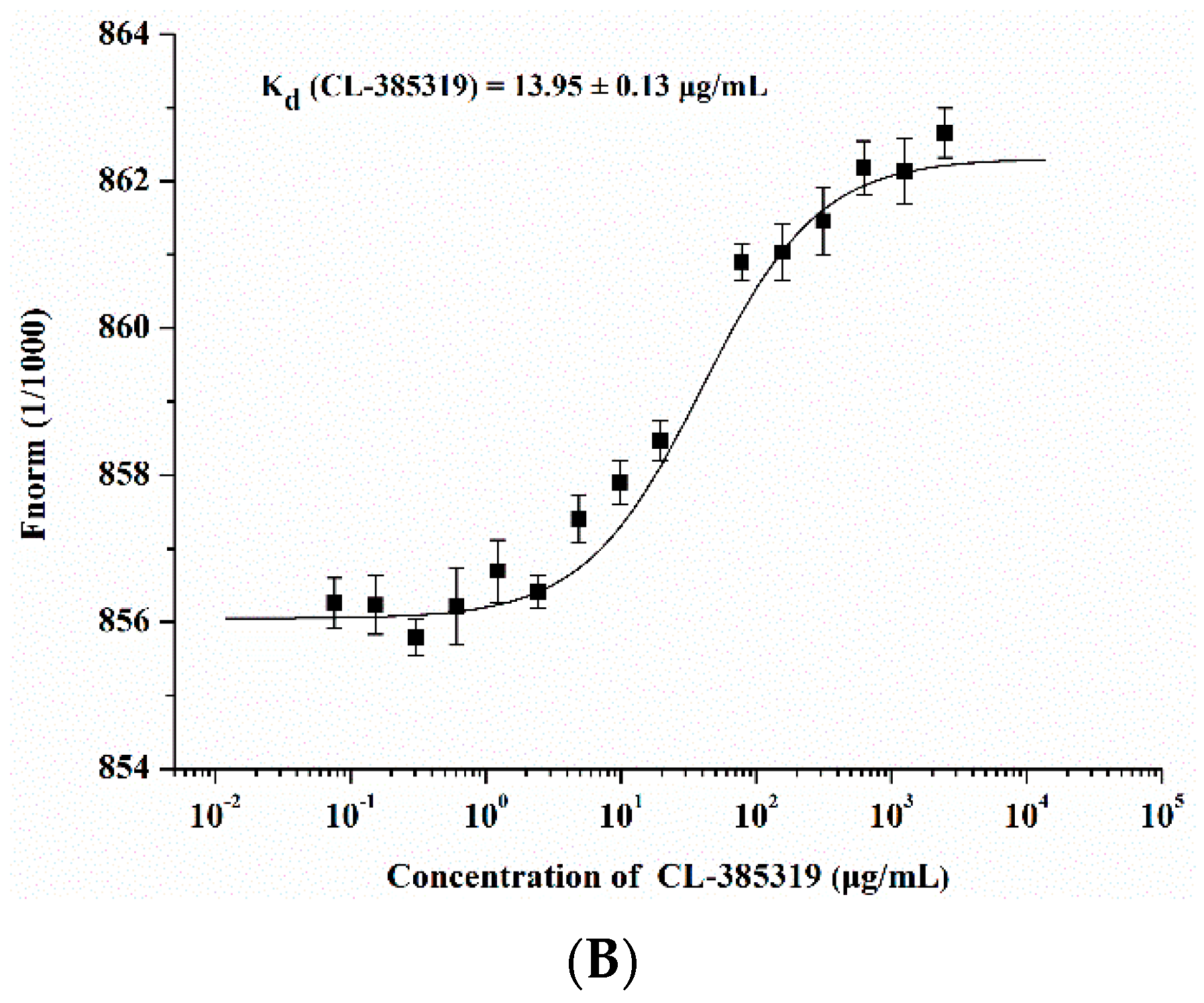
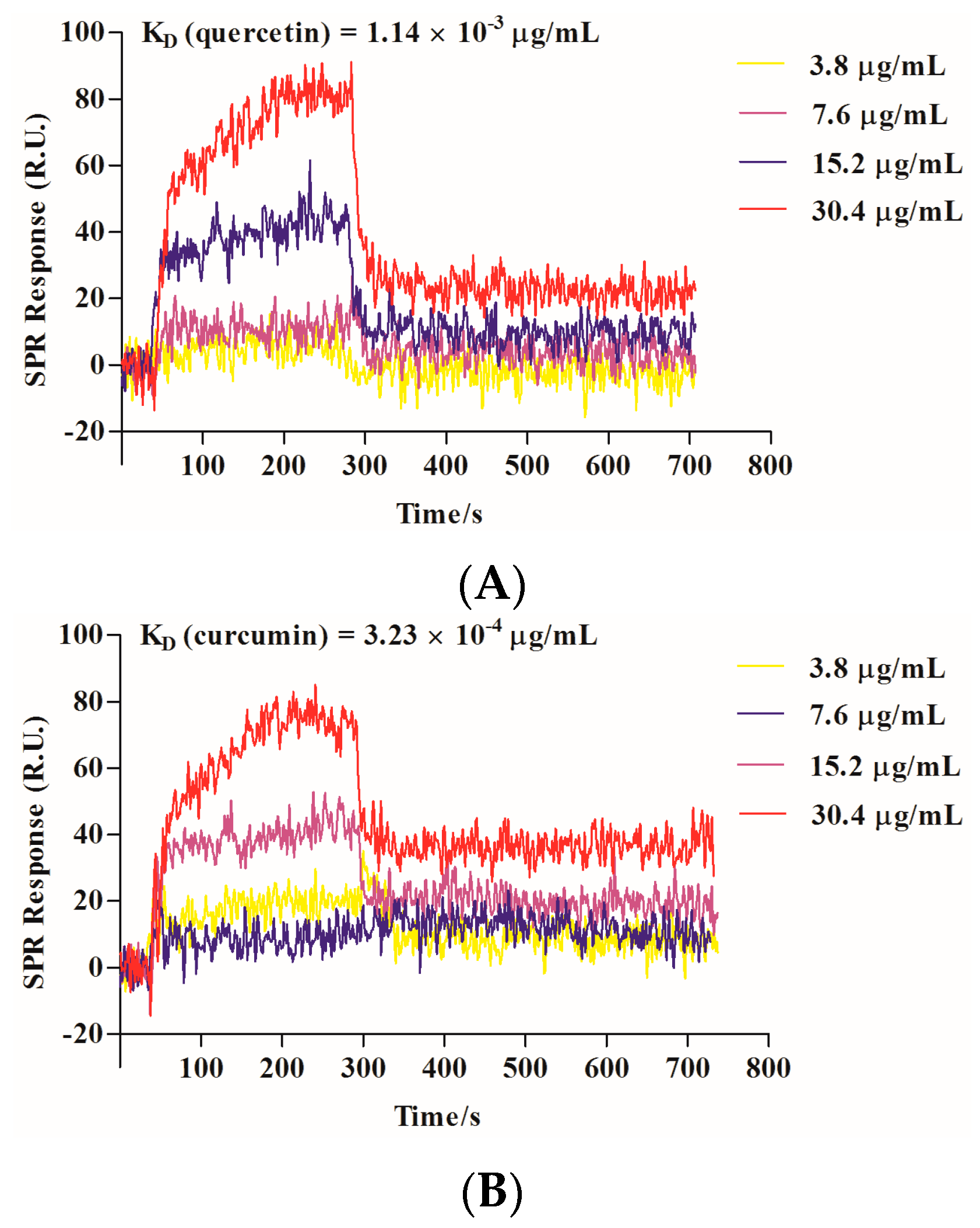
2.3. Quercetin Showed Binding Affinity to Influenza HA Protein
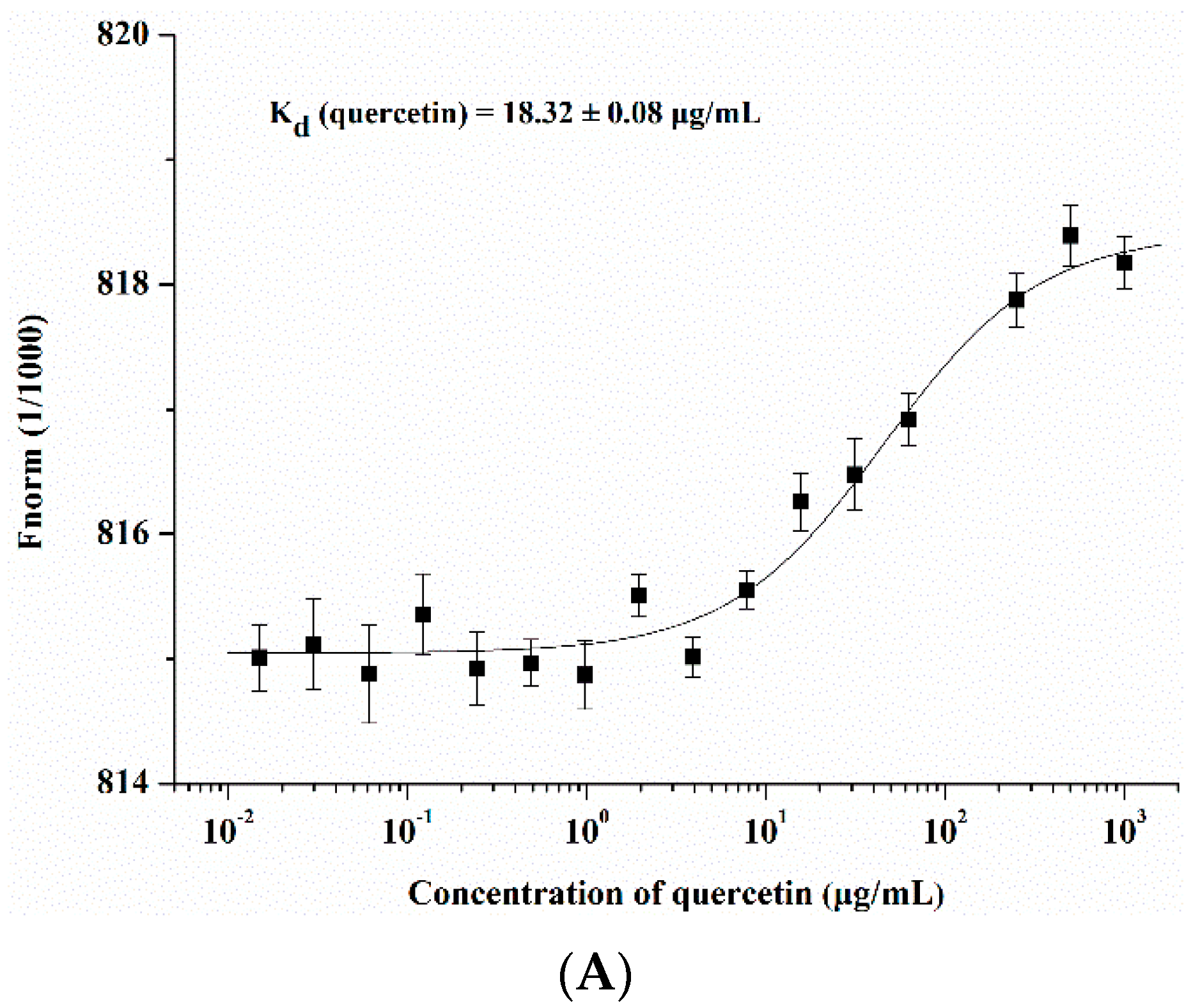
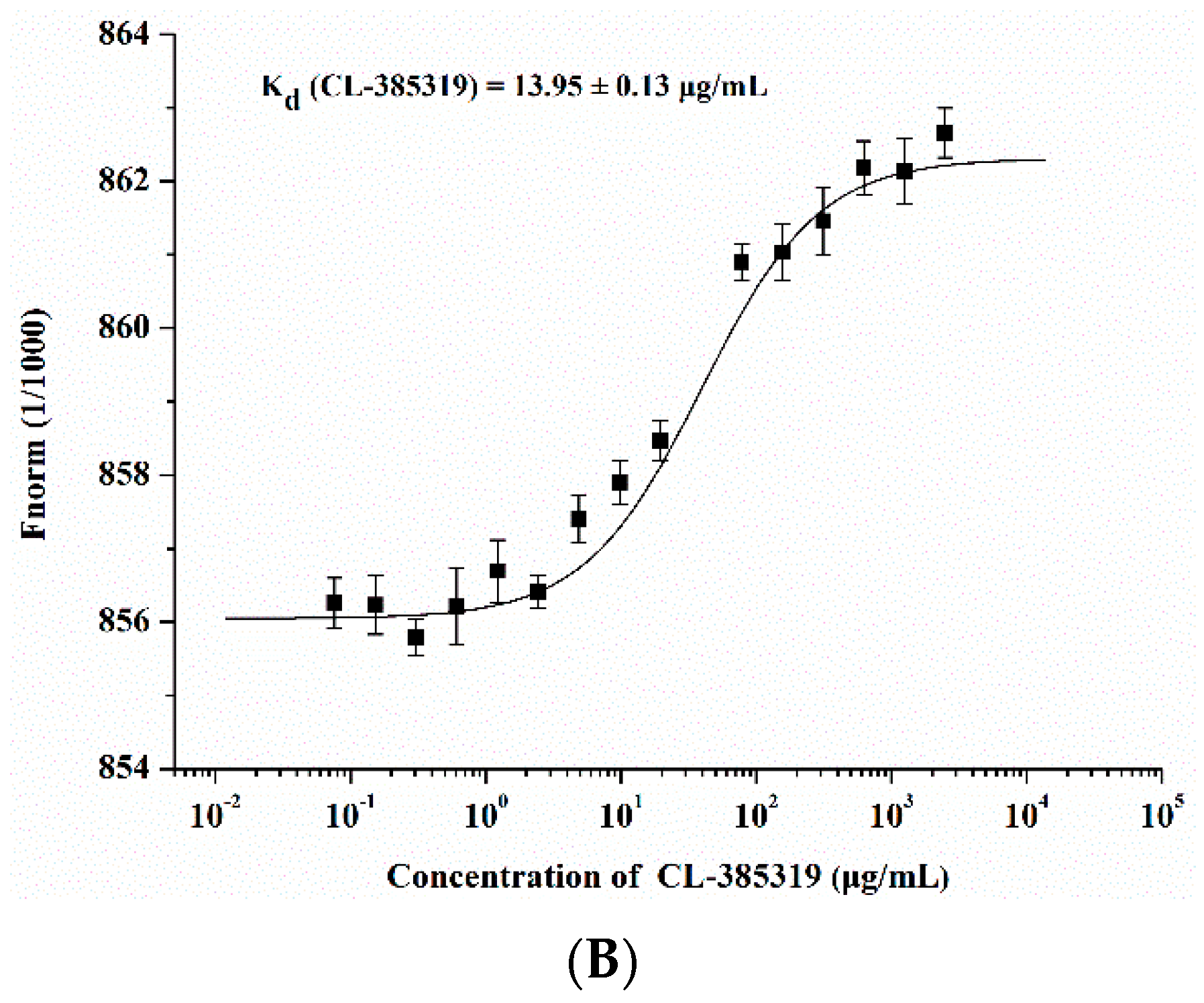
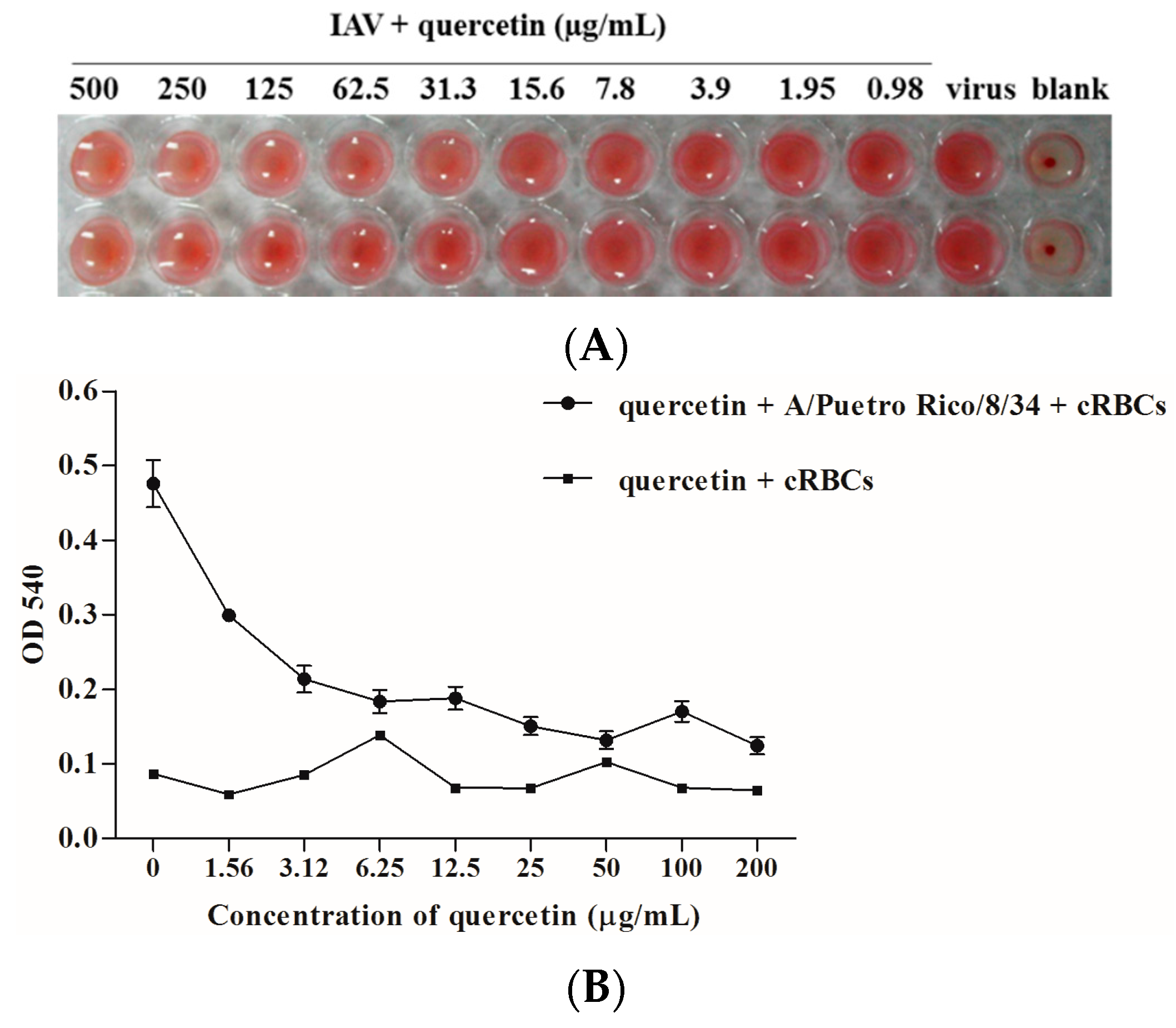
2.4. Quercetin Inhibited Influenza Virus HA Mediated Hemolysis
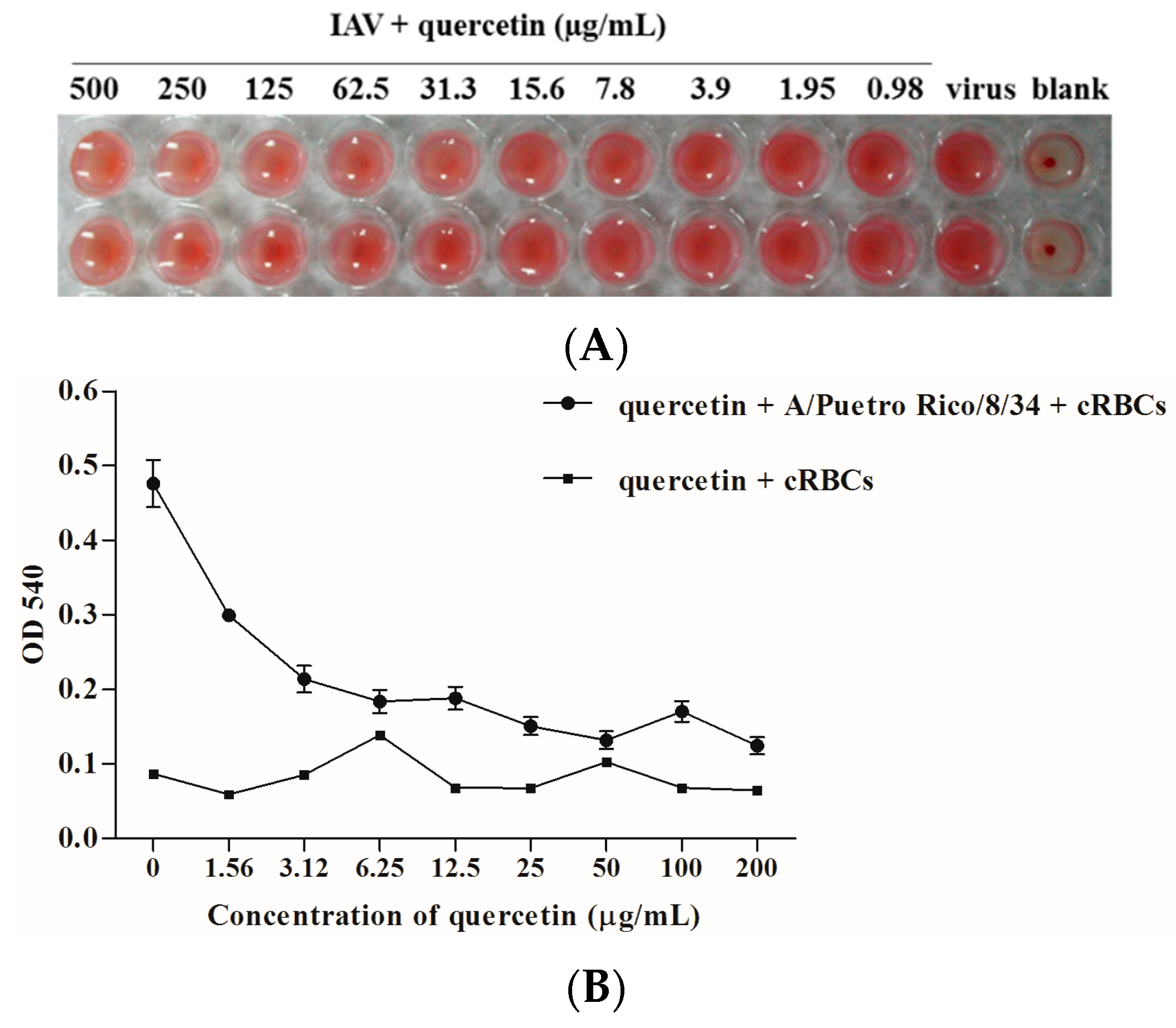
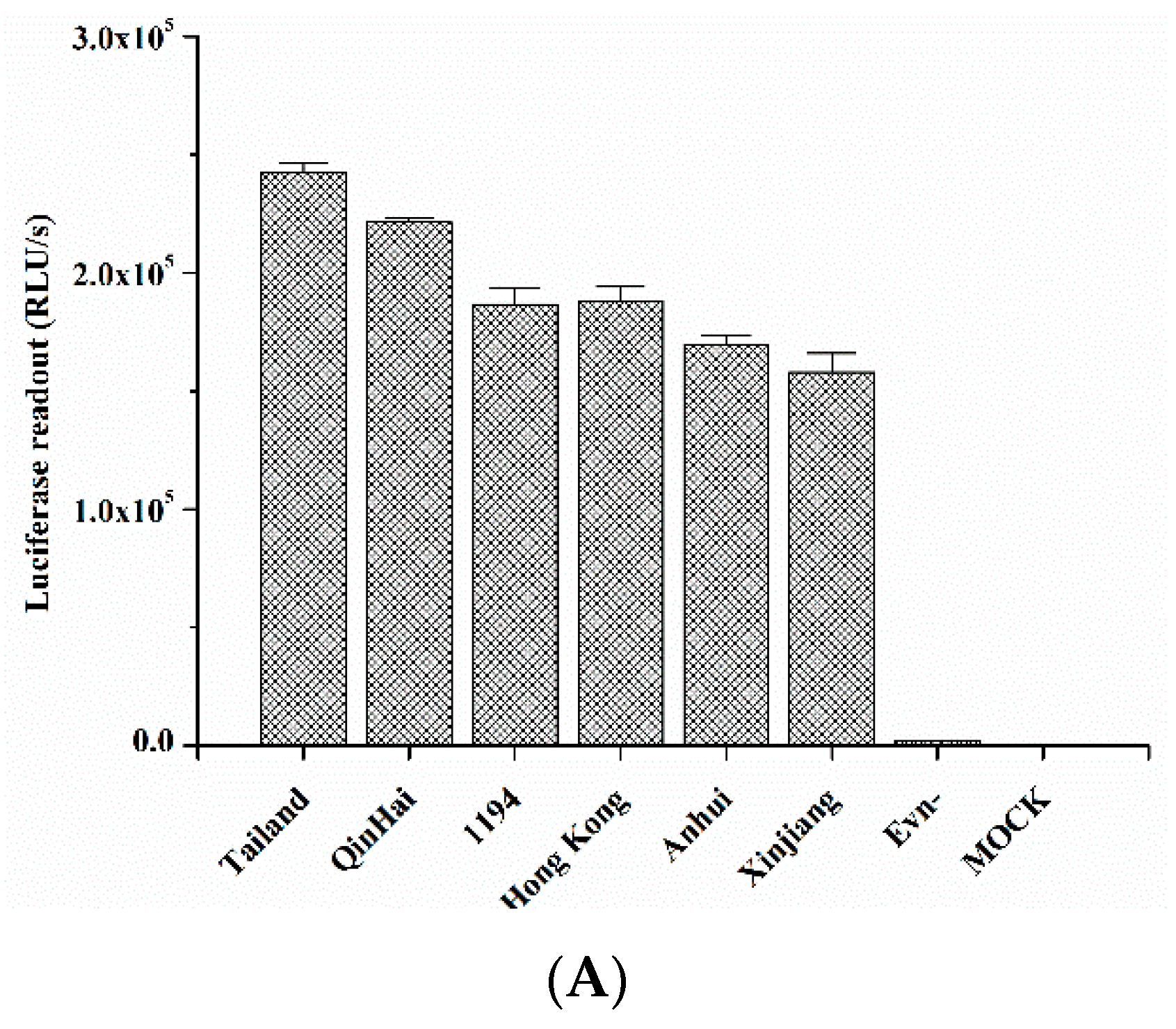
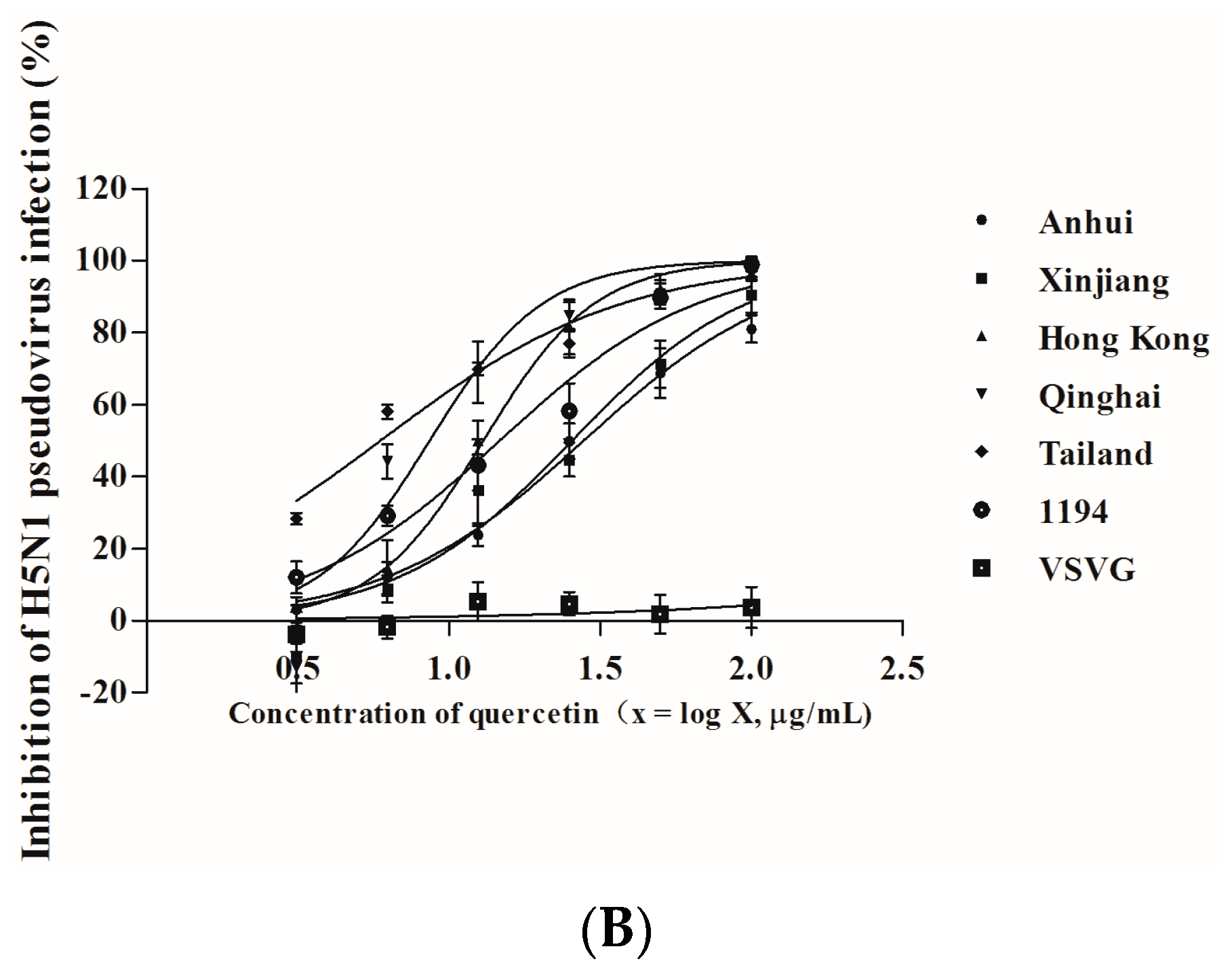
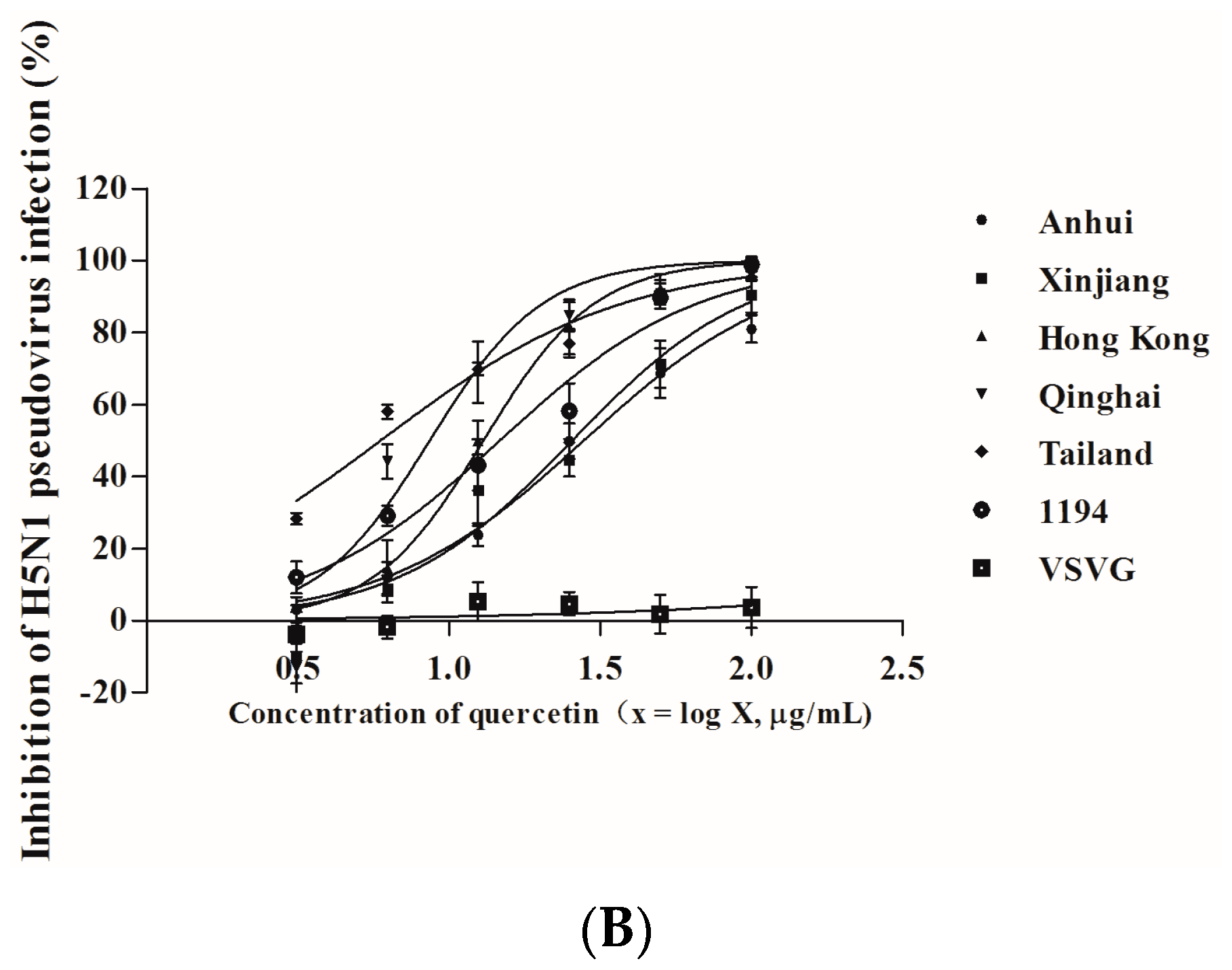
2.5. Quercetin Inhibited Entry of H5N1 Virus with Divergent Strains
| H5N1 Pseudovirus | Inhibitory Activity (Mean ± S.D.) a | |
|---|---|---|
| Quercetin | ||
| IC50 (µg/mL) | IC90 (µg/mL) | |
| A/Anhui/1/2005A | 31.47 ± 4.19 | 127.39 ± 16.37 |
| A/Xinjiang/1/2006 | 26.67 ± 4.49 | 92.27 ± 3.29 |
| A/Hong Kong/156/1997 | 18.90 ± 2.32 | 53.95 ± 1.84 |
| A/Qinghai/59/2005 | 12.27 ± 1.30 | 49.54 ± 4.51 |
| A/Thailand/Kan353/2004 | 11.89 ± 3.31 | 31.19 ± 4.82 |
| A/VietNam/1194/2004 | 17.35 ± 1.58 | 57.34 ± 4.71 |
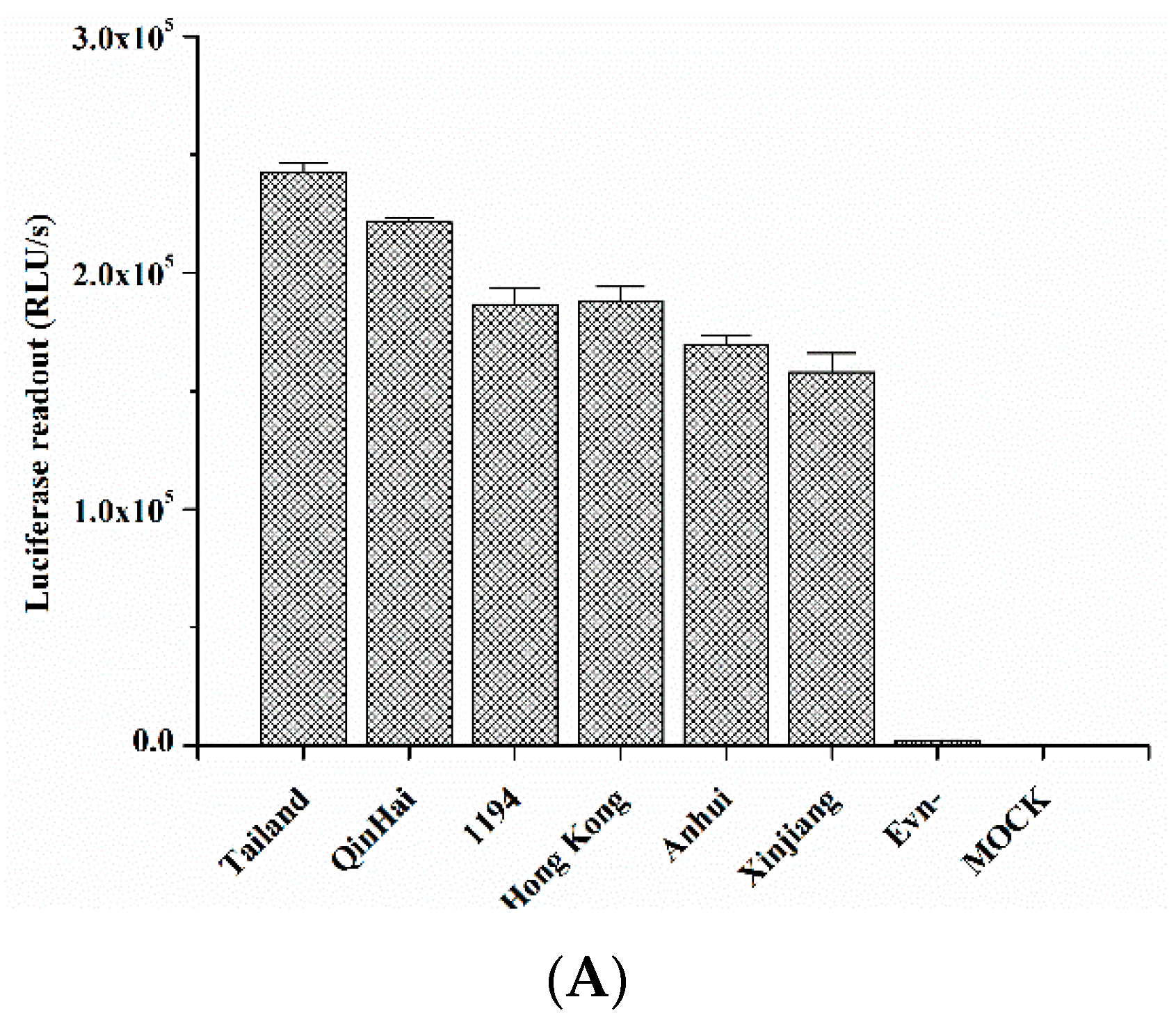
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cytopathic Effect (CPE) Reduction Assay
4.3. Quantitative Real-Time PCR
| Target Gene | Sequence |
|---|---|
| HA-Forward | 5′-TTCCCAAGATCCATCCGGCAA-3′ |
| HA-Reverse | 5′-CCTGCTCGAAGACAGCCACAACG-3′ |
| GAPDH-Forward | 5′-AGGGCAATGCCAGCCCCAGCG-3′ |
| GAPDH-Reverse | 5′-AGGCGTCGGAGGGCCCCCTC-3′ |
4.4. Indirect Immunofluorescence Microscopy
4.5. Time-of-Addition Assay
4.6. Surface Plasmon Resonance (SPR) Analysis
4.7. Microscale Thermophoresis (MST) Protein Binding Study
4.8. Hemagglutination Inhibition (HI) Assay
4.9. Hemolysis Inhibition Assay
4.10. HA Pseudovirus Neutralization Assay
4.11. Cytotoxicity Test
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, N.; Van Tin, N.; Sato, Y.; Thach, H.N.; Katano, H.; Diep, P.H.; Kumasaka, T.; Thuy, N.T.; Hasegawa, H.; San, L.T.; et al. Pathological study of archival lung tissues from five fatal cases of avian H5N1 influenza in Vietnam. Mod. Pathol. 2013, 26, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, L.; Zhou, M.; Chen, Z.; Li, F.; Wu, H.; Xiang, N.; Chen, E.; Tang, F.; Wang, D.; et al. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N. Engl. J. Med. 2014, 370, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. S4), D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Leonov, H.; Astrahan, P.; Krugliak, M.; Arkin, I.T. How do aminoadamantanes block the influenza M2 channel, and how does resistance develop? J. Am. Chem. Soc. 2011, 133, 9903–9911. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, N.; Pitiriga, V.; Gennimata, V.; Tsakris, A. A review of neuraminidase inhibitor susceptibility in influenza strains. Expert Rev. AntiInfect. Ther. 2014, 12, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Komeda, T.; Ishii, S.; Itoh, Y.; Ariyasu, Y.; Sanekata, M.; Yoshikawa, T.; Shimada, J. Post-marketing safety and effectiveness evaluation of the intravenous anti-influenza neuraminidase inhibitor peramivir. II: A pediatric drug use investigation. J. Infect. Chemother. 2015, 21, 194–201. [Google Scholar] [CrossRef] [PubMed]
- McKimm-Breschkin, J.L. Resistance of influenza viruses to neuraminidase inhibitors—A review. Antiviral. Res. 2000, 47, 1–17. [Google Scholar] [CrossRef]
- De Jong, M.D.; Tran, T.T.; Truong, H.K.; Vo, M.H.; Smith, G.J.; Nguyen, V.C.; Bach, V.C.; Phan, T.Q.; Do, Q.H.; Guan, Y.; et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med. 2005, 353, 2667–2672. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.M.; Kiso, M.; Someya, K.; Sakai, Y.T.; Nguyen, T.H.; Nguyen, K.H.; Pham, N.D.; Ngyen, H.H.; Yamada, S.; Muramoto, Y.; et al. Avian flu: Isolation of drug-resistant H5N1 virus. Nature 2005, 437. [Google Scholar] [CrossRef] [PubMed]
- Sheu, T.G.; Fry, A.M.; Garten, R.J.; Deyde, V.M.; Shwe, T.; Bullion, L.; Peebles, P.J.; Li, Y.; Klimov, A.I.; Gubareva, L.V. Dual resistance to adamantanes and oseltamivir among seasonal influenza A(H1N1) viruses: 2008–2010. J. Infect. Dis. 2011, 203, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, M.; Shen, X.; Liu, S. Influenza A virus entry inhibitors targeting the hemagglutinin. Viruses 2013, 5, 352–373. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sivaramakrishna, R.P.; Ludwig, K.; Korte, T.; Bottcher, C.; Herrmann, A. Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: Stability and energetics of the hemagglutinin. Biochim. Biophys. Acta 2003, 1614, 3–13. [Google Scholar] [CrossRef]
- Yang, Z.F.; Bai, L.P.; Huang, W.B.; Li, X.Z.; Zhao, S.S.; Zhong, N.S.; Jiang, Z.H. Comparison of in vitro antiviral activity of tea polyphenols against influenza A and B viruses and structure-activity relationship analysis. Fitoterapia 2014, 93, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.Y.; Youn, H.; Song, C.S.; Kang, S.C.; Bae, J.J.; Kim, H.T.; Lee, K.M.; Eom, T.H.; Kim, I.S.; Kwak, J.H. Antiviral effect of flavonol glycosides isolated from the leaf of Zanthoxylum piperitum on influenza virus. J. Microbiol. 2014, 52, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.Y.; Chang, H.W.; Lin, C.F.; Liu, C.J.; Hsieh, C.F.; Horng, J.T. Characterization of the anti-influenza activity of the Chinese herbal plant Paeonia lactiflora. Viruses 2014, 6, 1861–1875. [Google Scholar] [CrossRef] [PubMed]
- Grienke, U.; Schmidtke, M.; von Grafenstein, S.; Kirchmair, J.; Liedl, K.R.; Rollinger, J.M. Influenza neuraminidase: A druggable target for natural products. Nat. Prod. Rep. 2012, 29, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Wang, Y.F.; Xu, J.; Gu, Q.; Liu, H.B.; Xiao, P.G.; Zhou, J.; Liu, Y.; Yang, Z.; Su, H. Anti-influenza agents from traditional Chinese medicine. Nat. Prod. Rep. 2010, 27, 1758–1780. [Google Scholar] [CrossRef] [PubMed]
- Harwood, M.; Danielewska-Nikiel, B.; Borzelleca, J.F.; Flamm, G.W.; Williams, G.M.; Lines, T.C. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem. Toxicol. 2007, 45, 2179–2205. [Google Scholar] [CrossRef] [PubMed]
- Delgado, L.; Fernandes, I.; Gonzalez-Manzano, S.; de Freitas, V.; Mateus, N.; Santos-Buelga, C. Anti-proliferative effects of quercetin and catechin metabolites. Food Funct. 2014, 5, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Robaszkiewicz, A.; Balcerczyk, A.; Bartosz, G. Antioxidative and prooxidative effects of quercetin on A549 cells. Cell Biol. Int. 2007, 31, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, Z. Quercetin in a lotus leaves extract may be responsible for antibacterial activity. Arch. Pharm. Res. 2008, 31, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Dajas, F. Life or death: Neuroprotective and anticancer effects of quercetin. J. Ethnopharmacol. 2012, 143, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Song, J.H.; Park, K.S.; Kwon, D.H. Inhibitory effects of quercetin 3-rhamnoside on influenza A virus replication. Eur. J. Pharm. Sci. 2009, 37, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Uchide, N.; Toyoda, H. Antioxidant therapy as a potential approach to severe influenza-associated complications. Molecules 2011, 16, 2032–2052. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Si, L.; Wang, Y.; Wu, Y.; Yu, F.; Jiao, P.; Shi, Y.; Wang, H.; Xiao, S.; Fu, G.; et al. Discovery of pentacyclic triterpenoids as potential entry inhibitors of influenza viruses. J. Med. Chem. 2014, 57, 10058–10071. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.L.; Mizushina, Y.; Wang, S.Y.; Chuang, D.Y.; Nadar, M.; Hsu, W.L. Structure-activity relationship analysis of curcumin analogues on anti-influenza virus activity. FEBS J. 2013, 280, 5829–5840. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, R.; Zhang, R.; Chan, C.C.; Xi, B.; Zhu, Z.; Yang, J.; Poon, V.K.; Zhou, J.; Chen, M.; et al. CL-385319 inhibits H5N1 avian influenza A virus infection by blocking viral entry. Eur. J. Pharmacol. 2011, 660, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, S.; Jiang, S. HIV entry inhibitors targeting GP41: From polypeptides to small-molecule compounds. Curr. Pharm. Des. 2007, 13, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Este, J.A.; Telenti, A. HIV entry inhibitors. Lancet 2007, 370, 81–88. [Google Scholar] [CrossRef]
- Xia, S.; Liu, Q.; Wang, Q.; Sun, Z.; Su, S.; Du, L.; Ying, T.; Lu, L.; Jiang, S. Middle East respiratory syndrome coronavirus (MERS-CoV) entry inhibitors targeting spike protein. Virus Res. 2014, 194, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, J.; Kakui, M.; Iwasaki, H.; Sugimoto, H.; Fujiwara, T.; Hattori, N. Identification of amino acids of influenza virus HA responsible for resistance to a fusion inhibitor, Stachyflin. Microbiol. Immunol. 2000, 44, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, L.; Tan, S.; Jin, H.; Qiu, J.; Mao, Q.; Li, R.; Xia, C.; Jiang, Z.H.; Jiang, S.; et al. A natural theaflavins preparation inhibits HIV-1 infection by targeting the entry step: Potential applications for preventing HIV-1 infection. Fitoterapia 2012, 83, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Chen, Q.; Zhang, W.; Ren, S.; Guo, Y.; Li, Y. Structure-activity relationships of saponin derivatives: A series of entry inhibitors for highly pathogenic H5N1 influenza virus. Eur. J. Med. Chem. 2012, 53, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Jin, L.; Zhao, G.; Sun, S.; Li, J.; Yu, H.; Li, Y.; Zheng, B.J.; Liddington, R.C.; Zhou, Y.; et al. Identification and structural characterization of a broadly neutralizing antibody targeting a novel conserved epitope on the influenza virus H5N1 hemagglutinin. J. Virol. 2013, 87, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.S.; Wei, J.; Luo, G.; Cianci, C.; Danetz, S.; Torri, A.; Tiley, L.; Krystal, M.; Yu, K.L.; Huang, S.; et al. An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg. Med. Chem. Lett. 2001, 11, 2393–2396. [Google Scholar] [CrossRef]
- Nguyen, B.; Tanious, F.A.; Wilson, W.D. Biosensor-surface plasmon resonance: Quantitative analysis of small molecule-nucleic acid interactions. Methods 2007, 42, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.A.; Dijkman, P.M.; Lea, W.A.; van den Bogaart, G.; Jerabek-Willemsen, M.; Lazic, A.; Joseph, J.S.; Srinivasan, P.; Baaske, P.; Simeonov, A.; et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Ilyushina, N.A.; Hay, A.; Yilmaz, N.; Boon, A.C.; Webster, R.G.; Govorkova, E.A. Oseltamivir-ribavirin combination therapy for highly pathogenic H5N1 influenza virus infection in mice. Antimicrob. Agents Chemother. 2008, 52, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Govorkova, E.A.; Fang, H.B.; Tan, M.; Webster, R.G. Neuraminidase inhibitor-rimantadine combinations exert additive and synergistic anti-influenza virus effects in MDCK cells. Antimicrob. Agents Chemother. 2004, 48, 4855–4863. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpionts. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar]
- Khare, D.; Godbole, N.M.; Pawar, S.D.; Mohan, V.; Pandey, G.; Gupta, S.; Kumar, D.; Dhole, T.N.; Godbole, M.M. Calcitriol [1, 25[OH]2 D3] pre- and post-treatment suppresses inflammatory response to influenza A (H1N1) infection in human lung A549 epithelial cells. Eur. J. Nutr. 2013, 52, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Zu, M.; Yang, F.; Zhou, W.; Liu, A.; Du, G.; Zheng, L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antiviral. Res. 2012, 94, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wu, W.K.; Li, Z.J.; Wong, H.P.; Tai, E.K.; Li, H.T.; Wu, Y.C.; Cho, C.H. E series of prostaglandin receptor 2-mediated activation of extracellular signal-regulated kinase/activator protein-1 signaling is required for the mitogenic action of prostaglandin E2 in esophageal squamous-cell carcinoma. J. Pharmacol. Exp. Ther. 2008, 327, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Torri, A.; Harte, W.E.; Danetz, S.; Cianci, C.; Tiley, L.; Day, S.; Mullaney, D.; Yu, K.L.; Ouellet, C.; et al. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J. Virol. 1997, 71, 4062–4070. [Google Scholar] [PubMed]
- Basu, A.; Antanasijevic, A.; Wang, M.; Li, B.; Mills, D.M.; Ames, J.A.; Nash, P.J.; Williams, J.D.; Peet, N.P.; Moir, D.T.; et al. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza A virus fusion. J. Virol. 2014, 88, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, G.; Zhang, X.; Liu, Z.; Yu, H.; Zheng, B.J.; Zhou, Y.; Jiang, S. Development of a safe and convenient neutralization assay for rapid screening of influenza HA-specific neutralizing monoclonal antibodies. Biochem. Biophys. Res. Commun. 2010, 397, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Hrincius, E.R.; Korte, V.; Mazur, I.; Droebner, K.; Poetter, A.; Dreschers, S.; Schmolke, M.; Planz, O.; Ludwig, S. A polyphenol rich plant extract, CYSTUS052, exerts anti influenza virus activity in cell culture without toxic side effects or the tendency to induce viral resistance. Antiviral. Res. 2007, 76, 38–47. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Li, R.; Li, X.; He, J.; Jiang, S.; Liu, S.; Yang, J. Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry. Viruses 2016, 8, 6. https://doi.org/10.3390/v8010006
Wu W, Li R, Li X, He J, Jiang S, Liu S, Yang J. Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry. Viruses. 2016; 8(1):6. https://doi.org/10.3390/v8010006
Chicago/Turabian StyleWu, Wenjiao, Richan Li, Xianglian Li, Jian He, Shibo Jiang, Shuwen Liu, and Jie Yang. 2016. "Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry" Viruses 8, no. 1: 6. https://doi.org/10.3390/v8010006
APA StyleWu, W., Li, R., Li, X., He, J., Jiang, S., Liu, S., & Yang, J. (2016). Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry. Viruses, 8(1), 6. https://doi.org/10.3390/v8010006