
Bone Marrow Gene Therapy for HIV/AIDS


Abstract
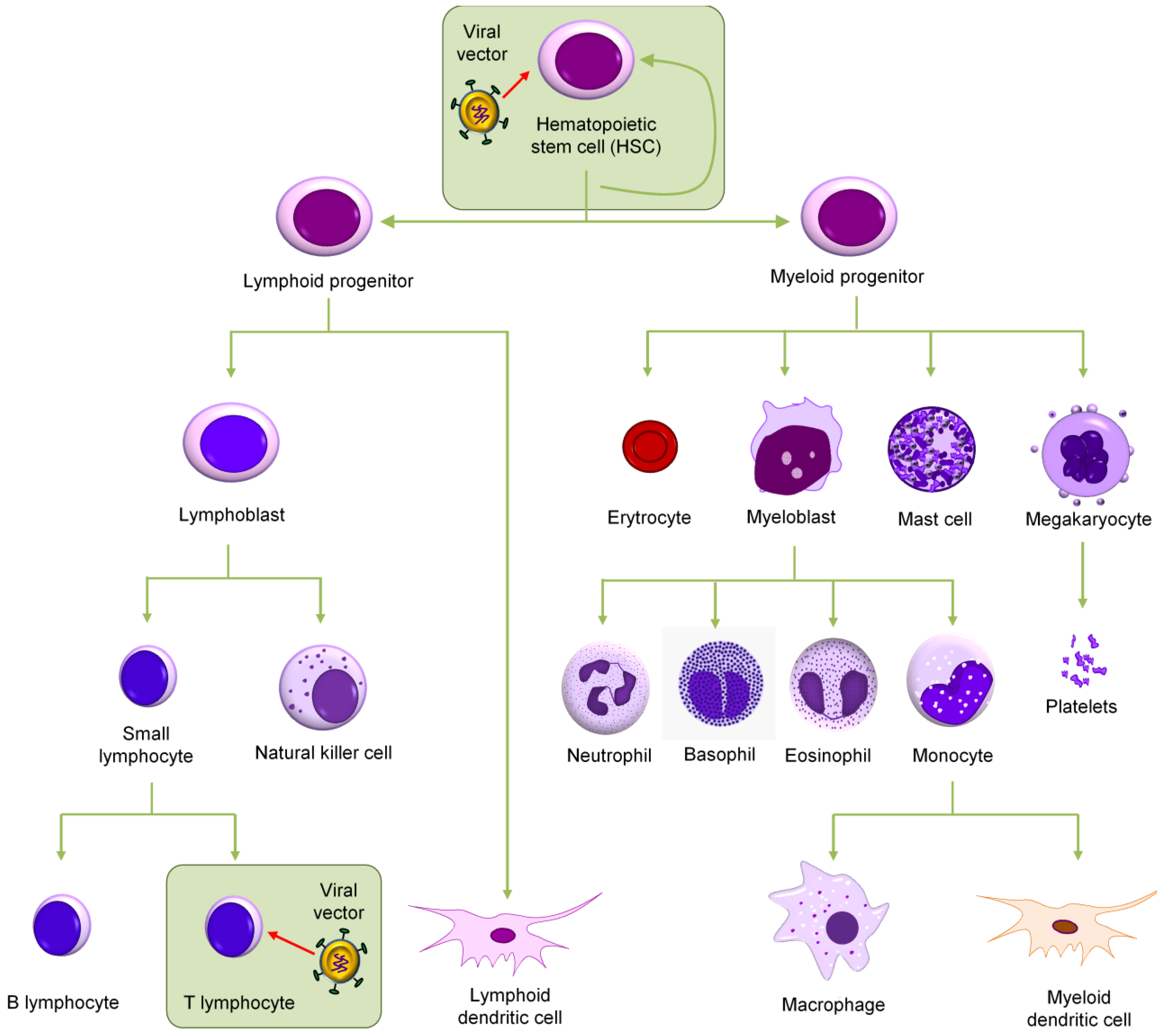
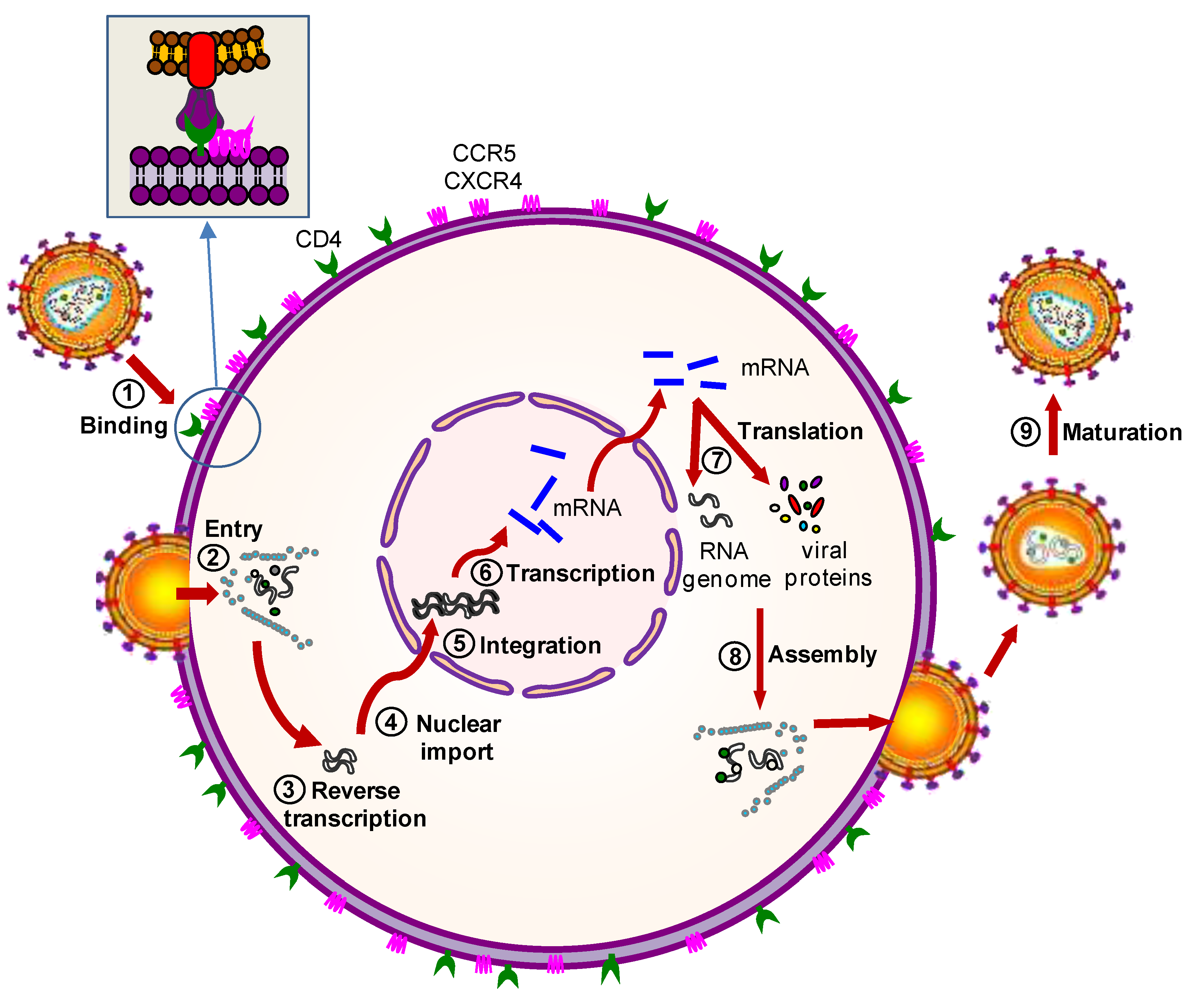
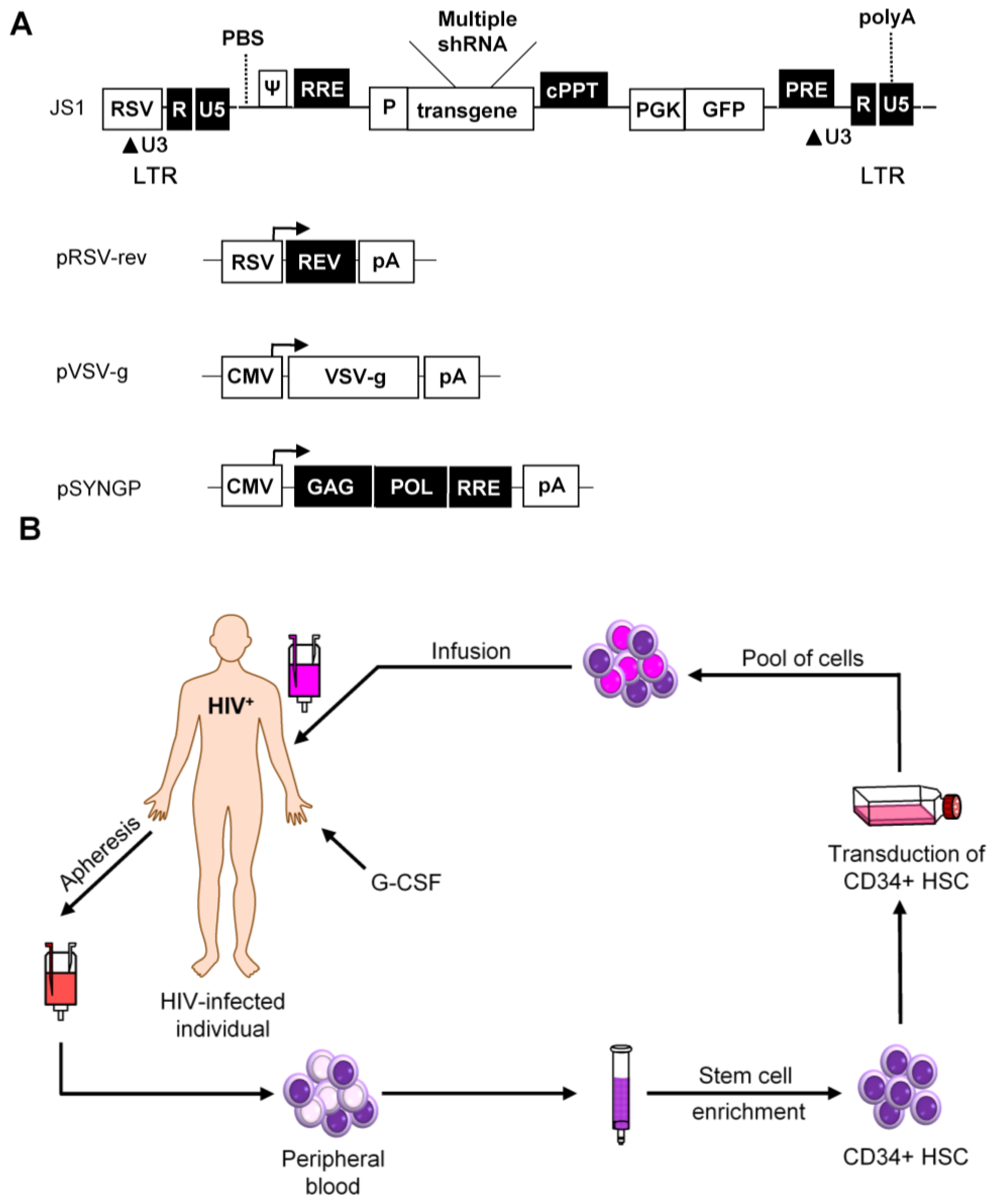
:1. Introduction

2. Selection of Therapeutic Targets
2.1. Artificial T Cell Receptors
2.2. Intracellular Immunization
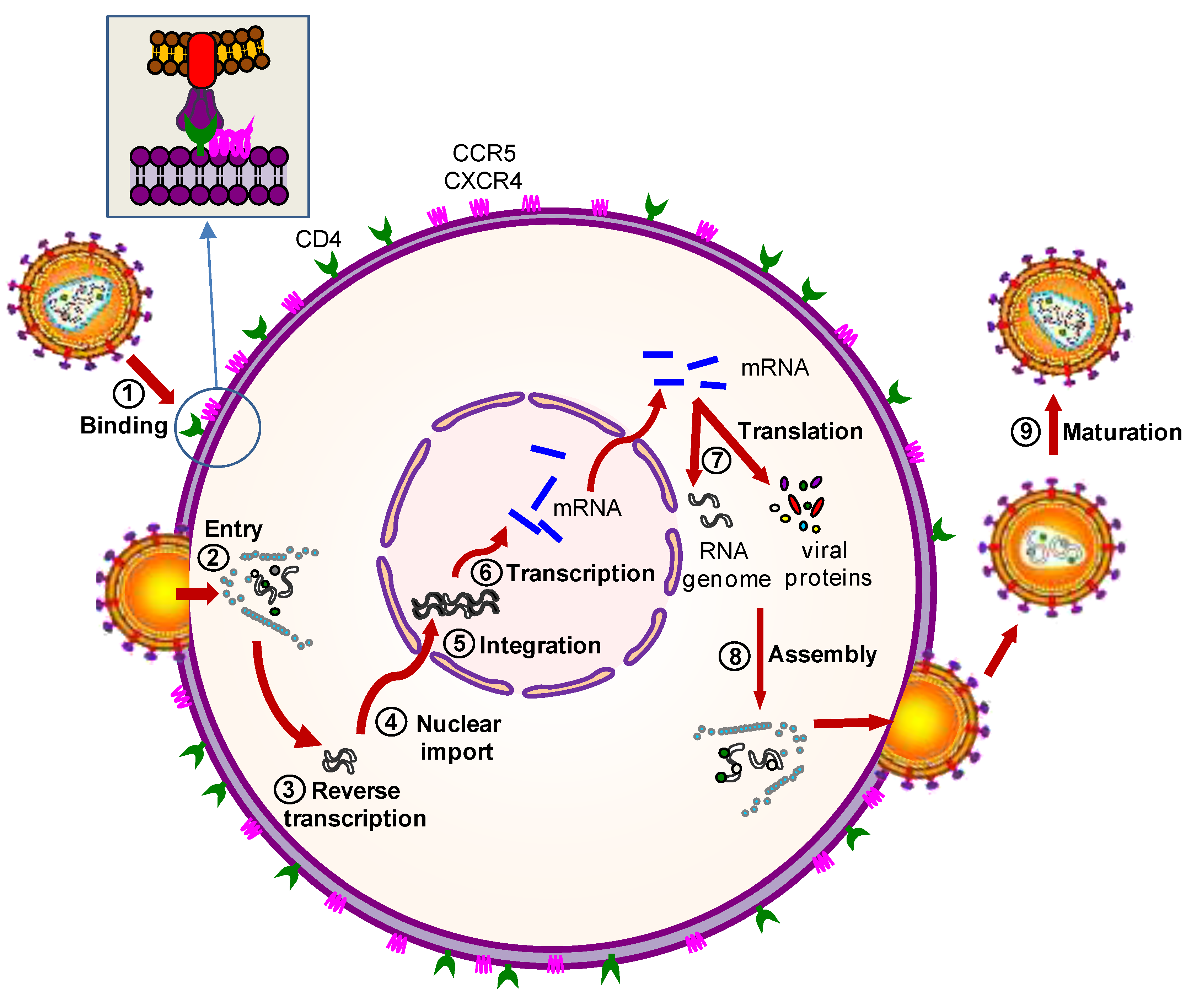
2.2.1. Protein-Based Therapies
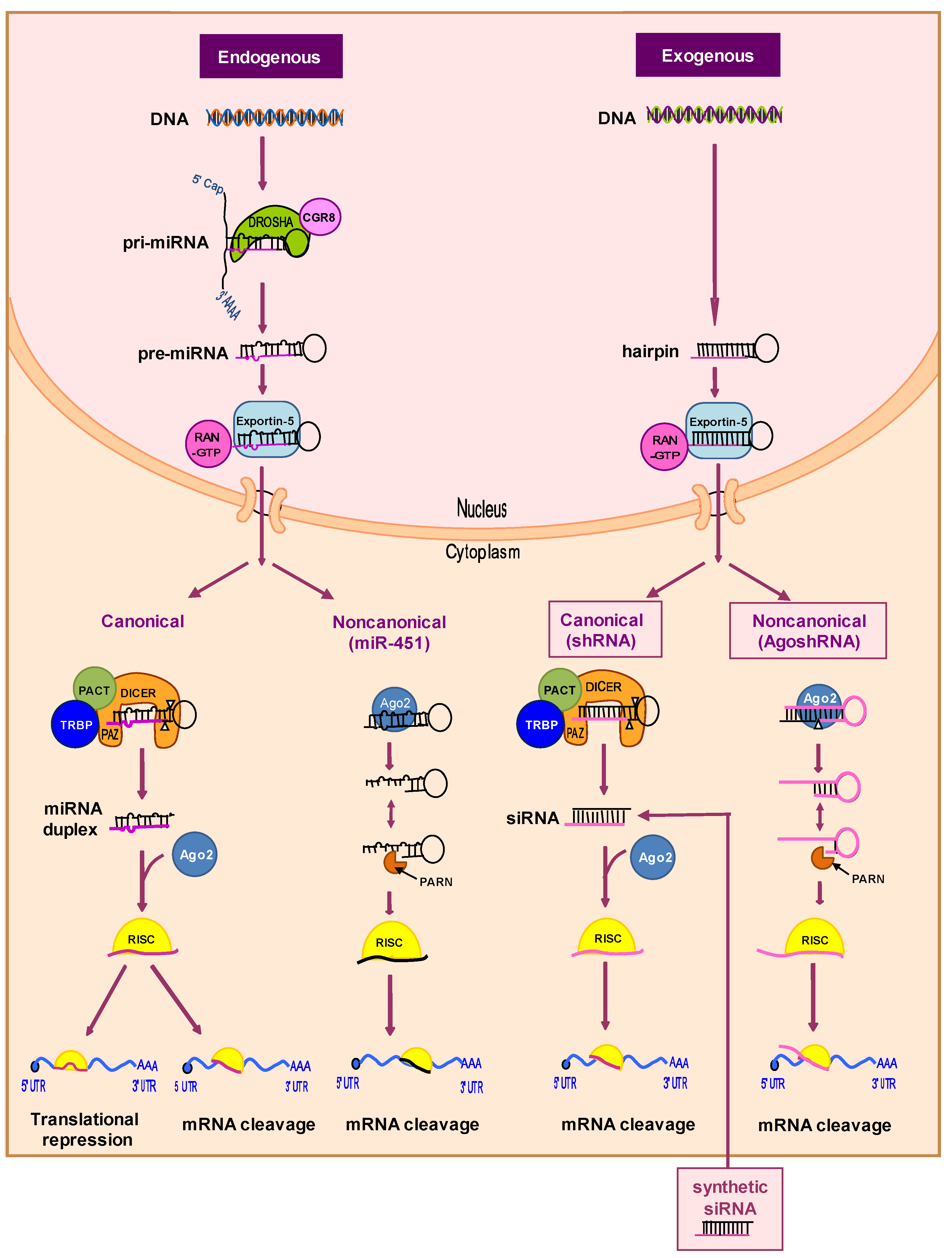
2.2.2. RNA-Based Therapies
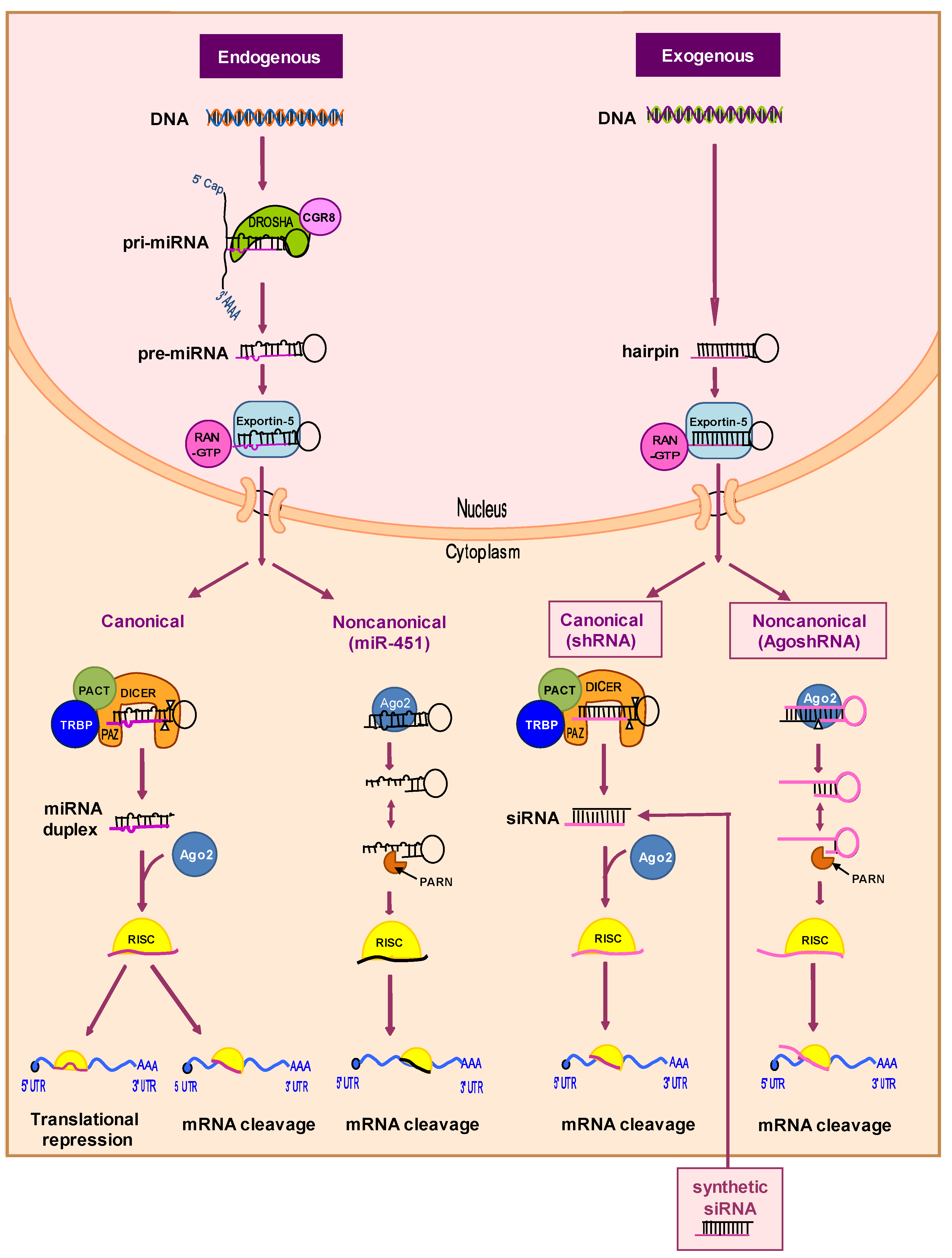
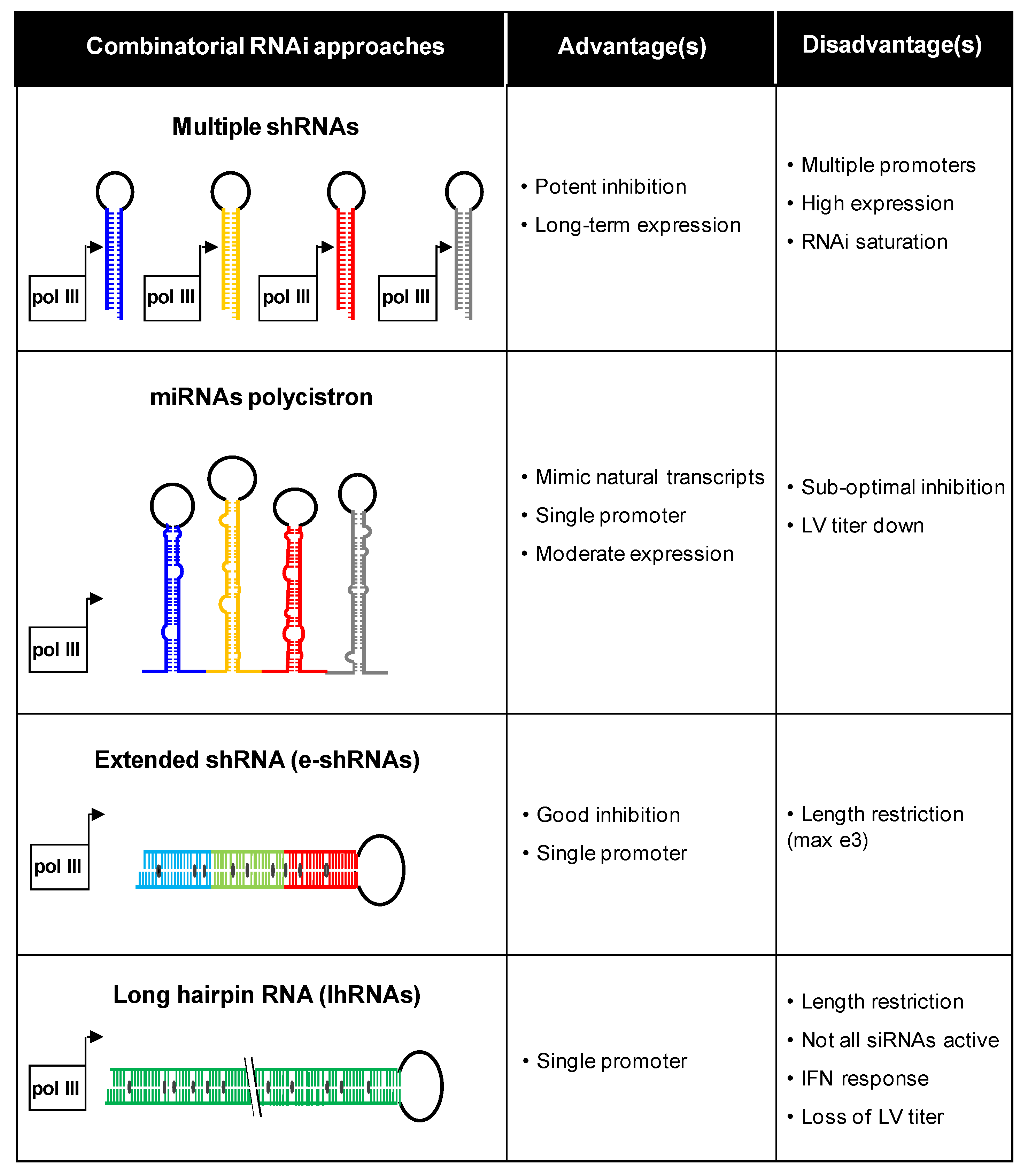
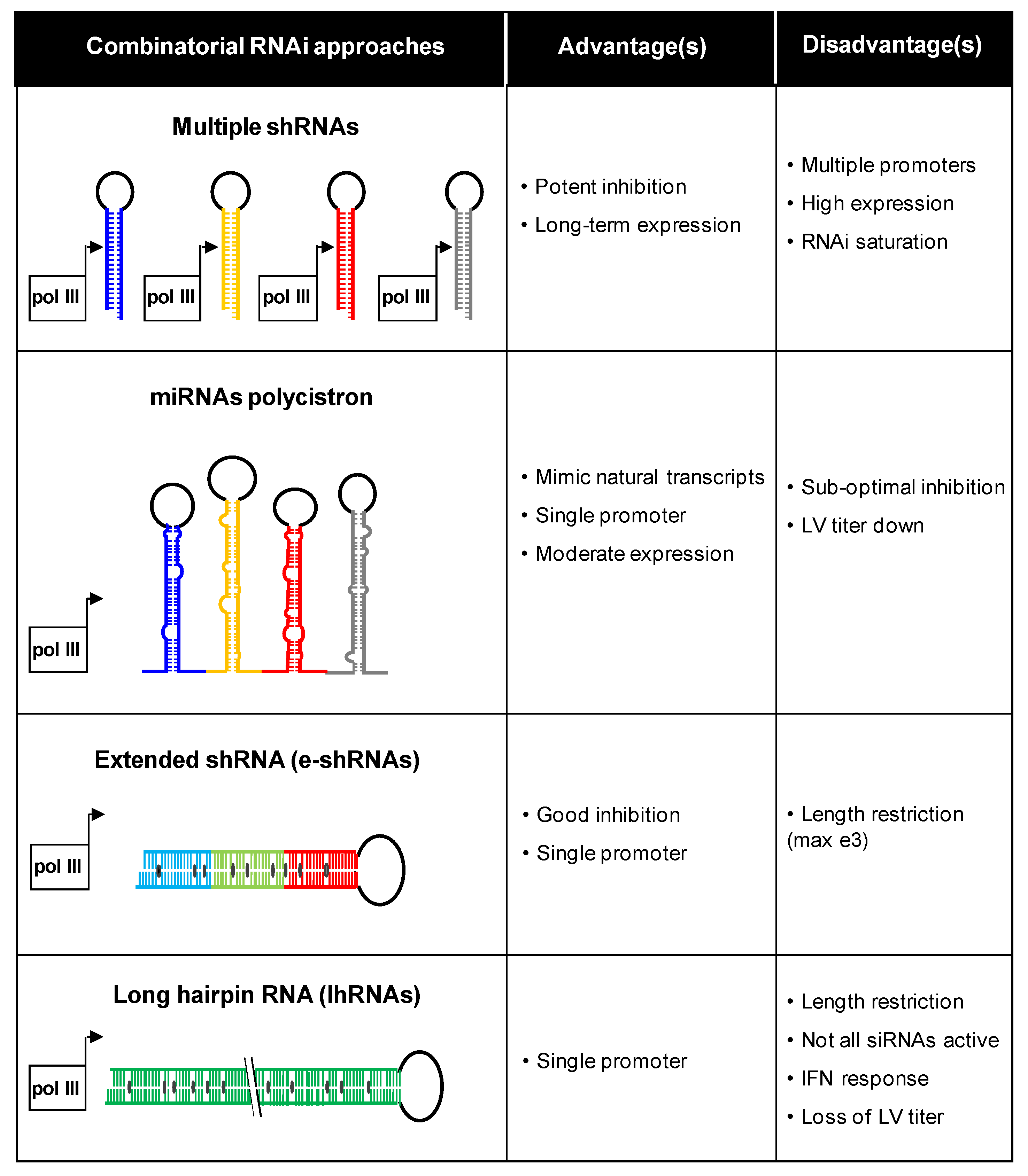
3. Combinatorial Approaches
4. Vector Choice
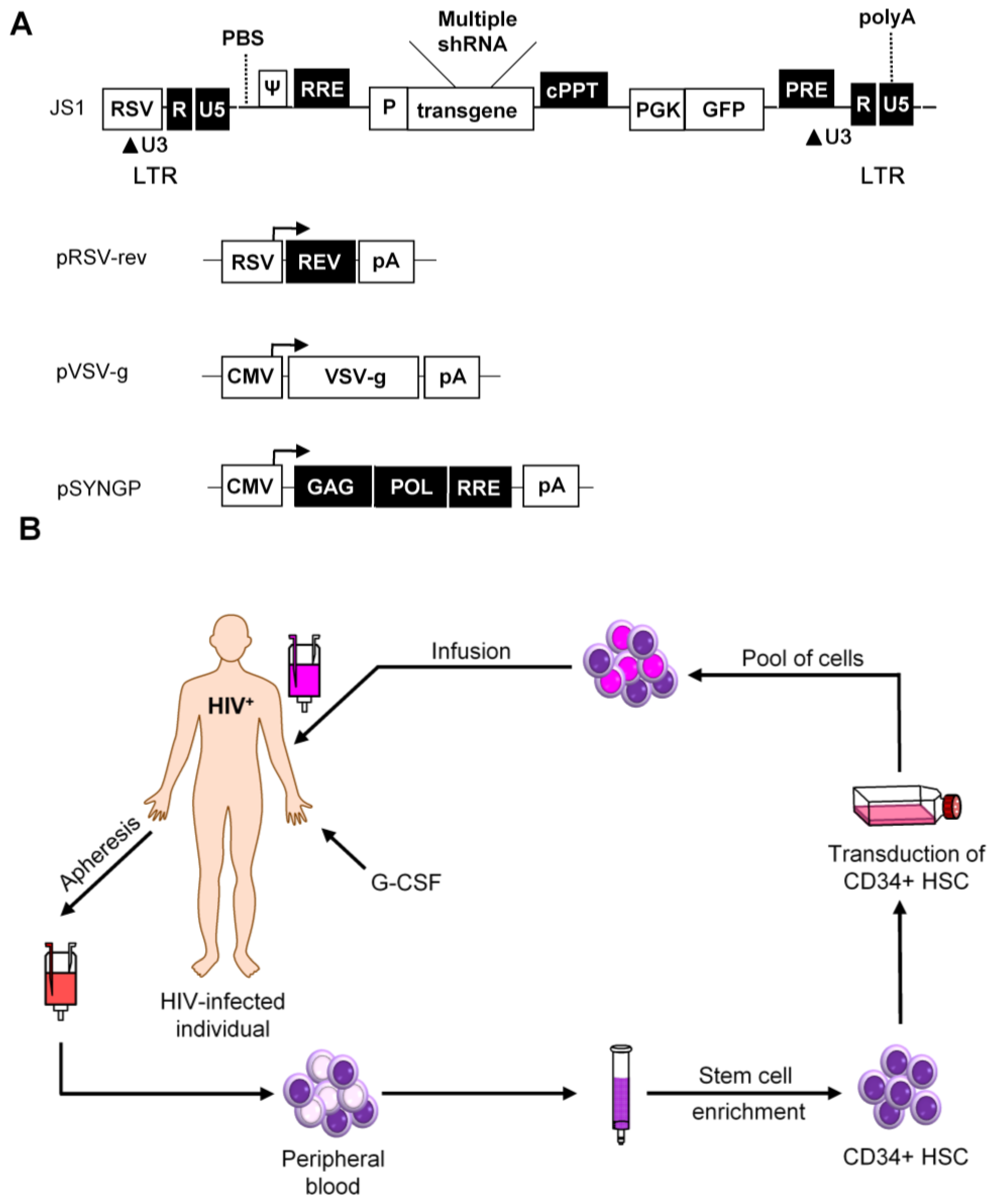
5. Preclinical Safety and Efficacy Tests
6. Clinical Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Therapy Mechanism | Phase | Reference(s) |
|---|---|---|
| Rev-responsive element decoy (Rev protein) | Pilot | [80] |
| Trans-dominant Rev (Rev protein) | I–II | [142] |
| Ribozyme (Tat/Rev mRNA) | II | [69,143] |
| NCT00074997 | ||
| NCT00002221 | ||
| II | NCT01177059 | |
| Combinatorial trans-dominant Rev (Rev protein) and antisense (Pol mRNA) | I–II | NCT00003942 |
| Combinatorial strategy: fusion inhibitor C46 (Env protein) and shRNA (CCR5) | I–II | NCT01734850 |
| Combinatorial strategy: shRNA (Tat/Rev mRNA), TAR decoy (Tat protein) and ribozyme (CCR5) | Pilot | [102] NCT00569985 |
| NCT01153646 |
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Reports on the Global AIDS Epidemic; UNAIDS: Geneva, Switzerland, 2013. [Google Scholar]
- Gray, G.E.; Allen, M.; Moodie, Z.; Churchyard, G.; Bekker, L.G.; Nchabeleng, M.; Mlisana, K.; Metch, B.; De, B.G.; Latka, M.H.; et al. Safety and efficacy of the HVTN 503/Phambili study of a clade-B-based HIV-1 vaccine in South Africa: A double-blind, randomised, placebo-controlled test-of-concept phase 2b study. Lancet Infect. Dis. 2011, 11, 507–515. [Google Scholar] [CrossRef]
- Hammer, S.M.; Sobieszczyk, M.E.; Janes, H.; Karuna, S.T.; Mulligan, M.J.; Grove, D.; Koblin, B.A.; Buchbinder, S.P.; Keefer, M.C.; Tomaras, G.D.; et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N. Engl. J. Med. 2013, 369, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.F.; Spindler, J.; Shao, W.; Yu, S.; Anderson, E.M.; O’Shea, A.; Rehm, C.; Poethke, C.; Kovacs, N.; Mellors, J.W.; et al. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004010. [Google Scholar] [CrossRef] [PubMed]
- Katlama, C.; Deeks, S.G.; Autran, B.; Martinez-Picado, J.; van, L.J.; Rouzioux, C.; Miller, M.; Vella, S.; Schmitz, J.E.; Ahlers, J.; et al. Barriers to a cure for HIV: New ways to target and eradicate HIV-1 reservoirs. Lancet 2013, 381, 2109–2117. [Google Scholar] [CrossRef]
- Llibre, J.M.; Buzon, M.J.; Massanella, M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Domingo, P.; Gatell, J.M.; Larrouse, M.; Gutierrez, M.; et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: A randomized 48-week study. Antivir. Ther. 2012, 17, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Perno, C.F.; Moyle, G.; Tsoukas, C.; Ratanasuwan, W.; Gatell, J.; Schechter, M. Overcoming resistance to existing therapies in HIV-infected patients: The role of new antiretroviral drugs. J. Med. Virol. 2008, 80, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Thiel, E. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: An update after 3 years and the search for patient No. 2. AIDS 2011, 25, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D. Gene therapy. Intracellular immunization. Nature 1988, 335, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Zheng, J.H.; Follenzi, A.; Dilorenzo, T.; Sango, K.; Hyman, J.; Chen, K.; Piechocka-Trocha, A.; Brander, C.; Hooijberg, E.; et al. Lentiviral vectors encoding human immunodeficiency virus type 1 (HIV-1)-specific T-cell receptor genes efficiently convert peripheral blood CD8 T lymphocytes into cytotoxic T lymphocytes with potent in vitro and in vivo HIV-1-specific inhibitory activity. J. Virol. 2008, 82, 3078–3089. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [PubMed]
- Walker, R.E.; Bechtel, C.M.; Natarajan, V.; Baseler, M.; Hege, K.M.; Metcalf, J.A.; Stevens, R.; Hazen, A.; Blaese, R.M.; Chen, C.C.; et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood 2000, 96, 467–474. [Google Scholar] [PubMed]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Bernstein, W.B.; Aronson, N.E.; Schlienger, K.; Cotte, J.; Perfetto, S.; Humphries, M.J.; Ratto-Kim, S.; Birx, D.L.; Steffens, C.; et al. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat. Med. 2002, 8, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Vatakis, D.N.; Arumugam, B.; Kim, S.G.; Bristol, G.; Yang, O.; Zack, J.A. Introduction of exogenous T-cell receptors into human hematopoietic progenitors results in exclusion of endogenous T-cell receptor expression. Mol. Ther. 2013, 21, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bohnlein, S.; Hauber, J.; Cullen, B.R. Functional dissection of the HIV-1 Rev trans-activator-derivation of a trans-dominant repressor of Rev function. Cell 1989, 58, 205–214. [Google Scholar] [CrossRef]
- Malim, M.H.; Freimuth, W.W.; Liu, J.; Boyle, T.J.; Lyerly, H.K.; Cullen, B.R.; Nabel, G.J. Stable expression of transdominant Rev protein in human T cells inhibits human immunodeficiency virus replication. J. Exp. Med. 1992, 176, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Bahner, I.; Sumiyoshi, T.; Kagoda, M.; Swartout, R.; Peterson, D.; Pepper, K.; Dorey, F.; Reiser, J.; Kohn, D.B. Lentiviral vector transduction of a dominant-negative Rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol. Ther. 2007, 15, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Walker, R.; Carter, C.S.; Natarajan, V.; Tavel, J.A.; Bechtel, C.; Herpin, B.; Muul, L.; Zheng, Z.; Jagannatha, S.; et al. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum. Gene Ther. 2005, 16, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Ranga, U.; Woffendin, C.; Verma, S.; Xu, L.; June, C.H.; Bishop, D.K.; Nabel, G.J. Enhanced T cell engraftment after retroviral delivery of an antiviral gene in HIV-infected individuals. Proc. Natl. Acad. Sci. USA 1998, 95, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, A.; Lin, M.H.; Sivakumaran, H.; Li, D.; Kershaw, M.H.; Harrich, D. A mutant Tat protein provides strong protection from HIV-1 infection in human CD4+ T cells. Hum. Gene Ther. 2013, 24, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Alberici, L.; Gubinelli, F.; Lupo, R.; Olgiati, C.; Rizzardi, G.P.; Bovolenta, C. The F12-Vif derivative Chim3 inhibits HIV-1 replication in CD4+ T lymphocytes and CD34+-derived macrophages by blocking HIV-1 DNA integration. Blood 2009, 113, 3443–3452. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.C., Jr.; Khan, M.A.; Kao, S.; Goila-Gaur, R.; Miyagi, E.; Strebel, K. Identification of dominant negative human immunodeficiency virus type 1 Vif mutants that interfere with the functional inactivation of APOBEC3G by virus-encoded Vif. J. Virol. 2010, 84, 5201–5211. [Google Scholar] [CrossRef] [PubMed]
- Cara, A.; Rybak, S.M.; Newton, D.L.; Crowley, R.; Rottschafer, S.E.; Reitz, M.S., Jr.; Gusella, G.L. Inhibition of HIV-1 replication by combined expression of gag dominant negative mutant and a human ribonuclease in a tightly controlled HIV-1 inducible vector. Gene Ther. 1998, 5, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Trono, D.; Feinberg, M.B.; Baltimore, D. HIV-1 Gag mutants can dominantly interfere with the replication of the wild-type virus. Cell 1989, 59, 113–120. [Google Scholar] [CrossRef]
- Cano, J.; Kalpana, G.V. Inhibition of early stages of HIV-1 assembly by INI1/hSNF5 transdominant negative mutant S6. J. Virol. 2011, 85, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.R.; Xu, W.; Mau, J.K.; Goodwin, C.D.; Suhasini, M.; Tang, H.; Frimpong, K.; Rose, D.W.; Wong-Staal, F. Inhibition of HIV replication by dominant negative mutants of Sam68, a functional homolog of HIV-1 Rev. Nat. Med. 1999, 5, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Marsh, K.; Soros, V.; Cochrane, A. Selective translational repression of HIV-1 RNA by Sam68DeltaC occurs by altering PABP1 binding to unspliced viral RNA. Retrovirology 2008, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Sorin, M.; Cano, J.; Das, S.; Mathew, S.; Wu, X.; Davies, K.P.; Shi, X.; Cheng, S.W.; Ott, D.; Kalpana, G.V. Recruitment of a SAP18-HDAC1 complex into HIV-1 virions and its requirement for viral replication. PLoS Pathog. 2009, 5, e1000463. [Google Scholar] [CrossRef] [PubMed]
- Daar, E.S.; Ho, D.D. Relative resistance of primary HIV-1 isolates to neutralization by soluble CD4. Am. J. Med. 1991, 90, 22S–26S. [Google Scholar] [CrossRef]
- Falkenhagen, A.; Ameli, M.; Asad, S.; Read, S.E.; Joshi, S. A novel gene therapy strategy using secreted multifunctional anti-HIV proteins to confer protection to gene-modified and unmodified target cells. Gene Ther. 2014, 21, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Baler-Bitterlich, G.; Ragheb, J.A.; Wong-Staal, F.; Gallo, R.C.; Anderson, W.F. Further evaluation of soluble CD4 as an anti-HIV type 1 gene therapy: Demonstration of protection of primary human peripheral blood lymphocytes from infection by HIV type 1. AIDS Res. Hum. Retroviruses 1994, 10, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Egerer, L.; Volk, A.; Kahle, J.; Kimpel, J.; Brauer, F.; Hermann, F.G.; von, L.D. Secreted antiviral entry inhibitory (SAVE) peptides for gene therapy of HIV infection. Mol. Ther. 2011, 19, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, M.; Brandenburg, G.; Martinius, H.; Schult-Dietrich, P.; Melikyan, G.; Kunert, R.; Baum, C.; Choi, I.; Alexandrov, A.; von, L.D. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J. Virol. 2004, 78, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Riley, J.L.; Carroll, R.G.; von, L.D.; June, C.H. Suppression of HIV-1 infection in primary CD4 T cells transduced with a self-inactivating lentiviral vector encoding a membrane expressed GP41-derived fusion inhibitor. Clin. Immunol. 2005, 115, 26–32. [Google Scholar] [CrossRef] [PubMed]
- van Lunzen, J.; Glaunsinger, T.; Stahmer, I.; von Baehr, V.; Baum, C.; Schilz, A.; Kuehlcke, K.; Naundorf, S.; Martinius, H.; Hermann, F.; et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 2007, 15, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, J.; Silva, F.; Freitas-Vieira, A.; Santa-Marta, M.; Malho, R.; Yang, X.; Gabuzda, D.; Barbas, C., III. Functional neutralization of HIV-1 VIF protein by intracellular immunization inhibits reverse transcription and viral replication. J. Biol. Chem. 2002, 277, 32036–32045. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Ishikawa, T.; Okui, N.; Kobayashi, N.; Kanda, T.; Shimada, T.; Miyake, K.; Yoshiike, K. Inhibition of replication of HIV-1 at both early and late stages of the viral life cycle by single-chain antibody against viral integrase. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1999, 20, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Levy-Mintz, P.; Duan, L.; Zhang, H.; Hu, B.; Dornadula, G.; Zhu, M.; Kulkosky, J.; Bizub-Bender, D.; Skalka, A.M.; Pomerantz, R.J. Intracellular expression of single-chain variable fragments to inhibit early stages of the viral life cycle by targeting human immunodeficiency virus type 1 integrase. J. Virol. 1996, 70, 8821–8832. [Google Scholar] [PubMed]
- Mhashilkar, A.M.; Bagley, J.; Chen, S.Y.; Szilvay, A.M.; Helland, D.G.; Marasco, W.A. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. EMBO J. 1995, 14, 1542–1551. [Google Scholar] [PubMed]
- Shaheen, F.; Duan, L.; Zhu, M.; Bagasra, O.; Pomerantz, R.J. Targeting human immunodeficiency virus type 1 reverse transcriptase by intracellular expression of single-chain variable fragments to inhibit early stages of the viral life cycle. J. Virol. 1996, 70, 3392–3400. [Google Scholar] [PubMed]
- Braun, S.E.; Taube, R.; Zhu, Q.; Wong, F.E.; Murakami, A.; Kamau, E.; Dwyer, M.; Qiu, G.; Daigle, J.; Carville, A.; et al. In vivo selection of CD4+ T cells transduced with a γ-retroviral vector expressing a single-chain intrabody targeting HIV-1 tat. Hum. Gene Ther. 2012, 23, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Schroers, R.; Davis, C.M.; Wagner, H.J.; Chen, S.Y. Lentiviral transduction of human T-lymphocytes with a RANTES intrakine inhibits human immunodeficiency virus type 1 infection. Gene Ther. 2002, 9, 889–897. [Google Scholar] [PubMed]
- Simmons, G.; Reeves, J.D.; Hibbitts, S.; Stine, J.T.; Gray, P.W.; Proudfoot, A.E.; Clapham, P.R. Co-receptor use by HIV and inhibition of HIV infection by chemokine receptor ligands. Immunol. Rev. 2000, 177, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.G.; Zhang, X.; Torti, F.; Chen, S.Y. Anti-HIV type 1 activity of wild-type and functional defective RANTES intrakine in primary human lymphocytes. Hum. Gene Ther. 1998, 9, 2005–2018. [Google Scholar] [CrossRef] [PubMed]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors CCR5 and CXCR4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Wang, J.; Tilton, J.C.; Miller, J.C.; Kim, K.A.; Rebar, E.J.; Sherrill-Mix, S.A.; Patro, S.C.; Secreto, A.J.; Jordan, A.P.; et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011, 7, e1002020. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wang, J.; Crain, K.; Fearns, C.; Kim, K.A.; Hua, K.L.; Gregory, P.D.; Holmes, M.C.; Torbett, B.E. Zinc-finger nuclease editing of human CXCR4 promotes HIV-1 CD4+ T cell resistance and enrichment. Mol. Ther. 2012, 20, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wang, P.; Ding, D.; Li, L.; Wang, H.; Ma, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, X.; et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013, 41, 7771–7782. [Google Scholar] [CrossRef] [PubMed]
- Badia, R.; Pauls, E.; Riveira-Munoz, E.; Clotet, B.; Este, J.A.; Ballana, E. Zinc-finger endonuclease targeting PSIP-1 inhibits HIV-1 integration. Antimicrob. Agents Chemother. 2014, 58, 4318–4327. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Goncalves, M.A. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Fadel, H.J.; Morrison, J.H.; Saenz, D.T.; Fuchs, J.R.; Kvaratskhelia, M.; Ekker, S.C.; Poeschla, E.M. TALEN knockout of the PSIP1 gene in human cells: Analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J. Virol. 2014, 88, 9704–9717. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Towers, G.J.; Qasim, W. Gene therapy strategies to exploit TRIM derived restriction factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-De-Castillia, C.; Grutter, C.; Martinetti, G.; Mazzucchelli, L.; Grutter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Invest. 2009, 119, 3035–3047. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Wang, L.; Joyce, M.G.; Yang, Z.Y.; Balazs, A.B.; Cheng, C.; Ko, S.Y.; Kong, W.P.; Rudicell, R.S.; Georgiev, I.S.; et al. Broadly neutralizing human immunodeficiency virus type 1 antibody gene transfer protects non-human primates from mucosal simian-human immunodeficiency virus infection. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.R.; Kattenhorn, L.M.; Kondur, H.R.; von, S.M.; Dorfman, T.; Chiang, J.J.; Haworth, K.G.; Decker, J.M.; Alpert, M.D.; Bailey, C.C.; et al. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature 2015, 519, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Sanders, R.W. Gene therapy as a vaccine for HIV-1. Expert Opin. Biol. Ther. 2012, 12, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.B.; Chen, J.; Hong, C.M.; Rao, D.S.; Yang, L.; Baltimore, D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 2012, 481, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.B.; Ouyang, Y.; Hong, C.M.; Chen, J.; Nguyen, S.M.; Rao, D.S.; An, D.S.; Baltimore, D. Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission. Nat. Med. 2014, 20, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Humeau, L.M.; Binder, G.K.; Lu, X.; Slepushkin, V.; Merling, R.; Echeagaray, P.; Pereira, M.; Slepushkina, T.; Barnett, S.; Dropulic, L.K.; et al. Efficient lentiviral vector-mediated control of HIV-1 replication in CD4 lymphocytes from diverse HIV+ infected patients grouped according to CD4 count and viral load. Mol. Ther. 2004, 9, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, Q.; Binder, G.K.; Chen, Z.; Slepushkina, T.; Rossi, J.; Dropulic, B. Antisense-mediated inhibition of human immunodeficiency virus (HIV) replication by use of an HIV type 1-based vector results in severely attenuated mutants incapable of developing resistance. J. Virol. 2004, 78, 7079–7088. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.; Baker, B.F.; Cook, P.D.; Zounes, M.; Buckheit, R.W., Jr.; Germany, J.; Ecker, D.J. Inhibition of HIV-LTR gene expression by oligonucleotides targeted to the TAR element. Nucleic Acids Res. 1991, 19, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Mitsuyasu, R.T.; Rosenblatt, J.D.; Ngok, F.K.; Bakker, A.; Cole, S.; Chorn, N.; Lin, L.S.; Bristol, G.; Boyd, M.P.; et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: Myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum. Gene Ther. 2004, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Michienzi, A.; Castanotto, D.; Lee, N.; Li, S.; Zaia, J.A.; Rossi, J.J. RNA-mediated inhibition of HIV in a gene therapy setting. Ann. N. Y. Acad. Sci. 2003, 1002, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Wong-Staal, F.; Poeschla, E.M.; Looney, D.J. A controlled, Phase 1 clinical trial to evaluate the safety and effects in HIV-1 infected humans of autologous lymphocytes transduced with a ribozyme that cleaves HIV-1 RNA. Hum. Gene Ther. 1998, 9, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, G.; Maijgren-Steffensson, C.; Ahrlund-Richter, L. Efficacy and mode of action of hammerhead and hairpin ribozymes against various HIV-1 target sites. Mol. Ther. 2004, 10, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Yamada, O.; Kraus, G.; Luznik, L.; Yu, M.; Wong-Staal, F. A chimeric human immunodeficiency virus type 1 (HIV-1) minimal Rev response element-ribozyme molecule exhibits dual antiviral function and inhibits cell-cell transmission of HIV-1. J. Virol. 1996, 70, 1596–1601. [Google Scholar] [PubMed]
- Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Analysis of trans-acting response decoy RNA-mediated inhibition of human immunodeficiency virus type 1 transactivation. J. Virol. 1991, 65, 6811–6816. [Google Scholar] [PubMed]
- Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 1990, 63, 601–608. [Google Scholar] [CrossRef]
- Lee, T.C.; Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Overexpression of RRE-derived sequences inhibits HIV-1 replication in CEM cells. New Biol. 1992, 4, 66–74. [Google Scholar] [PubMed]
- Michienzi, A.; Li, S.; Zaia, J.A.; Rossi, J.J. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc. Natl. Acad. Sci. USA 2002, 99, 14047–14052. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Banda, N.; Lee, N.S.; Rossi, J.; Akkina, R. RNA-based anti-HIV-1 gene therapeutic constructs in SCID-hu mouse model. Mol. Ther. 2002, 6, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Li, M.J.; Remling, L.; Rossi, J.; Akkina, R. Lentiviral transduction of Tar Decoy and CCR5 ribozyme into CD34+ progenitor cells and derivation of HIV-1 resistant T cells and macrophages. AIDS Res. Ther. 2004, 1. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34+ cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Banerjea, A.; Akkina, R. Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides 2003, 13, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Liu, X.; He, J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Ther. 2005, 12, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antiviral Res. 2011, 89, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Surabhi, R.M.; Gaynor, R.B. RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus type 1 replication. J. Virol. 2002, 76, 12963–12973. [Google Scholar] [CrossRef] [PubMed]
- Von Eije, K.J.; Ter Brake, O.; Berkhout, B. Stringent testing identifies highly potent and escape-proof anti-HIV short hairpin RNAs. J. Gene Med. 2009, 11, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, E.M.; Vink, M.; Haasnoot, P.C.; Das, A.T.; Berkhout, B. A conditionally replicating HIV-based vector that stably expresses an antiviral shRNA against HIV-1 replication. Mol. Ther. 2006, 14, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Kamata, M.; Kittipongdaja, P.; Chen, K.N.; Kim, S.; Pang, S.; Boyer, J.; Qin, F.X.; An, D.S.; Chen, I.S. Characterization of a potent non-cytotoxic shRNA directed to the HIV-1 co-receptor CCR5. Genet. Vaccines Ther. 2009, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [PubMed]
- Berkhout, B.; Liu, Y.P. Towards improved shRNA and miRNA reagents as inhibitors of HIV-1 replication. Future Microbiol. 2014, 9, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Schopman, N.C.; Berkhout, B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013, 41, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Karg, M.; Harwig, A.; Herrera-Carrillo, E.; Jongejan, A.; van kampen, A.; Berkhout, B. Mechanistic insights on the dicer-independent Ago2-mediated processing of AgoshRNAs. RNA Biol. 2014, 12, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.; Van, D.R.; Carpio, L.; Guendel, I.; Kehn-Hall, K.; Chevalier, S.; Narayanan, A.; Luu, T.; Lee, N.; Klase, Z.; et al. Absence of DICER in monocytes and its regulation by HIV-1. J. Biol. Chem. 2010, 285, 31930–31943. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Herrera-Carrillo, E.; Jongejan, A.; van kampen, A.; Berkhout, B. Deep sequence analysis of AgoshRNA processing reveals 3′ A addition and trimming. Mol. Ther. Nucleic Acids 2015, 4. in press. [Google Scholar] [CrossRef]
- Gu, S.; Jin, L.; Zhang, Y.; Huang, Y.; Zhang, F.; Valdmanis, P.N.; Kay, M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of Dicer processing in vivo. Cell 2012, 151, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Dueck, A.; Ziegler, C.; Eichner, A.; Berezikov, E.; Meister, G. MicroRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012, 40, 9850–9862. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Harwig, A.; Liu, Y.P.; Berkhout, B. Probing the shRNA characteristics that hinder Dicer recognition and consequently allow Ago-mediated processing and AgoshRNA activity. RNA 2014, 20, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Harwig, A.; Berkhout, B. Towards optimization of AgoshRNA molecules that use a non-canonical RNAi pathway: Variations in the top and bottom base pairs. RNA Biol. 2015, 12, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Akkina, R. Human immunodeficiency virus type 1 restriction by human-rhesus chimeric tripartite motif 5α (TRIM 5α) in CD34+ cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum. Gene Ther. 2008, 19, 217–228. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34+ cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; ‘t Hooft, K.; Liu, Y.P.; Centlivre, M.; von Eije, K.J.; Berkhout, B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol. Ther. 2008, 16, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Haasnoot, J.; Ter Brake, O.; Berkhout, B.; Konstantinova, P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008, 36, 2811–2824. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.L.; Ahmed, I.; Steel, L.F. RNA polymerase III can drive polycistronic expression of functional interfering RNAs designed to resemble microRNAs. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Bharaj, P.; Abraham, S.; Ma, H.; Yi, G.; Ye, C.; Dang, Y.; Manjunath, N.; Wu, H.; Shankar, P. Multiplexing seven miRNA-based shRNAs to suppress HIV replication. Mol. Ther. 2015, 23, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; von Eije, K.J.; Schopman, N.C.; Westerink, J.T.; Ter Brake, O.; Haasnoot, J.; Berkhout, B. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol. Ther. 2009, 17, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Haasnoot, J.; Berkhout, B. Design of extended short hairpin RNAs for HIV-1 inhibition. Nucleic Acids Res. 2007, 35, 5683–5693. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, P.; de Vries, W.; Haasnoot, J.; Ter Brake, O.; de Haan, P.; Berkhout, B. Inhibition of human immunodeficiency virus type 1 by RNA interference using long-hairpin RNA. Gene Ther. 2006, 13, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.; Arbuthnot, P.; Weinberg, M.S. Deriving four functional anti-HIV siRNAs from a single Pol III-generated transcript comprising two adjacent long hairpin RNA precursors. Nucleic Acids Res. 2010, 38, 6652–6663. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Berkhout, B. Combinatorial RNAi strategies against HIV-1 and other escape-prone viruses. Int. J. BioSci. Technol. 2008, 1, 1–10. [Google Scholar]
- Boutimah, F.; Eekels, J.J.; Liu, Y.P.; Berkhout, B. Antiviral strategies combining antiretroviral drugs with RNAi-mediated attack on HIV-1 and cellular co-factors. Antiviral Res. 2013, 98, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Schopman, N.C.; Braun, A.; Berkhout, B. Directed HIV-1 evolution of protease inhibitor resistance by second-generation short hairpin RNAs. Antimicrob. Agents Chemother. 2012, 56, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Li, M.J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and efficacy of a lentiviral vector containing three anti-HIV genes—CCR5 ribozyme, Tat-Rev siRNA, and TAR decoy—In SCID-hu mouse-derived T cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E.; Chen, R.X.; McGee, J.; Nacey, C.; Pollard, R.B.; Abedi, M.; Bauer, G.; Nolta, J.A.; Anderson, J.S. Generation of an HIV-1-resistant immune system with CD34+ hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J. Virol. 2012, 86, 5719–5729. [Google Scholar] [CrossRef] [PubMed]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von, L.D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Neff, C.P.; Zhou, J.; Remling, L.; Kuruvilla, J.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Rossi, J.J.; Akkina, R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4+ T cell decline in humanized mice. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Current progress in the development of RNAi-based therapeutics for HIV-1. Gene Ther. 2011, 18, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS. Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein, G. J. Virol. 1996, 70, 2581–2585. [Google Scholar] [PubMed]
- Trono, D. HIV accessory proteins: Leading roles for the supporting cast. Cell 1995, 82, 189–192. [Google Scholar] [CrossRef]
- Miyoshi, H.; Blomer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [PubMed]
- Bukovsky, A.A.; Song, J.P.; Naldini, L. Interaction of human immunodeficiency virus-derived vectors with wild-type virus in transduced cells. J. Virol. 1999, 73, 7087–7092. [Google Scholar] [PubMed]
- Liu, Y.P.; Berkhout, B. Lentiviral delivery of RNAi effectors against HIV-1. Curr. Top. Med. Chem. 2009, 9, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Vink, M.A.; Westerink, J.T.; Ramirez de, A.E.; Konstantinova, P.; Ter Brake, O.; Berkhout, B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010, 16, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Berkhout, B. Lentiviral vectors that carry anti-HIV shRNAs: Problems and solutions. J. Gene Med. 2007, 9, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; von Eije, K.J.; Berkhout, B. Probing the sequence space available for HIV-1 evolution. AIDS 2008, 22, 1875–1877. [Google Scholar] [CrossRef] [PubMed]
- Von Eije, K.J.; Ter Brake, O.; Berkhout, B. Human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J. Virol. 2008, 82, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Pasternak, A.O.; Schut, A.M.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. A competitive cell growth assay for the detection of subtle effects of gene transduction on cell proliferation. Gene Ther. 2012, 19, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Centlivre, M.; Legrand, N.; Klamer, S.; Liu, Y.P.; Eije, K.J.; Bohne, M.; Rijnstra, E.S.; Weijer, K.; Blom, B.; Voermans, C.; et al. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol. Ther. Nucleic Acids 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Zinc finger nuclease-mediated CCR5 knockout hematopoietic stem cell transplantation controls HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Liu, Y.P.; Berkhout, B. The impact of unprotected T cells in RNAi-based gene therapy for HIV-AIDS. Mol. Ther. 2014, 22, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. The impact of HIV-1 genetic diversity on the efficacy of a combinatorial RNAi-based gene therapy. Gene Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Binder-Scholl, G.; Mukherjee, R.; Brady, T.; Rebello, T.; Humeau, L.; Kalos, M.; Papasavvas, E.; Montaner, L.J.; et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 2013, 121, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Daw, S.; Wynn, R.; Wallace, H. Management of relapsed and refractory classical Hodgkin lymphoma in children and adolescents. Br. J. Haematol. 2011, 152, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.J.; Horning, S.J. Autologous hematopoietic cell transplantation in Hodgkin’s disease. Biol. Blood Marrow Transpl. 2000, 6, 289–300. [Google Scholar] [CrossRef]
- Kang, E.M.; De, W.M.; Malech, H.; Morgan, R.A.; Carter, C.; Leitman, S.F.; Childs, R.; Barrett, A.J.; Little, R.; Tisdale, J.F. Gene therapy-based treatment for HIV-positive patients with malignancies. J. Hematother. Stem. Cell Res. 2002, 11, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Savkovic, B.; MacPherson, J.L.; Zaunders, J.; Kelleher, A.D.; Knop, A.E.; Pond, S.; Evans, L.; Symonds, G.; Murray, J.M. T-lymphocyte perturbation following large-scale apheresis and hematopoietic stem cell transplantation in HIV-infected individuals. Clin. Immunol. 2012, 144, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-Carrillo, E.; Berkhout, B. Bone Marrow Gene Therapy for HIV/AIDS. Viruses 2015, 7, 3910-3936. https://doi.org/10.3390/v7072804
Herrera-Carrillo E, Berkhout B. Bone Marrow Gene Therapy for HIV/AIDS. Viruses. 2015; 7(7):3910-3936. https://doi.org/10.3390/v7072804
Chicago/Turabian StyleHerrera-Carrillo, Elena, and Ben Berkhout. 2015. "Bone Marrow Gene Therapy for HIV/AIDS" Viruses 7, no. 7: 3910-3936. https://doi.org/10.3390/v7072804
APA StyleHerrera-Carrillo, E., & Berkhout, B. (2015). Bone Marrow Gene Therapy for HIV/AIDS. Viruses, 7(7), 3910-3936. https://doi.org/10.3390/v7072804