
Genome, Proteome and Structure of a T7-Like Bacteriophage of the Kiwifruit Canker Phytopathogen Pseudomonas syringae pv. actinidiae

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials, Bacterial Strains and Culture Conditions
2.2. Phage Lysate Preparation
2.3. Phage Genome Sequencing, Assembly and Annotation
2.4. Purification of φPsa17 for Proteomics
2.5. Proteomics of φPsa17
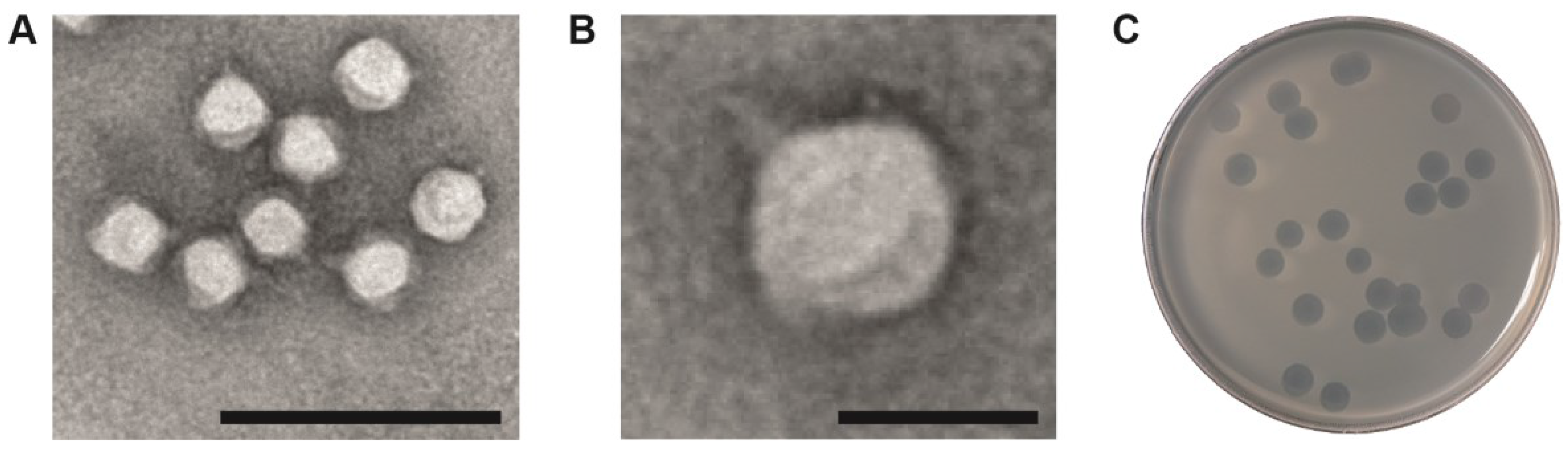
2.6. Purification of φPsa17 for Cryo-Electron Microscopy
2.7. Transmission Electron Microscopy and Cryo-Electron Microscopy
3. Results and Discussion
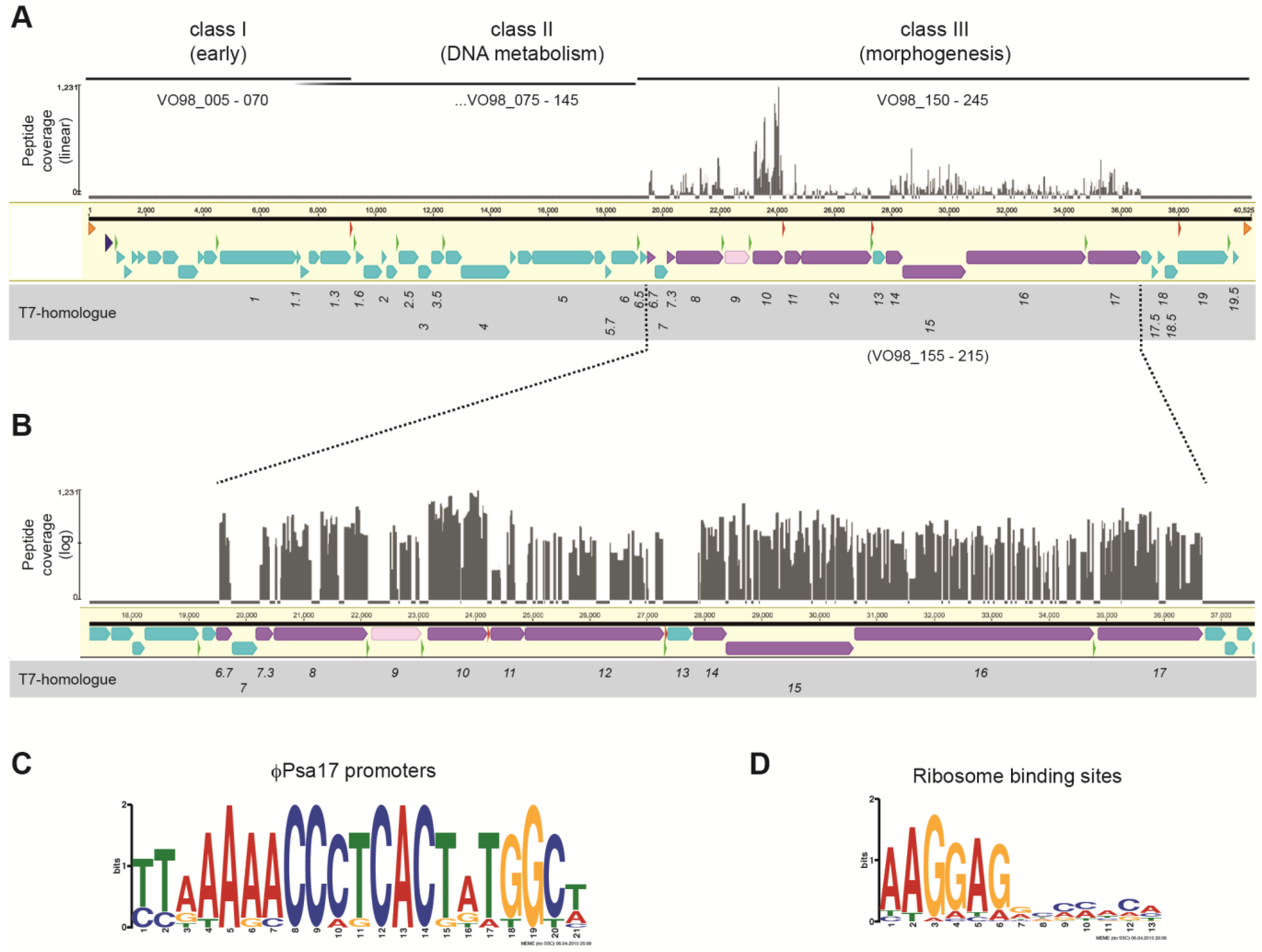
3.1. Podovirus φPsa17 Genome Sequence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag | T7 Homologue | Description | Start | End | Length (bp) | Domains or PHA# |
|---|---|---|---|---|---|---|
| VO98_005 | hypothetical | 993 | 1262 | 270 | ||
| VO98_010 | hypothetical | 1259 | 1513 | 255 | ||
| VO98_015 | hypothetical | 1513 | 1710 | 198 | ||
| VO98_020 | hypothetical | 1715 | 1990 | 276 | ||
| VO98_025 | hypothetical | 2078 | 2557 | 480 | ||
| VO98_030 | hypothetical | 2588 | 3124 | 537 | ||
| VO98_035 | hypothetical | 3121 | 3825 | 705 | Pfam13640 | |
| VO98_040 | hypothetical | 3825 | 4028 | 204 | ||
| VO98_045 | hypothetical | 4030 | 4470 | 441 | ||
| VO98_050 | gp1 | DNA-directed RNA polymerase (EC 2.7.7.6) | 4598 | 7255 | 2658 | PHA00452 |
| VO98_055 | gp1.1 | hypothetical | 7269 | 7406 | 138 | |
| VO98_060 | hypothetical | 7403 | 7675 | 273 | ||
| VO98_065 | hypothetical | 7675 | 8061 | 387 | ||
| VO98_070 | gp1.3 | ATP-dependent DNA ligase | 8073 | 9137 | 1065 | PHA00454 |
| VO98_075 | gp1.6 | hypothetical | 9331 | 9588 | 258 | PHA00455 |
| VO98_080 | hypothetical | 9585 | 10,232 | 648 | ABC_ATPase superfamily | |
| VO98_085 | gp2 | host RNA polymerase inhibitor | 10,229 | 10,396 | 168 | PHA00457 |
| VO98_090 | hypothetical | 10,393 | 10,758 | 366 | ||
| VO98_095 | gp2.5 | T7-like ssDNA-binding | 10,812 | 11,513 | 702 | PHA00458 |
| VO98_100 | gp3 | T7-like endonuclease (EC 3.1.21.2) | 11,513 | 11,956 | 444 | Phage_endo_I superfamily |
| VO98_105 | gp3.5 | lysozyme, N-acetylmuramoyl-L-alanine amidase (EC 3.5.1.28) | 11,959 | 12,399 | 441 | PGRP superfamily |
| VO98_110 | hypothetical | 12,469 | 13,008 | 540 | PolyA_pol superfamily | |
| VO98_115 | gp4 | T7-like DNA primase/helicase | 12,995 | 14,686 | 1692 | |
| VO98_120 | hypothetical | 14,705 | 14,902 | 198 | ||
| VO98_125 | hypothetical | 14,971 | 15,480 | 510 | ||
| VO98_130 | gp5 | T7-like DNA Polymerase (EC 2.7.7.7) | 15,491 | 17,638 | 2148 | DNA_pol_A superfamily |
| VO98_135 | hypothetical | 17,651 | 18,031 | 381 | ||
| VO98_140 | gp5.7 | hypothetical | 18,024 | 18,233 | 210 | PHA00422 |
| VO98_145 | gp6 | exonuclease | 18,230 | 19,174 | 945 | PHA00439 |
| VO98_150 | gp6.5 | hypothetical | 19,243 | 19,485 | 243 | DUF2717 |
| VO98_155 | gp6.7 | T7 virion protein | 19,488 | 19,760 | 273 | PHA00441 |
| VO98_160 | gp7 | hypothetical | 19,757 | 20,200 | 444 | PHA01807 |
| VO98_165 | gp7.3 | tail assembly | 20,172 | 20,474 | 303 | PHA00437 |
| VO98_170 | gp8 | collar/T7-like head-to-tail connector | 20,489 | 22,120 | 1632 | PHA00670 |
| VO98_175 | gp9 | capsid and scaffold | 22,189 | 23,064 | 876 | PHA00435 |
| VO98_180 | gp10 | major capsid protein | 23,164 | 24,207 | 1044 | PHA00201 (PHA02004 superfamily) |
| VO98_185 | gp11 | T7-like tail tubular protein A | 24,271 | 24,858 | 588 | PHA00428 |
| VO98_190 | gp12 | T7-like tail tubular protein B | 24,868 | 27,294 | 2427 | |
| VO98_195 | gp13 | protein inside capsid A | 27,353 | 27,787 | 435 | PHA00432 |
| VO98_200 | gp14 | protein inside capsid B | 27,798 | 28,385 | 588 | PHA00101 |
| VO98_205 | gp15 | protein inside capsid C | 28,378 | 30,594 | 2217 | PHA00431 |
| VO98_210 | gp16 | protein inside capsid D | 30,607 | 34,785 | 4179 | PHA00638 |
| VO98_215 | gp17 | tail fibre | 34,848 | 36,677 | 1830 | PHA00430 |
| VO98_220 | hypothetical | 36,717 | 37,073 | 357 | ||
| VO98_225 | gp17.5 | holin, class II | 37,073 | 37,288 | 216 | PHA00426 |
| VO98_230 | gp18 | T7-like DNA packaging protein A, small terminase subunit | 37,285 | 37,542 | 258 | PHA00425 |
| VO98_235 | gp18.5 | endopeptidase (EC 3.4.-.-), lambda Rz-like | 37,542 | 37,991 | 450 | PHA00276 |
| VO98_240 | gp19 | DNA packaging, large terminase subunit | 37,991 | 39,739 | 1749 | Pfam03237 |
| VO98_245 | gp19.5 | hypothetical | 39,919 | 40,092 | 174 |
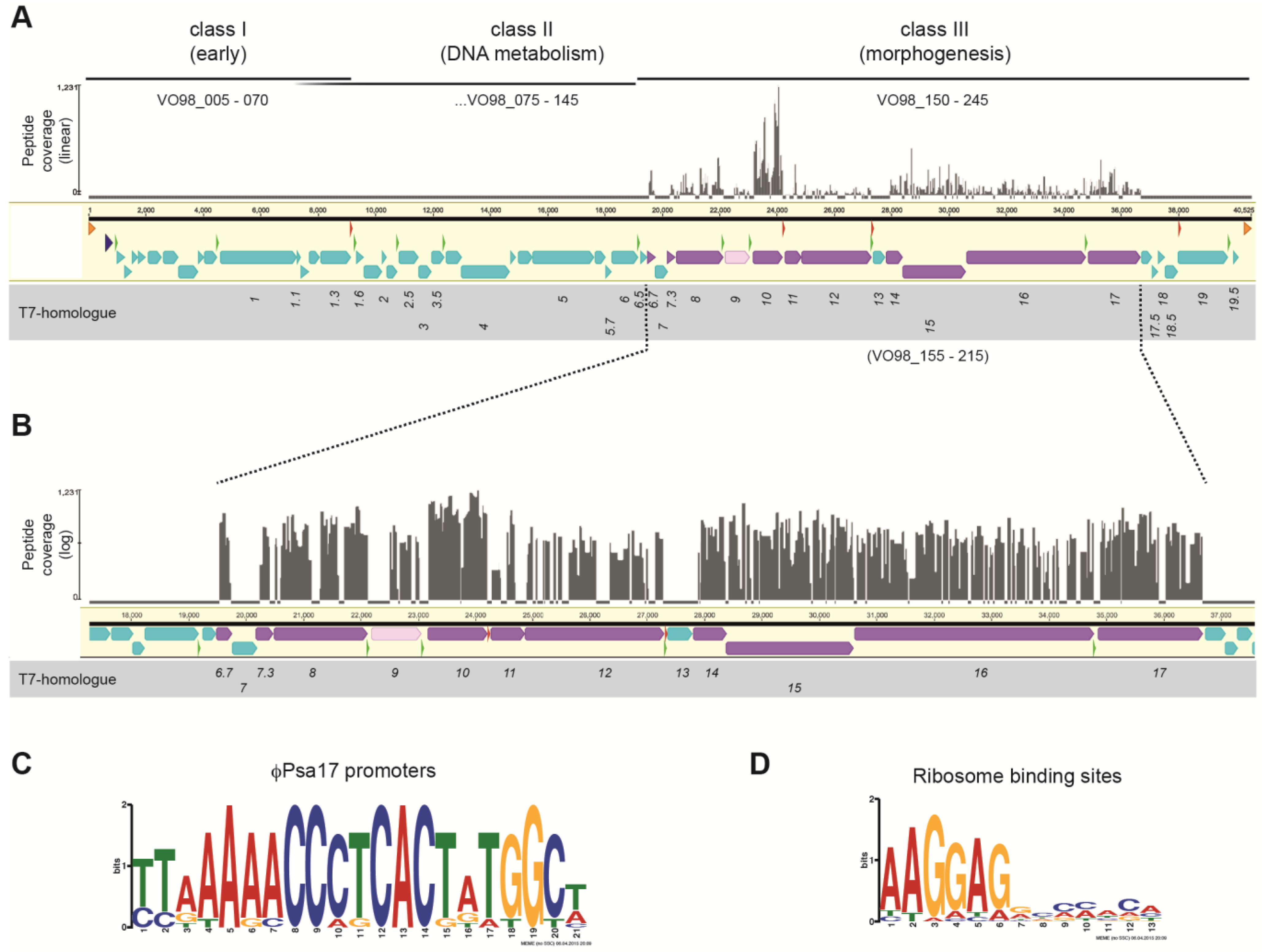
3.2. Transcriptional Organisation of the φPsa17 Genome
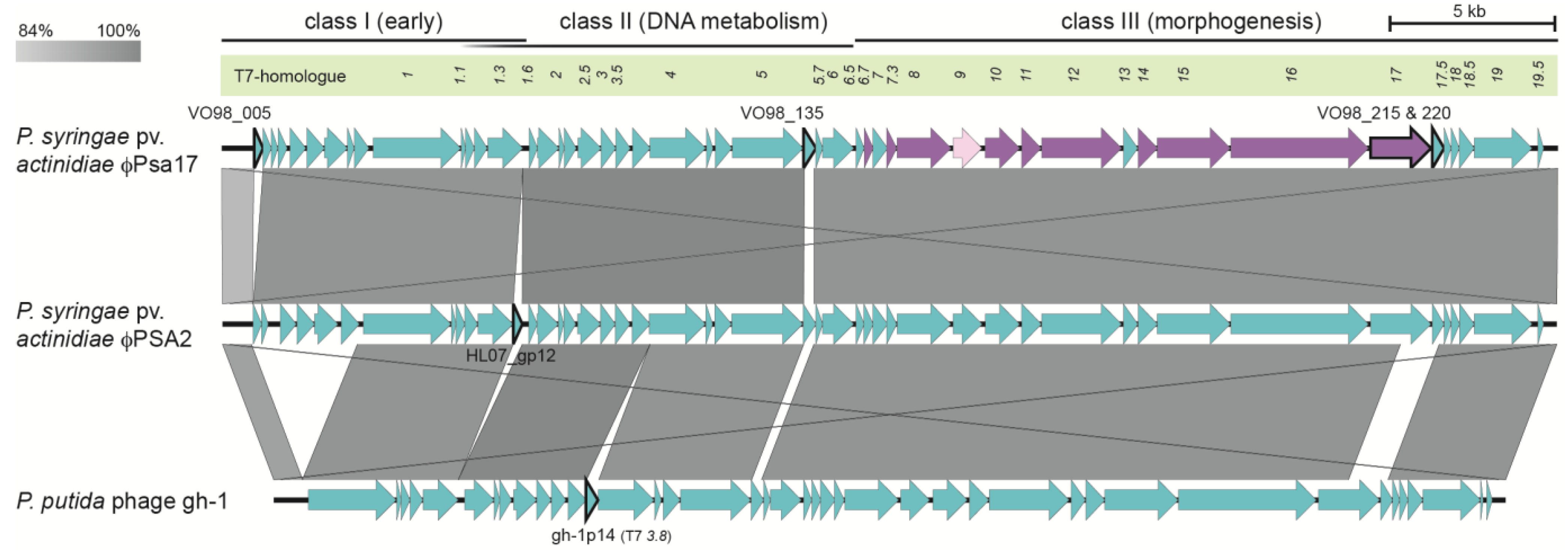
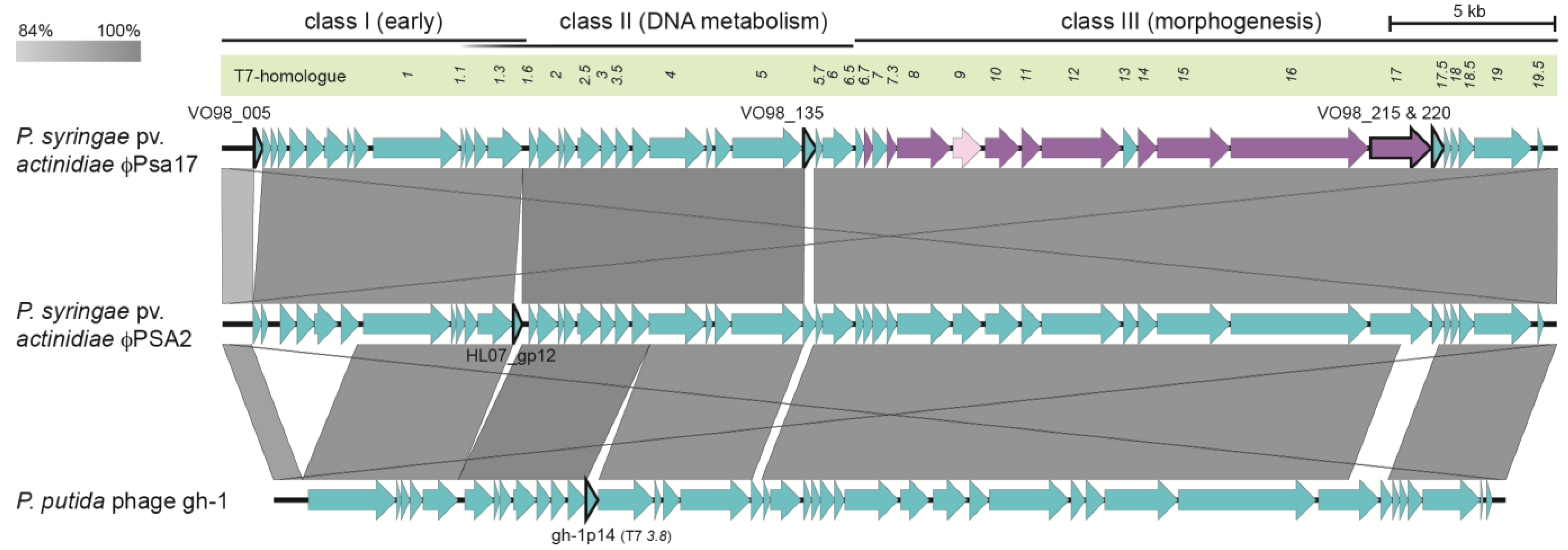
3.3. φPsa17 is a T7likevirus Similar to φPSA2 and gh-1
3.4. Structural Proteome of φPsa17
| Locus Tag | Description/T7 Homologue | Unique Peptides b | Total Peptides | Total Mascot | Total Amanda | Total Sequest | Total PSMs c | Coverage (%) |
|---|---|---|---|---|---|---|---|---|
| VO98_155 | virion protein, gp6.7 | 16 | 18 | 16 | 14 | 18 | 426 | 80.00 |
| VO98_165 | tail assembly protein, gp7.3 | 8 | 10 | 10 | 6 | 10 | 140 | 60.00 |
| VO98_170 | collar/T7-like head-to-tail joining protein, gp8 | 72 | 112 | 101 | 70 | 110 | 2998 | 85.08 |
| VO98_175 | capsid and scaffold, gp9a | 20 | 27 | 24 | 16 | 25 | 468 | 52.92 |
| VO98_180 | major capsid protein, gp10 | 151 | 215 | 192 | 69 | 195 | 7780 | 99.14 |
| VO98_185 | tail protein/T7-like tail tubular protein A, gp11 | 14 | 18 | 16 | 12 | 15 | 560 | 56.92 |
| VO98_190 | tail protein/T7-like tail tubular protein B, gp12 | 42 | 56 | 53 | 45 | 54 | 1366 | 71.66 |
| VO98_200 | protein inside capsid B, gp14 | 30 | 64 | 56 | 34 | 61 | 866 | 84.10 |
| VO98_205 | protein inside capsid C, gp15 | 141 | 221 | 196 | 131 | 201 | 4467 | 95.66 |
| VO98_210 | protein inside capsid D, gp16 | 153 | 232 | 209 | 165 | 222 | 4282 | 86.28 |
| VO98_215 | tail fibres, gp17 | 79 | 108 | 93 | 66 | 103 | 3155 | 92.28 |
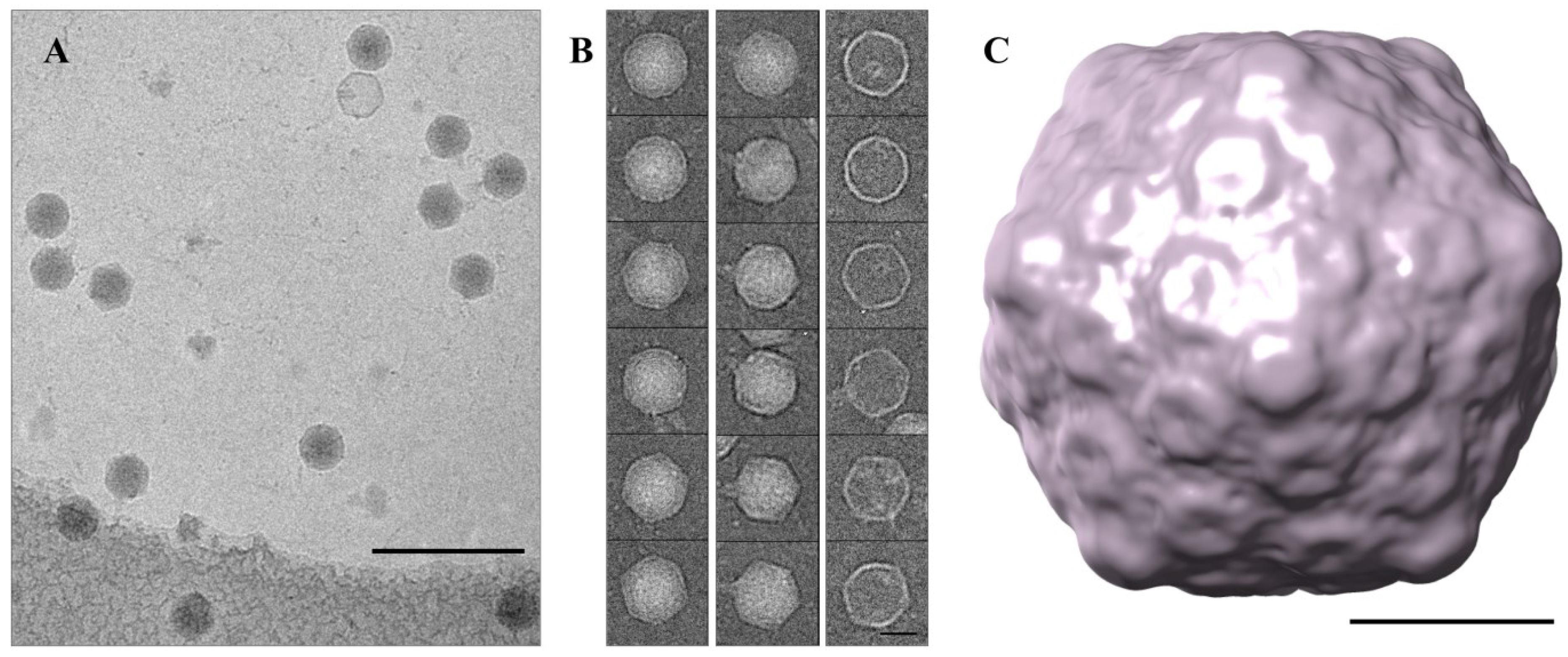
3.5. Cryo-TEM Structure of φPsa17
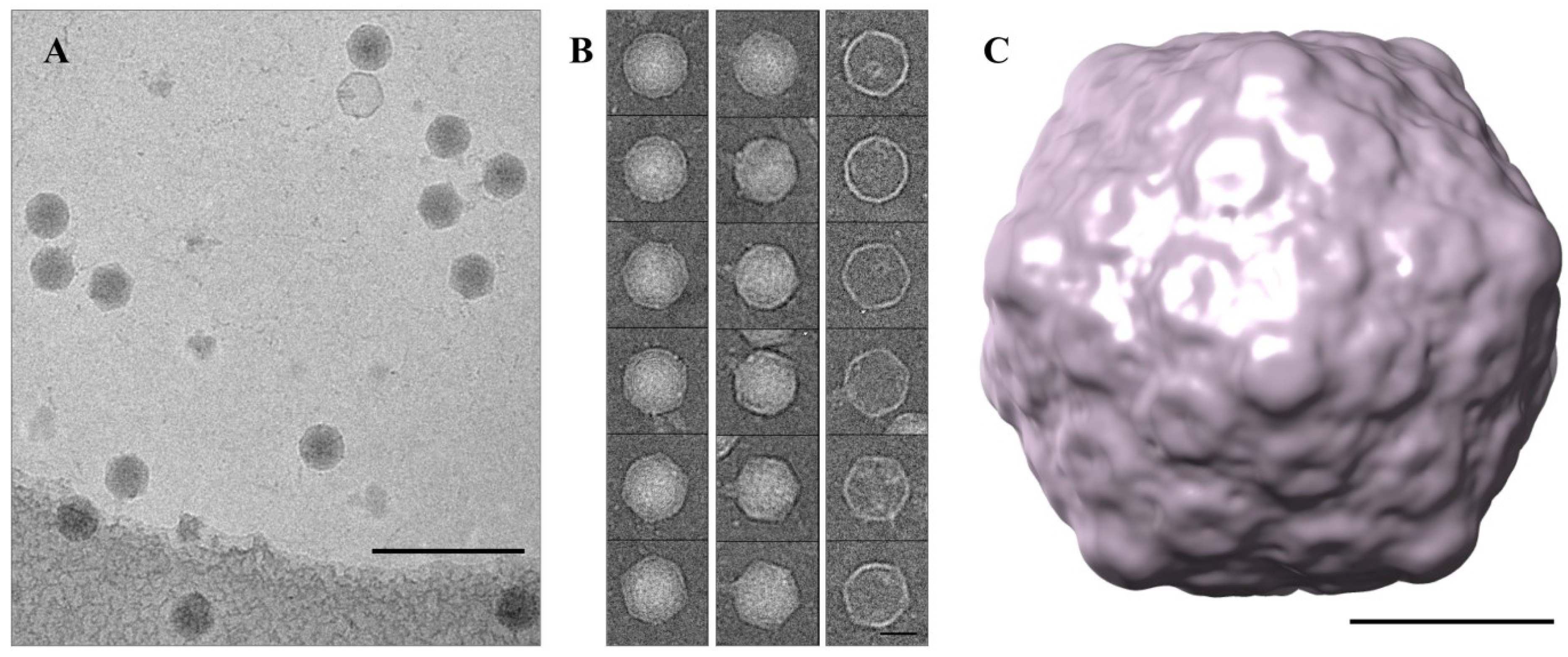
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scortichini, M.; Marcelletti, S.; Ferrante, P.; Petriccione, M.; Firrao, G. Pseudomonas syringae pv. actinidiae: A re-emerging, multi-faceted, pandemic pathogen. Mol. Plant Pathol. 2012, 13, 631–640. [Google Scholar]
- Takikawa, Y.; Serizawa, S.; Ichikawa, T.; Tsuyumu, S.; Goto, M. Pseudomonas syringae pv. actinidae pv. nov.: The causal bacterium of canker of kiwifruit in Japan. Ann. Phytopathol. Soc. Jpn. 1989, 55, 437–444. [Google Scholar]
- Koh, J.; Cha, B.; Chung, H.; Lee, D. Outbreak and spread of bacterial canker in kiwifruit. Korean J. Plant Pathol. 1994, 10, 68–72. [Google Scholar]
- Liang, Y.; Zhang, X.; Tian, C.; Gao, A.; Wang, P. Pathogenic identification of kiwifruit bacterial canker in Shaanxi. J. Northwest For. Coll. 2000, 15, 37–39. [Google Scholar]
- Scortichini, M. Occurrence of Pseudomonas syringae pv. actinidiae on kiwifruit in Italy. Plant Pathol. 1994, 43, 1035–1038. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, X.; Liu, S. Identification of the pathogenic bacterium for bacterial canker on Actinidia in Sichuan. J. Southwest Agric. Univ. 1992. [Google Scholar] [CrossRef]
- Abelleira, A.; López, M.M.; Peñalver, J.; Aguín, O.; Mansilla, J.P.; Picoaga, A.; García, M.J. First report of bacterial canker of kiwifruit caused by Pseudomonas syringae pv. actinidiae in Spain. Plant Dis. 2011, 95, 1583–1583. [Google Scholar] [CrossRef]
- ProMED-mail. 25 March 2011. Bacterial canker, kiwifruit—Chile: first report (O’Higgins, Maule). Int. Soc. Infect. Dis. ProMED-mail: 20110325.0940. Accessed 1 November 2013
- Balestra, G.M.; Renzi, M.; Mazzaglia, A. First report of bacterial canker of Actinidia delicosa caused by Pseudomonas syringae pv. actinidiae in Portugal. New Dis. Rep. 2010, 22, 10. [Google Scholar] [CrossRef]
- Balestra, G.M.; Renzi, M.; Mazzaglia, A. First report of Pseudomonas syringae pv. actinidiae on kiwifruit plants in Spain. New Dis. Rep. 2011, 24, 10. [Google Scholar] [CrossRef]
- Bastas, K.K.; Karakaya, A. First report of bacterial canker of kiwifruit caused by Pseudomonas syringae pv. actinidiae in Turkey. Plant Dis. 2012, 96, 452–452. [Google Scholar] [CrossRef]
- Ferrante, P.; Scortichini, M. Identification of Pseudomonas syringae pv. actinidiae as causal agent of bacterial canker of yellow kiwifruit (Actinidia chinensis Planchon) in central Italy. J. Phytopathol. 2009, 157, 768–770. [Google Scholar]
- Koh, Y.; Kim, G.; Jung, J.; Lee, Y.; Hur, J. Outbreak of bacterial canker on Hort16A (Actinidia chinensis Planchon) caused by Pseudomonas syringae pv. actinidiae in Korea. N. Zeal. J. Crop Hortic. Sci. 2010, 38, 275–282. [Google Scholar] [CrossRef]
- Vanneste, J.L.; Poliakoff, F.; Audusseau, C.; Cornish, D.A.; Paillard, S.; Rivoal, C.; Yu, J. First report of Pseudomonas syringae pv. actinidiae, the causal agent of bacterial canker of kiwifruit in France. Plant Dis. 2011, 95, 1311–1311. [Google Scholar]
- Ferrante, P.; Scortichini, M. Molecular and phenotypic features of Pseudomonas syringae pv. actinidiae isolated during recent epidemics of bacterial canker on yellow kiwifruit (Actinidia chinensis) in central Italy. Plant Pathol. 2010, 59, 954–962. [Google Scholar]
- Everett, K.R.; Taylor, R.K.; Romberg, M.K.; Rees-George, J.; Fullerton, R.A.; Vanneste, J.L.; Manning, M.A. First report of Pseudomonas syringae pv. actinidiae causing kiwifruit bacterial canker in New Zealand. Australas. Plant Dis. Notes 2011, 6, 67–71. [Google Scholar] [CrossRef]
- Kiwifruit Vine Health. Available online: http://www.kvh.org.nz/ (accessed on 5 May 2013).
- Scortichini, M.; Ferrante, P.; Marcelletti, S.; Petriccione, M. Omics, epidemiology and integrated approach for the coexistence with bacterial canker of kiwifruit, caused by Pseudomonas syringae pv. actinidiae. Ital. J. Agron. 2014, 9, 163. [Google Scholar] [CrossRef]
- Renzi, M.; Copini, P.; Taddei, A.R.; Rossetti, A.; Gallipoli, L.; Mazzaglia, A.; Balestra, G.M. Bacterial canker on kiwifruit in Italy: Anatomical changes in the wood and in the primary infection sites. Phytopathology 2012, 102, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.I.; Stockwell, P.A.; Black, M.A.; Day, R.C.; Lamont, I.L.; Poulter, R.T. Pseudomonas syringae pv. actinidiae from recent outbreaks of kiwifruit bacterial canker belong to different clones that originated in China. PLoS ONE 2013, 8, e57464. [Google Scholar]
- Chapman, J.R.; Taylor, R.K.; Weir, B.S.; Romberg, M.K.; Vanneste, J.L.; Luck, J.; Alexander, B.J. Phylogenetic relationships among global populations of Pseudomonas syringae pv. actinidiae. Phytopathology 2012, 102, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Marcelletti, S.; Ferrante, P.; Petriccione, M.; Firrao, G.; Scortichini, M. Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species. PLoS ONE 2011, 6, e27297. [Google Scholar]
- Mazzaglia, A.; Studholme, D.J.; Taratufolo, M.C.; Cai, R.; Almeida, N.F.; Goodman, T.; Guttman, D.S.; Vinatzer, B.A.; Balestra, G.M. Pseudomonas syringae pv. actinidiae (PSA) isolates from recent bacterial canker of kiwifruit outbreaks belong to the same genetic lineage. PLoS ONE 2012, 7, e36518. [Google Scholar]
- McCann, H.C.; Rikkerink, E.H.; Bertels, F.; Fiers, M.; Lu, A.; Rees-George, J.; Andersen, M.T.; Gleave, A.P.; Haubold, B.; Wohlers, M.W.; et al. Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog. 2013, 9, e1003503. [Google Scholar]
- Frampton, R.A.; Pitman, A.R.; Fineran, P.C. Advances in bacteriophage-mediated control of plant pathogens. Int. J. Microbiol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Balogh, B.; Jones, J.B.; Iriarte, F.B.; Momol, M.T. Phage therapy for plant disease control. Curr. Pharm. Biotechnol. 2010, 11, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Frampton, R.A.; Taylor, C.; Holguin Moreno, A.V.; Visnovsky, S.B.; Petty, N.K.; Pitman, A.R.; Fineran, P.C. Identification of bacteriophages for biocontrol of the kiwifruit canker phytopathogen Pseudomonas syringae pv. actinidiae. Appl. Environ. Microbiol. 2014, 80, 2216–2228. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Petty, N.K.; Salmond, G.P.C. Transduction: Host DNA transfer by bacteriophages. In The Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Elsevier: Oxford, UK, 2009. [Google Scholar]
- Petty, N.K.; Evans, T.J.; Fineran, P.C.; Salmond, G.P. Biotechnological exploitation of bacteriophage research. Trends Biotechnol. 2007, 25, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinf. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2013, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Extract Upstream DNA. Available online: https://lfz.corefacility.ca/extractUpStreamDNA/ (assessed on 7 April 2015).
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Lesnik, E.A.; Sampath, R.; Levene, H.B.; Henderson, T.J.; McNeil, J.A.; Ecker, D.J. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 2001, 29, 3583–3594. [Google Scholar] [CrossRef] [PubMed]
- Macke, T.J.; Ecker, D.J.; Gutell, R.R.; Gautheret, D.; Case, D.A.; Sampath, R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001, 29, 4724–4735. [Google Scholar] [CrossRef] [PubMed]
- Gautheret, D.; Lambert, A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001, 313, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Salamov, A. Automatic Annotation of Microbial Genomes and Metagenomic Sequences. In Metagenomics and Its Applications in Agriculture, Biomedicine and Environmental Studies; Li, R.W., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2011; pp. 61–78. [Google Scholar]
- Reese, M.G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Ceyssens, P.J.; Robben, J. Phage proteomics: Applications of mass spectrometry. Methods Mol. Biol. 2009, 502, 239–251. [Google Scholar] [PubMed]
- Shevchenko, A.; Jensen, O.N.; Podtelejnikov, A.V.; Sagliocco, F.; Wilm, M.; Vorm, O.; Mortensen, P.; Shevchenko, A.; Boucherie, H.; Mann, M. Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. USA 1996, 93, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- MS Amanda, Mass spectrometry, Protein Chemistry Facility (IMP, IMBA & GMI). Available online: http://ms.imp.ac.at/?goto=msamanda (assessed on 25 May 2014).
- Percolator. Available online: http://per-colator.com (assessed on 25 May 2014).
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A.; Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003, 142, 334–347. [Google Scholar] [CrossRef]
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Molineux, I.J. The T7 Group. In The Bacteriophages, 2nd ed.; Calendar, R., Ed.; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- McLean, B.W.; Wiseman, S.L.; Kropinski, A.M. Functional analysis of sigma-70 consensus promoters in Pseudomonas aeruginosa and Escherichia coli. Can. J. Microbiol. 1997, 43, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Kovalyova, I.V.; Kropinski, A.M. The complete genomic sequence of lytic bacteriophage gh-1 infecting Pseudomonas putida—Evidence for close relationship to the T7 group. Virology 2003, 311, 305–315. [Google Scholar] [CrossRef]
- Studier, F.W. Gene 0.3 of bacteriophage T7 acts to overcome the DNA restriction system of the host. J. Mol. Biol. 1975, 94, 283–295. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Di Lallo, G.; Evangelisti, M.; Mancuso, F.; Ferrante, P.; Marcelletti, S.; Tinari, A.; Superti, F.; Migliore, L.; D’Addabbo, P.; Frezza, D.; et al. Isolation and partial characterization of bacteriophages infecting Pseudomonas syringae pv. actinidiae, causal agent of kiwifruit bacterial canker. J. Basic Microbiol. 2014, 54, 1210–1221. [Google Scholar]
- Ionel, A.; Velazquez-Muriel, J.A.; Luque, D.; Cuervo, A.; Caston, J.R.; Valpuesta, J.M.; Martin-Benito, J.; Carrascosa, J.L. Molecular rearrangements involved in the capsid shell maturation of bacteriophage T7. J. Biol. Chem. 2011, 286, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Z.; Fang, P.A.; Zhang, Q.; Wright, E.T.; Wu, W.; Zhang, C.; Vago, F.; Ren, Y.; Jakana, J.; et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. USA 2014, 111, E4606–E4614. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. The bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science 2013, 339, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Z.; Vago, F.; Ren, Y.; Wu, W.; Wright, E.T.; Serwer, P.; Jiang, W. Visualization of uncorrelated, tandem symmetry mismatches in the internal genome packaging apparatus of bacteriophage T7. Proc. Natl. Acad. Sci. USA 2013, 110, 6811–6816. [Google Scholar] [CrossRef] [PubMed]
- Agirrezabala, X.; Velazquez-Muriel, J.A.; Gomez-Puertas, P.; Scheres, S.H.; Carazo, J.M.; Carrascosa, J.L. Quasi-atomic model of bacteriophage T7 procapsid shell: Insights into the structure and evolution of a basic fold. Structure 2007, 15, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Agirrezabala, X.; Martin-Benito, J.; Caston, J.R.; Miranda, R.; Valpuesta, J.M.; Carrascosa, J.L. Maturation of phage T7 involves structural modification of both shell and inner core components. EMBO J. 2005, 24, 3820–3829. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, P.B.; Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003, 333, 721–745. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frampton, R.A.; Acedo, E.L.; Young, V.L.; Chen, D.; Tong, B.; Taylor, C.; Easingwood, R.A.; Pitman, A.R.; Kleffmann, T.; Bostina, M.; et al. Genome, Proteome and Structure of a T7-Like Bacteriophage of the Kiwifruit Canker Phytopathogen Pseudomonas syringae pv. actinidiae. Viruses 2015, 7, 3361-3379. https://doi.org/10.3390/v7072776
Frampton RA, Acedo EL, Young VL, Chen D, Tong B, Taylor C, Easingwood RA, Pitman AR, Kleffmann T, Bostina M, et al. Genome, Proteome and Structure of a T7-Like Bacteriophage of the Kiwifruit Canker Phytopathogen Pseudomonas syringae pv. actinidiae. Viruses. 2015; 7(7):3361-3379. https://doi.org/10.3390/v7072776
Chicago/Turabian StyleFrampton, Rebekah A., Elena Lopez Acedo, Vivienne L. Young, Danni Chen, Brian Tong, Corinda Taylor, Richard A. Easingwood, Andrew R. Pitman, Torsten Kleffmann, Mihnea Bostina, and et al. 2015. "Genome, Proteome and Structure of a T7-Like Bacteriophage of the Kiwifruit Canker Phytopathogen Pseudomonas syringae pv. actinidiae" Viruses 7, no. 7: 3361-3379. https://doi.org/10.3390/v7072776
APA StyleFrampton, R. A., Acedo, E. L., Young, V. L., Chen, D., Tong, B., Taylor, C., Easingwood, R. A., Pitman, A. R., Kleffmann, T., Bostina, M., & Fineran, P. C. (2015). Genome, Proteome and Structure of a T7-Like Bacteriophage of the Kiwifruit Canker Phytopathogen Pseudomonas syringae pv. actinidiae. Viruses, 7(7), 3361-3379. https://doi.org/10.3390/v7072776