
CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape

Abstract
:1. Introduction
2. Strategies to Down Regulate and Inhibit Synthesis of CCR5
- ZFNs are engineered proteins with zinc finger domains that can bind to targeted regions of DNA and conduct gene editing via double strand DNA breaks. Locations of DNA breakage can undergo either non homologous end joining or homologous recombination by insertion of donor DNA, both of which lead to mutations of the gene [10,11]. Tebas et al. 2014 report that they safely used ZFN to modify CCR5 of autologous CD4+ T cells used to treat HIV positive patients [12]. A trial currently recruiting participants uses ZFN to modify CCR5 in CD4+ T cells with increasing doses of cyclophosphamide to promote engraftment (NIH clinical trial NCT01543152).
- Recently, more advanced techniques in gene suppression have been established. For example TALENs can successfully target sites in the CCR5 loci with less cytotoxicity compared to ZFNs [13]. Similar to ZFNs the TALENs binding domains recognize and cleave specific DNA by using the previously fused endonuclease part of this complex. This can be carried by adenoviruses. Unlike ZFNs, TALENs recognize only one nucleotide instead of three [14]. Mock et al. 2015report the use of TALEN to knockout CCR5. This technique was shown to protect CCR5 T cells from R5-tropic HIV [15]. However, it should be noted that Mock et al. used transient transfection methods and only report one long term (12 days) exposure to HIV which showed incomplete suppression of HIV replication [15].
- To combat hostile agents, bacteria harbor an effective defense mechanism: the CRISPR/Cas9 system. It works as an intracellular defense system against plasmids or viral DNA by causing site specific double strand breaks. The CRISPR/Cas9 system was adapted as a molecular tool to break down single human genes. In fact, it has been successfully tested in human cells. There, Kim et al. were able to affect 18% of the CCR5 genes, a percentage that may be essential for a successful clinical use [16].
- siRNAs are small pieces of synthetically derived RNA that guide an endonuclease to cleave a targeted site in mRNA. siRNAs are exogenously synthesized, fragile (21-23-mer short), and prone to quick degradation. They need to be administered in high dosages to reach the targeted RNA of interest. siRNAs have been used to target CCR5 in several studies, however, they resulted in an incomplete inhibition of HIV-1 and off-target effects [17,18]. Even though siRNAs are target specific, viral escape mutants have been documented to make their use less than ideal for clinical applications [19,20].
- shRNAs differ from siRNAs by virtue of a more stable secondary structure (hairpin loop). Such structure enables researchers to use only a small dose of it to reach the target. Also, shRNAs can be expressed in the target cell’s nucleus via a gene cassette . Lentiviral vectors can efficiently express shRNAs. In fact, they have recently been shown to inhibit HIV in human cells and animal models [21,22,23]. There is currently an open clinical trial that employs a lentiviral vector to express shRNA to CCR5 in combination with C46 (NIH clinical trial NCT01734850).
- Translation at the mRNA level can be inhibited by antisense RNAs; single stranded complementary RNAs. Li et al. 2006 report the downregulation of CCR5 by recombinant adenovirus expressing antisense CCR5 RNA [24]. However, the authors note that the vector is only transiently expressed and, if used for treatment, would require frequent dosing due to elimination by the host’s immune system. Therefore, making this technique less than an ideal gene therapy.
- Ribozymes are small catalytic RNA molecules that can act like protein enzymes and be engineered to target specific RNA sequences [25,26,27,28]. Three clinical trials have positively showed the safety, feasibility, and long term stability of using ribozymes targeted to tat and tat-vpr HIV elements [27,29,30]. However, the transduction efficiency left room for improvement. Also, none of the trial participants underwent myeloablation. Since then advances have been made in gene transfer technology. Currently, myeloablation is being tested in conjunction with gene therapy to treat HIV. DiGiusto et al. 2010 reports a combinatorial approach that includes Tat/Rev shRNA, Tat activation-response region (TAR) decoy, and CCR5 ribozyme used to genetically modify autologous peripheral blood derived CD34+ HSC from AIDS patients [31]. Reports from this ongoing clinical trial revealed that the stability of CCR5 ribozyme was maintained up to 24 months and noted the need for improvement of transduction processes (NIH clinical trial NCT00569985).
3. Strategies to Inhibit Cell Surface Expression of CCR5
- Intrakines are intracellular chemokines capable of targeting newly synthesized CCR5 in the endoplasmic reticulum by blocking transport of CCR5 to the cell surface [7,8,9]. Probably one of the first attempts to inhibit the use of chemokine co-receptors to generate HIV resistant cells was published in 1997. Here, the group used intrakines against CCR5 [32]. However, the major problem in this approach was reported to be an incomplete inhibition of CCR5.
- In contrast to the use of intrakines, the use of intrabodies provided a more complete inhibition of CCR5. Intrabody is an intracellular single chain variable fragment antibody (scFv) that can bind to a protein of interest potentially rendering it dysfunctional. Steinberger et al. 2000 developed a CCR5 specific intrabody capable of blocking surface expression of CCR5, thereby protecting gene modified cells from HIV infection [33].
4. Differences between the “Berlin” and the “Essen Patient”

{kind=link}
| Berlin patient | Essen patient | |
|---|---|---|
| Age, sex | 40 years, male | 27 years, male |
| Malignancy | acute myeloid leukemia | anaplastic large T-cell lymphoma |
| Time between infection and ART | 7 years | 3 years |
| Time between infection and Tx | 12 years | 5 years |
| Tx regimen | intermediate intensity | myeloablative + 12 Gy TBI |
| Immunosuppression | ATG, CSA, MTX, MMF | ATG, CSA, MTX, |
| GVHD | max. grade 1 (skin) | max. grade 1–2 (skin) |
| Engraftment | day +11 | day +39 |
| ART discontinuation | on day of Tx | 7 days before Tx |
| V3 sequence | CIRPNNNTRKGIHIGPGRAFYTTGEIIGDIRQAHC | CTRPNNNTRKGIPLGPGKVFYAT-EIIRDIRKAYC |
| >3 months prior Tx * | ||
| X4 prediction ** | ||
| 3months prior Tx | capable | intermediate |
| Immediate prior Tx | nd | capable |
5. HIV Tropism and CCR5 Suppression
5.1. Entry Inhibitors of HIV
5.2. CCR5 Gene Therapy of HIV Disease
5.3. Problems Unsolved: Alternative Chemokine Receptors
6. Size of the Reservoir and Probability of Rebound
- Two HIV+ patients received allogeneic stem cell transplantation (CCR5 wild type graft). Both received antiretroviral medication—2.5 and 4.3 years after transplantation. Both developed a continuous sero-deconversion concerning anti-HIV antibodies indicating that no significant replication had occurred in this time. Otherwise the anti-HIV antibodies would have still been measurable. Furthermore, tissue samples and outgrowth assays from peripheral blood were all negative. It has been speculated that allogeneic transplantation led to elimination of the viral reservoir by turnover of latently infected cells in combination with (a postulated) cytotoxic effect from graft T-cells against the reservoirs (graft versus HIV effect). However, both patients rebounded with HIV after three and seven weeks, respectively. Interestingly, the time between discontinuation of medication and rebound was unusually long, indicating that both patients had a very small reservoir but had not eradicated the virus and that latently infected cells “hid” in undetectable niches [47,48].
- A perinatal infected child received antiviral therapy rapidly (30 h) after delivery and was taken off medication after 18 months. Surprisingly, the child was found without HIV replication after ART discontinuation. Occasionally very small traces of virus material were detected but no replication competent virus was found. The patient displayed no immunological reaction in terms of anti-HIV antibody production. It was assumed that early initiation of ART led to a minimized reservoir and that the immune system was able to control this small number of infected cells. However, approximately 27.6 months after discontinuation of ART the child was found with active HIV replication [49].
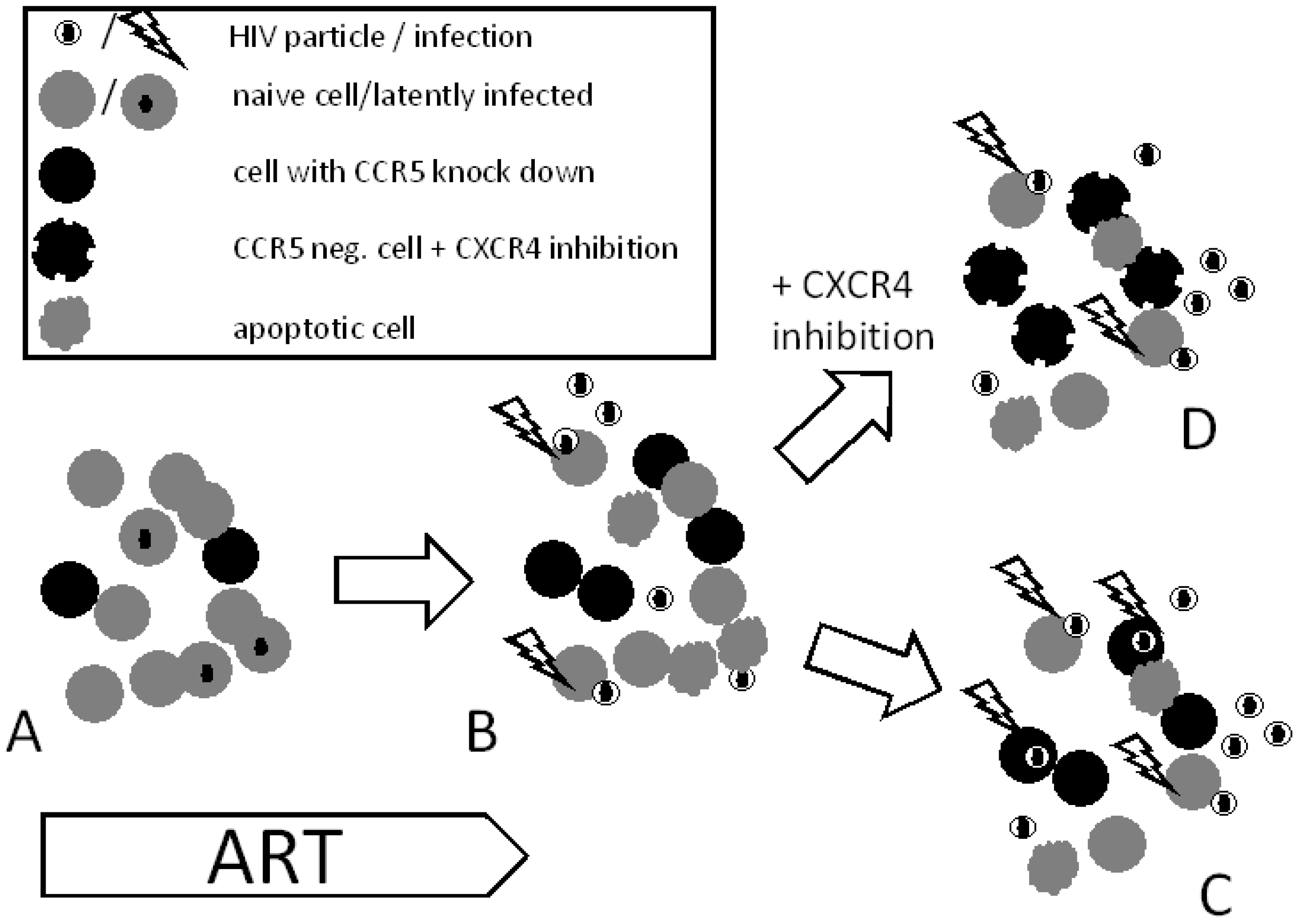
7. Strategies to Minimize the Viral Reservoir
7.1. Chemotherapy
7.2. Using HIV Cytopathic Effects
7.3. Agents Toxic to Viral Reservoir
7.4. Gene Therapy
8. Strategies to Overcome Viral Rebound
8.1. Additional HIV Entry Inhibition
8.2. CXCR4 Blockage
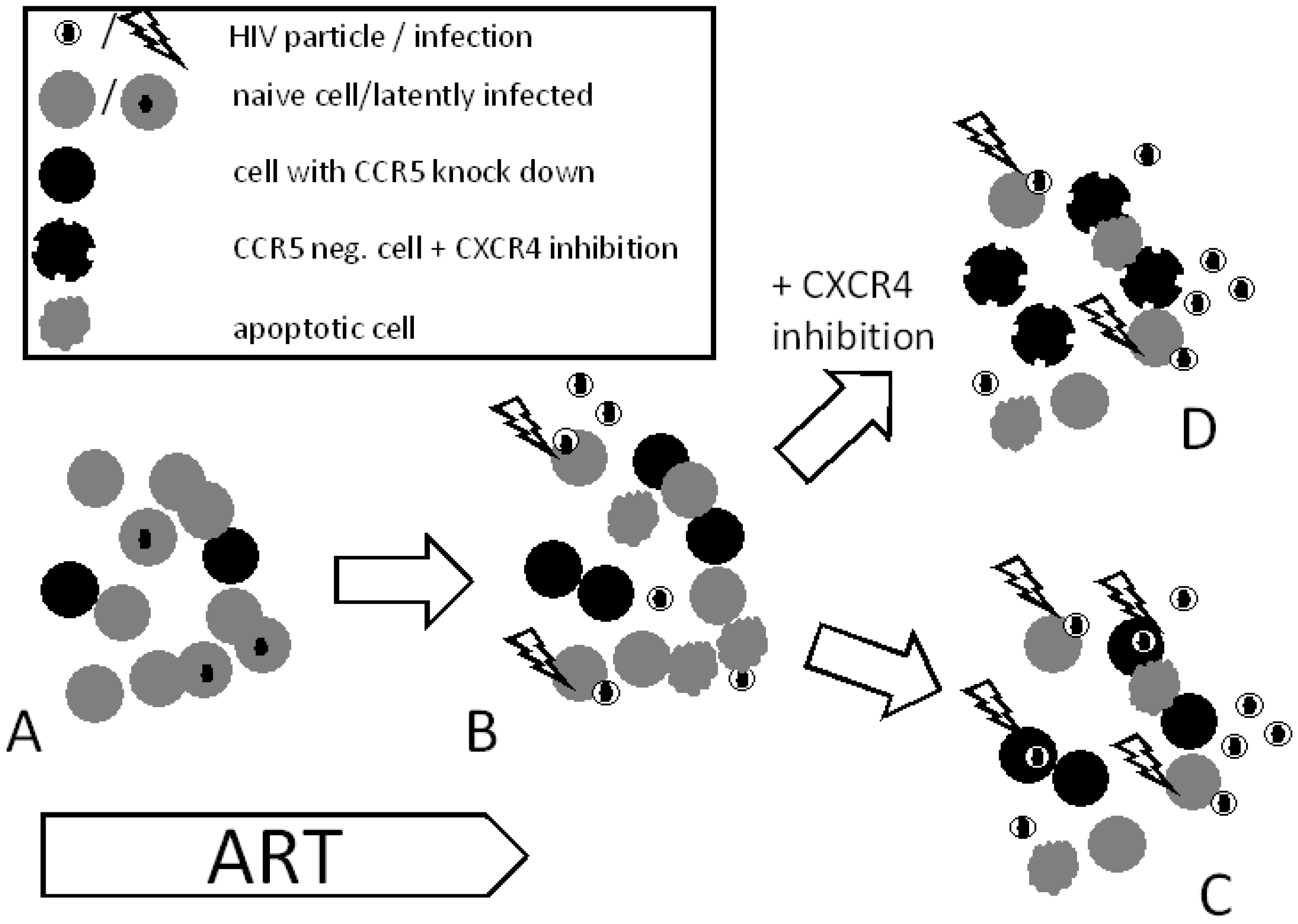
8.3. Chemokine Receptor Down-regulation
9. Summary and Outlook
Author Contributions
Conflicts of Interest
References
- Doms, R.W. Beyond receptor expression: The influence of receptor conformation, density, and affinity in HIV-1 infection. Virology 2000, 276, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A. Thirty years on: HIV receptor gymnastics and the prevention of infection. BMC Biol. 2013, 11, e57. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, G.; Tresoldi, E.; Bjorndal, A.; Fredriksson, R.; Colognesi, C.; Deng, H.K.; Malnati, M.S.; Plebani, A.; Siccardi, A.G.; Littman, D.R.; et al. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 1997, 3, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Nazari, R.; Joshi, S. CCR5 as target for HIV-1 gene therapy. Curr. Gene Ther. 2008, 8, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Schroers, R.; Davis, C.M.; Wagner, H.J.; Chen, S.Y. Lentiviral transduction of human T-lymphocytes with a RANTES intrakine inhibits human immunodeficiency virus type 1 infection. Gene Ther. 2002, 9, 889–897. [Google Scholar] [PubMed]
- Bai, X.; Chen, J.D.; Yang, A.G.; Torti, F.; Chen, S.Y. Genetic co-inactivation of macrophage- and T-tropic HIV-1 chemokine coreceptors CCR-5 and CXCR-4 by intrakines. Gene Ther. 1998, 5, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Luis Abad, J.; Gonzalez, M.A.; del Real, G.; Mira, E.; Manes, S.; Serrano, F.; Bernad, A. Novel interfering bifunctional molecules against the CCR5 coreceptor are efficient inhibitors of HIV-1 infection. Mol. Ther. 2003, 8, 475–484. [Google Scholar] [CrossRef]
- Li, L.; Wu, L.P.; Chandrasegaran, S. Functional domains in Fok I restriction endonuclease. Proc. Natl. Acad. Sci. USA 1992, 89, 4275–4279. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Goncalves, M.A. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41, e63. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; Machowicz, R.; Hauber, I.; Horn, S.; Abramowski, P.; Berdien, B.; Hauber, J.; Fehse, B. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 2015, 43, 5560–5571. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A.; Gutierrez, A.; Armand-Ugon, M.; Blanco, J.; Parera, M.; Gomez, J.; Clotet, B.; Este, J.A. Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS 2002, 16, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.F.; An, D.S.; Chen, I.S.; Baltimore, D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. USA 2003, 100, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.P.; Levin, B.R.; Zhang, J.; Sahakyan, A.; Boyer, J.; Carroll, M.V.; Colon, J.C.; Keech, N.; Rezek, V.; Bristol, G.; et al. Engineering Cellular Resistance to HIV-1 Infection In Vivo Using a Dual Therapeutic Lentiviral Vector. Mol. Ther. Nucleic Acids 2015, 4, e236. [Google Scholar] [CrossRef] [PubMed]
- Wolstein, O.; Boyd, M.; Millington, M.; Impey, H.; Boyer, J.; Howe, A.; Delebecque, F.; Cornetta, K.; Rothe, M.; Baum, C.; et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014, 1, e11. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Kamata, M.; Kittipongdaja, P.; Chen, K.N.; Kim, S.; Pang, S.; Boyer, J.; Qin, F.X.; An, D.S.; Chen, I.S. Characterization of a potent non-cytotoxic shRNA directed to the HIV-1 co-receptor CCR5. Genet. Vaccines Ther. 2009, 7, e8. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yu, M.; Bai, L.; Bu, D.; Xu, X. Downregulation of CCR5 expression on cells by recombinant adenovirus containing antisense CCR5, a possible measure to prevent HIV-1 from entering target cells. J. Acquir. Immune Defic. Syndr. 2006, 43, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Sarver, N.; Cantin, E.M.; Chang, P.S.; Zaia, J.A.; Ladne, P.A.; Stephens, D.A.; Rossi, J.J. Ribozymes as potential anti-HIV-1 therapeutic agents. Science 1990, 247, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.J. Targeted cleavage: Tuneable cis-cleaving ribozymes. Proc. Natl. Acad. Sci. USA 2007, 104, 14881–14882. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Q.; Wang, L.; Gerlach, W.L.; Symonds, G. Target sequence-specific inhibition of HIV-1 replication by ribozymes directed to tat RNA. Nucleic Acids Res. 1995, 23, 2909–2913. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Mitsuyasu, R.T.; Rosenblatt, J.D.; Ngok, F.K.; Bakker, A.; Cole, S.; Chorn, N.; Lin, L.S.; Bristol, G.; Boyd, M.P.; et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: Myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum. Gene Ther. 2004, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.G.; Bai, X.; Huang, X.F.; Yao, C.; Chen, S. Phenotypic knockout of HIV type 1 chemokine coreceptor CCR-5 by intrakines as potential therapeutic approach for HIV-1 infection. Proc. Natl. Acad. Sci. USA 1997, 94, 11567–11572. [Google Scholar] [CrossRef] [PubMed]
- Steinberger, P.; Andris-Widhopf, J.; Buhler, B.; Torbett, B.E.; Barbas, C.F., 3rd. Functional deletion of the CCR5 receptor by intracellular immunization produces cells that are refractory to CCR5-dependent HIV-1 infection and cell fusion. Proc. Natl. Acad. Sci. USA 2000, 97, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Petz, L.D.; Redei, I.; Bryson, Y.; Regan, D.; Kurtzberg, J.; Shpall, E.; Gutman, J.; Querol, S.; Clark, P.; Tonai, R.; et al. Hematopoietic cell transplantation with cord blood for cure of HIV infections. Biol. Blood Marrow Transplant. 2013, 19, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hutter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Verheyen, J.; Beelen, D.W.; Horn, P.A.; Heinold, A.; Kaiser, R.; Trenschel, R.; Schadendorf, D.; Dittmer, U.; Esser, S. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N. Engl. J. Med. 2014, 371, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Zaia, J.A. Allogeneic haematopoietic stem cell transplantation in patients with human immunodeficiency virus: The experiences of more than 25 years. Clin. Exp. Immunol. 2011, 163, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Flepisi, B.T.; Bouic, P.; Sissolak, G.; Rosenkranz, B. Drug-drug interactions in HIV positive cancer patients. Biomed. Pharmacother. 2014, 68, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Fatkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; et al. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.; Braverman, M.S.; Taillon, B.E.; Desany, B.; James, I.; Harrigan, P.R.; Lewis, M.; Robertson, D.L. Detection of low-frequency pretherapy chemokine (CXC motif) receptor 4 (CXCR4)-using HIV-1 with ultra-deep pyrosequencing. AIDS 2009, 23, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.; Churchill, M.J.; Keane, N.; Sterjovski, J.; Ellett, A.M.; Purcell, D.F.; Poumbourios, P.; Kol, C.; Wang, B.; Saksena, N.K.; et al. Genetic and functional analysis of R5X4 human immunodeficiency virus type 1 envelope glycoproteins derived from two individuals homozygous for the CCR5delta32 allele. J. Virol. 2006, 80, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hanhauser, E.; Hu, Z.; Stellbrink, H.J.; Noah, C.; Martin, J.N.; Deeks, S.G.; Kuritzkes, D.R.; Pereyra, F. Viremic control and viral coreceptor usage in two HIV-1-infected persons homozygous for CCR5 Delta32. AIDS 2015, 29, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Cortes, F.H.; Passaes, C.P.; Bello, G.; Teixeira, S.L.; Vorsatz, C.; Babic, D.; Sharkey, M.; Grinsztejn, B.; Veloso, V.; Stevenson, M.; et al. HIV controllers with different viral load cutoff levels have distinct virologic and immunologic profiles. J. Acquir. Immune Defic. Syndr. 2015, 68, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hu, Z.; Li, J.Z.; Sciaranghella, G.; Busch, M.P.; Keating, S.M.; Gallien, S.; Lin, N.H.; Giguel, F.F.; Lavoie, L.; et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 2013, 207, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Gay, H.; Ziemniak, C.; Sanborn, K.B.; Somasundaran, M.; Rainwater-Lovett, K.; Mellors, J.W.; Rosenbloom, D.; Persaud, D. Viremic relapse after HIV-1 remission in a perinatally infected child. N. Engl. J. Med. 2015, 372, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Eisele, E.E.; Rabi, S.A.; Lai, J.; Chioma, S.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog. 2013, 9, e1003398. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J.A.; Forman, S.J. Transplantation in HIV-infected subjects: Is cure possible? Hematology Am. Soc. Hematol. Educ. Program 2013, 2013, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Margolis, D.M. Emerging strategies to deplete the HIV reservoir. Curr. Opin. Infect. Dis. 2014, 27, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Chirullo, B.; Sgarbanti, R.; Limongi, D.; Shytaj, I.L.; Alvarez, D.; Das, B.; Boe, A.; DaFonseca, S.; Chomont, N.; Liotta, L.; et al. A candidate anti-HIV reservoir compound, auranofin, exerts a selective 'anti-memory' effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis. 2013, 4, e944. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, I.; Hauber, I.; Hauber, J.; Buchholz, F. HIV-1 proviral DNA excision using an evolved recombinase. Science 2007, 316, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Hildinger, M.; Dittmar, M.T.; Schult-Dietrich, P.; Fehse, B.; Schnierle, B.S.; Thaler, S.; Stiegler, G.; Welker, R.; von Laer, D. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J. Virol. 2001, 75, 3038–3042. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Schiedlmeier, B.; Kuhlcke, K.; Verstegen, M.; Margison, G.P.; Li, Z.; Kamino, K.; Bohne, J.; Alexandrov, A.; Hermann, F.G.; et al. Towards hematopoietic stem cell-mediated protection against infection with human immunodeficiency virus. Gene Ther. 2006, 13, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Younan, P.M.; Polacino, P.; Kowalski, J.P.; Peterson, C.W.; Maurice, N.J.; Williams, N.P.; Ho, O.; Trobridge, G.D.; von Laer, D.; Prlic, M.; et al. Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood 2013, 122, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Van Lunzen, J.; Glaunsinger, T.; Stahmer, I.; von Baehr, V.; Baum, C.; Schilz, A.; Kuehlcke, K.; Naundorf, S.; Martinius, H.; Hermann, F.; et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 2007, 15, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Ledger, S.; Howe, A.; Ong, A.; Boyd, M.; Millington, M.; Savkovic, B.; Murray, J.; Symonds, G. Entry inhibitor gene modifieid T cells show selective survival advantage against HIV infection. Hum. Gene Ther. 2015. Submitted. [Google Scholar]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Li, M.J.; Palmer, B.; Remling, L.; Li, S.; Yam, P.; Yee, J.K.; Rossi, J.; Zaia, J.; Akkina, R. Safety and efficacy of a lentiviral vector containing three anti-HIV genes—CCR5 ribozyme, tat-rev siRNA, and TAR decoy—in SCID-hu mouse-derived T cells. Mol. Ther. 2007, 15, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Michienzi, A.; Li, S.; Zaia, J.A.; Rossi, J.J. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc. Natl. Acad. Sci. USA 2002, 99, 14047–14052. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Demosthenous, L.; Wiesner, G.; Link, E.; Westerman, D.A.; Came, N.; Ritchie, D.S.; Harrison, S.; Seymour, J.F.; Prince, H.M. Plerixafor plus pegfilgrastim is a safe, effective mobilization regimen for poor or adequate mobilizers of hematopoietic stem and progenitor cells: A phase I clinical trial. Bone Marrow Transplant. 2014, 49, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.T.; Jui, H.Y.; Huang, Y.H.; Su, M.Y.; Wu, Y.W.; Tseng, W.Y.; Hsu, M.C.; Chiang, B.L.; Wu, K.K.; Lee, C.M. CXCR4 Antagonist TG-0054 Mobilizes Mesenchymal Stem Cells, Attenuates Inflammation, and Preserves Cardiac Systolic Function in a Porcine Model of Myocardial Infarction. Cell Transplant. 2015, 24, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Reynes, J.; Portales, P.; Segondy, M.; Baillat, V.; Andre, P.; Avinens, O.; Picot, M.C.; Clot, J.; Eliaou, J.F.; Corbeau, P. CD4 T cell surface CCR5 density as a host factor in HIV-1 disease progression. AIDS 2001, 15, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Mettling, C.; Portales, P.; Reynes, J.; Clot, J.; Corbeau, P. Cell surface CCR5 density determines the postentry efficiency of R5 HIV-1 infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15590–15595. [Google Scholar] [CrossRef] [PubMed]
- Mahic, M.; Yaqub, S.; Johansson, C.C.; Tasken, K.; Aandahl, E.M. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J. Immunol. 2006, 177, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Cutler, C.; Multani, P.; Robbins, D.; Kim, H.T.; Le, T.; Hoggatt, J.; Pelus, L.M.; Desponts, C.; Chen, Y.B.; Rezner, B.; et al. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood 2013, 122, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hütter, G.; Bodor, J.; Ledger, S.; Boyd, M.; Millington, M.; Tsie, M.; Symonds, G. CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses 2015, 7, 4186-4203. https://doi.org/10.3390/v7082816
Hütter G, Bodor J, Ledger S, Boyd M, Millington M, Tsie M, Symonds G. CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses. 2015; 7(8):4186-4203. https://doi.org/10.3390/v7082816
Chicago/Turabian StyleHütter, Gero, Josef Bodor, Scott Ledger, Maureen Boyd, Michelle Millington, Marlene Tsie, and Geoff Symonds. 2015. "CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape" Viruses 7, no. 8: 4186-4203. https://doi.org/10.3390/v7082816
APA StyleHütter, G., Bodor, J., Ledger, S., Boyd, M., Millington, M., Tsie, M., & Symonds, G. (2015). CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses, 7(8), 4186-4203. https://doi.org/10.3390/v7082816