1. Introduction
Ebola virus disease (EVD) is one of the deadliest infectious diseases that has ravaged Central and Western Africa many times in the past, and has evoked global fear while testing the global community’s preparedness and response during the ongoing 2014 Western African epidemic [
1]. This is in part due to the high case-fatality ratio (CFR) of EVD, the general lack of control measures in resource-poor nations to curtail transmission, and a lack of approved medical countermeasures such as vaccines or antiviral therapeutics. The genus
Ebolavirus has five members with genetic diversity ranging from 25%–35%: Tai Forest virus (TAFV), Reston virus (RESTV), Sudan virus (SUDV), Ebola virus (EBOV), and Bundibugyo virus (BDBV) [
2]. There have been at least 41 filovirus disease outbreaks in humans during the period between 1967 and 18 September 2014. In addition, ebolavirus has emerged repeatedly in animal populations in Central and Western Africa, causing massive die-offs of gorillas and chimpanzees [
3]. The CFRs in major human outbreaks vary from 25% to 90% depending on the viral species, number of cases and other factors, such as access to healthcare [
1].
The ongoing EVD epidemic originated in Guinea in December 2013 and is the largest in recorded history, affecting multiple countries in Western Africa [
4]. According to an Ebola Response model constructed by the U.S. Centers for Disease Control and Prevention (CDC), if trends had continued without scale-up of effective interventions or changes in community behavior (e.g., early diagnosis and hospitalization in Ebola Treatment Units and reductions in unsafe burial practices), it was estimated that there would have been as many as 1.4 million cases by 20 January 2015 [
5]. Revised estimates by a different group predicted a similar trend in the absence of effective steps to curtail transmission [
6]. However, as of January 2015, the trend appears to have slowed as opposed to the exponential increase predicted by the model, despite the unprecedented epidemiological scale of this outbreak. The limited success in containment can be attributed to the effectiveness of some of the control measures put in place in affected countries [
7]. As of 17 May 2015 there have been 26,969 total cases (suspected, probable, and confirmed), 14,991 laboratory-confirmed cases, and 11,135 total deaths amounting to a case fatality ratio (CFR) of 41.2% in Western Africa, and a few imported and secondary cases in various other countries including the U.S [
8].
A recent study has suggested that the ongoing epidemic in Western Africa originated from a single zoonotic transmission event from a free-tailed bat (
Mops condylurus) to a two-year-old boy in Meliandou, Guinea, who is believed to have been the index case [
9]. Hence, active environmental biosurveillance can help predict/prevent the transmission of the virus from its natural reservoirs to humans [
10]. In addition, rapid, sensitive, safe and simple diagnostic tests may expedite case finding and the timely initiation of medical and outbreak control measures to interrupt viral transmission in resource constrained settings, such as Ebola Treatment Units. Such a measure can also help in preventing nosocomial infections, guiding triage and clinical decisions, aiding contact tracing and facilitating the early isolation of cases [
11]. Efforts to contain the EVD outbreak in Western Africa are currently hampered by cumbersome, slow, complex, and expensive diagnostic tests that impose a number of additional logistical challenges, including requirements for a high level of laboratory biosafety and staff expertise in using sophisticated instruments [
11].
Hence, many laboratories and private organizations are developing and deploying handheld tests for rapid detection of EBOV in clinical specimens to address this gap [
12]. Recently, Corgenix Medical Corporation received an Emergency Use Authorization (EUA) approval from the U.S. Food and Drug Administration (FDA) for its ReEBOV™ Antigen Rapid Test. In addition, several laboratory-developed immunoassays for different platforms (e.g., ELISA, MAGPIX) are currently in use for EBOV testing.
The current gold standard for EBOV detection is rRT-PCR molecular assays that can be run on various PCR instruments [(Applied Biosystems real time PCR systems, Joint Biological Agent Identification and Diagnostic System (JBAIDS), Light Cycler
etc.)] for real time detection of EBOV from serum and plasma. Several organizations, including the U.S. Department of Defense (DoD), have deployed detection kits based on PCR assays. Early PCR testing enhances the medical care of both infected and uninfected patients, and guides rational hospital infection control. Real-time RT-PCR tests are also valuable for correlating viral load to disease severity. Rapid detection tests can rule out non-EVD cases if other PCR assays are available to test for the etiologic agents of diseases that present with similar symptoms. Expanded PCR testing would also enable detecting co-infection in EVD patients and guide treatments accordingly. Additionally, they can be used for post-infection tracking of asymptomatic shedding in convalescent patients including documentation of virus in the semen and breast milk weeks after resolution of symptoms [
13].
The FDA has, thus far, approved seven molecular assays through the Emergency Use Authorization (EUA) process, and these include [
14]: (1) CDC Ebola Virus VP40 Real-time RT-PCR Assay; (2) CDC Ebola Virus NP Real-time RT-PCR Assay; (3) DoD EZ1 Real-time RT-PCR Assay based on VP40; (4) BioFire Defense LLC FilmArray NGDS BT-E Assay; (5) BioFire Defense LLC FilmArray Biothreat-E Test; (6) RealStar
® Ebolavirus RT-PCR Kit 1.0; and (7) LightMix
® Ebola Zaire rRT-PCR Test. Many other assays; e.g., OasigTM-rRT-PCR and the DoD EZ2 assay, both based on the NP gene, are in the EUA pipeline. However, many of the assays were developed using sequence information prior to the 2014 outbreak or from a limited number of sequences obtained early in the current outbreak. The assay signatures of preexisting assays have not been systematically and periodically evaluated against new genome/gene sequences that are deposited in GenBank in order to assess their performance against new viral lineages.
In a landmark genomic study, Gire
et al. sequenced 99 viral genomes in samples originating from 78 patients early in the 2014 Western African outbreak [
15]. Results based on in depth genome sequence analyses revealed a number crucial factors about viral transmission dynamics and genome plasticity of 2014 EBOV: (1) the finding that the potential source of the Western African variant is a central African lineage from 2004; (2) intra- and inter-host genetic variations; (3) sustained human-to-human transmission from the initial jump from a natural reservoir without additional inter host transmission; and (4) very high mutation rates typical of RNA viruses (8 × 10
−4 per base per year) that approaches the mutational rate of seasonal influenza [
16]. This study also highlighted the fact that many of the mutations alter RNA and protein sequences that are targets for diagnostics, vaccines and therapeutics and hence the need to redesign existing diagnostics (and therapeutics in the pipeline) as well as accelerate the development and approval of medical countermeasures undergoing clinical trials [
15]. A recent report has assessed the potential impact of genomic changes in EBOV on the efficacy of sequence-based candidate therapeutics [
17].
In this work, we have focused on the potential limitations of the ebolavirus molecular assays by assessing (1) signature erosion due to viral genetic changes [the term ‘signature-erosion’ is used here to signify potential false-positive or false-negative results in molecular assays due to mutations in the PCR primers/probe/amplicon target sequences (PCR signatures)] [
18,
19]; (2) the need for addressing genomic changes when re-optimizing old assays or designing new assays; (3) a critical need for capturing the genetic diversity of ebolavirus from geographically and temporally diverse isolates; and (4) a need for deep sequencing to capture the quasi-species evolution within individuals over time (e.g., longitudinal studies). Rapid generation of sequence data at the onset and throughout the course of an outbreak is critical for establishing confidence in the fielded diagnostic assays and, if required, for the rapid development of new assays or optimization of old assays. Deep sequencing would allow prediction and development of proactive signatures, in the event rare variants present in a quasi-species that have a predictable effect on existing signatures become prevalent. We draw the attention of the research community to newer approaches to molecular assay design that takes a whole genome approach rather than gene-centric approach and long term solutions to assay design that are proactive rather than reactive for new and reemerging threats. We also discuss mitigation strategies and the need for: crowd sourcing of data, analyses and rapid development of assay quality control measures, regulatory approvals and future technologies. There are other assay limitations such as time to result, sensitivity and specificity, and issues with sample collection and preparation that are not directly addressed here.
2. Materials and Methods
2.1. Viruses and Sequencing
Two different EBOV isolates used in this study were obtained from the Critical Reagents Program [
20] (Unified Culture Collection (UCC) [
21] These viruses included EBOV/H.sapiens-tc/COD/1995/Kikwit (UCC# R4368) and EBOV/H.sapiens-tc/SLE/2014/Makona (UCC# R4491). Total nucleic acid was purified from 200 µL cell culture supernatant in TRIzol (Life Technologies, Grand Island, NY, USA) using the EZ1 Virus 2.0 kit (Qiagen, Valencia, CA, USA) and the EZ1 robot (Qiagen) according to the manufacturer’s instructions. Nucleic acid was eluted in a final volume of 60 µL elution buffer.
The purified nucleic acid for each virus was amplified by whole transcriptome amplification (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Amplified cDNA was processed for sequencing on the MiSeq Desktop Sequencer (Illumina, San Diego, CA, USA) using the Apollo 324 System (Wafergen Biosystems, Fremont, CA, USA) and the PrepX ILMN DNA Library Kit (Wafergen Biosystems). After sequencing on the MiSeq, the resulting reads were processed using CLC Genomics Workbench (CLC Bio, Boston, MA, USA). Reads were quality trimmed and aligned against the EBOV reference sequence (Ebola virus/H.sapiens-tc/COD/1976/Yambuku-Mayinga; GenBank number NC_002549.1) or (Ebola virus/H.sapiens-wt/SLE/2014/Makona-G3831; GenBank number KM233103). The trimmed reads were then mapped against this initial consensus sequence to generate a final consensus sequence for each isolate used in this study. The GenBank accession # for the two isolates are: Zaire_1 (Makona) KR824525; Zaire_2 (Kikwit) KR824526. The sequence reads have been submitted to the SRA library under the project ID: PRJNA284630.
2.2. Real-Time RT-PCR Assay Evaluations
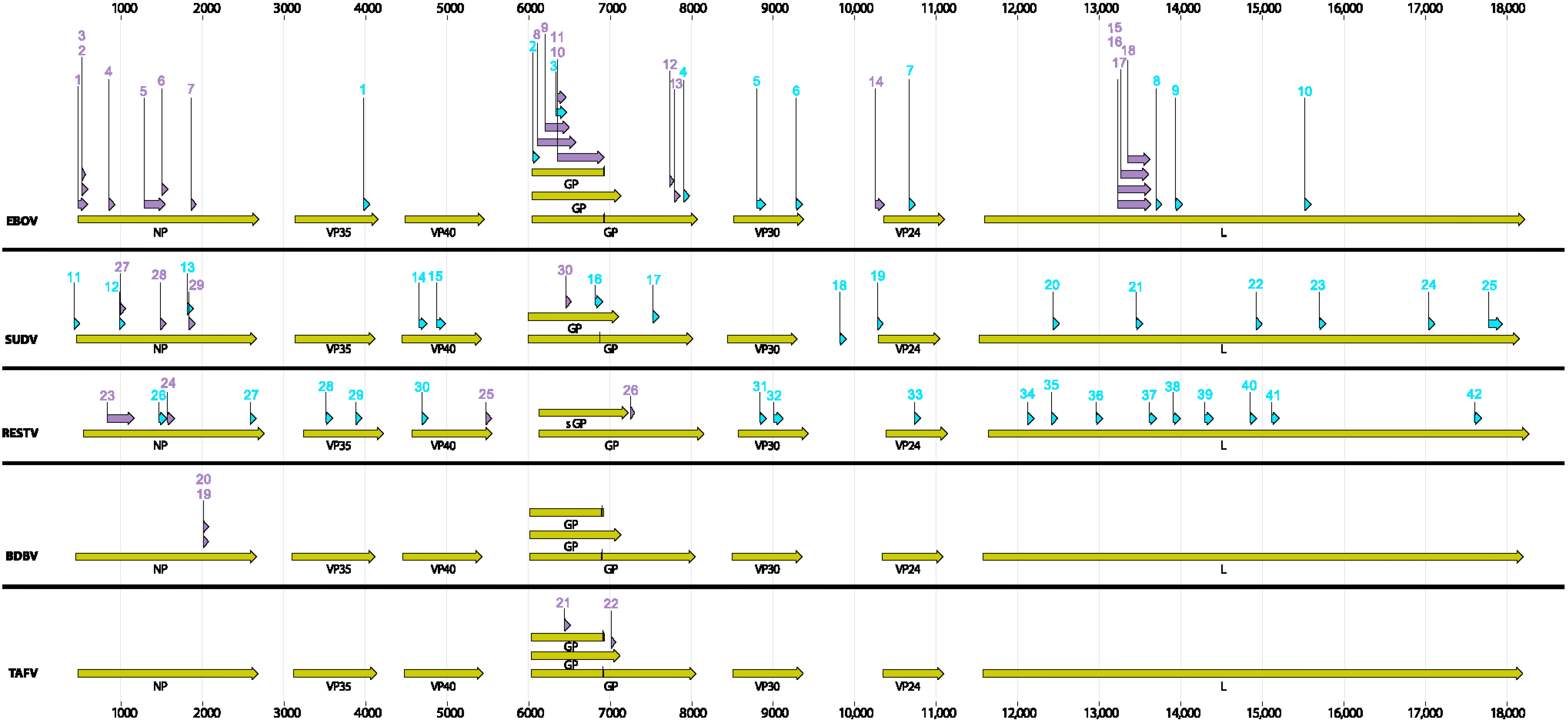
All EBOV assays, irrespective of the original design format (conventional end point, TaqMan probe or MGB probe,
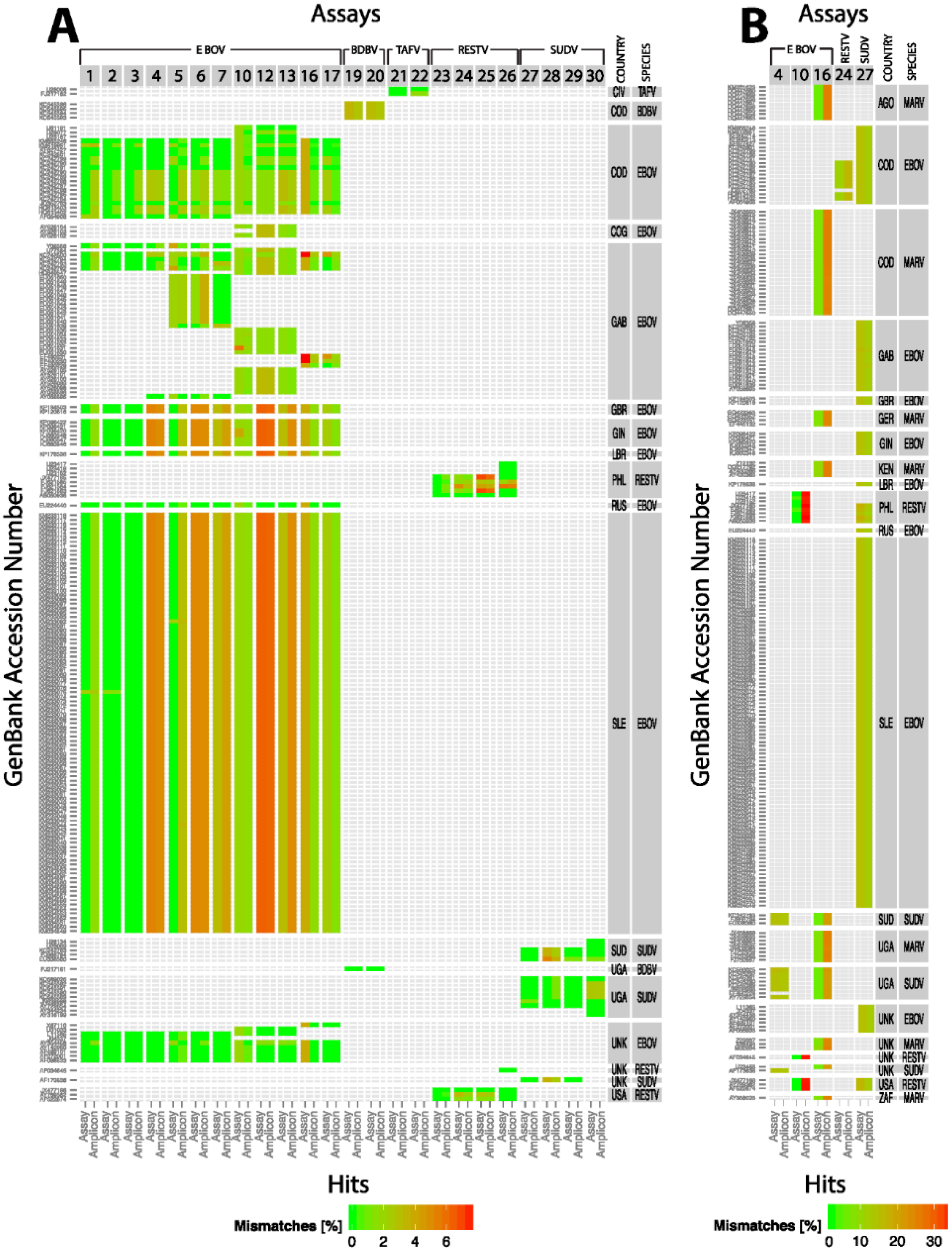
etc.), were tested in a SYBR Green assay format under one PCR conditions. The two templates used in the assays were: EBOV Kikwit (GB accession #KR824526) and Makona EBOV (2014 GB accession #KR824525). The mismatches if any in the primer binding sites in each of the templates are listed in
Table 2.
Primers for each assay were purchased from Life Technologies. All real-time RT-PCR assays were performed with primers at a final concentration of 1.0 µM and SYBR Green at 0.2× using the SuperScript One-Step RT-PCR Kit (Life Technologies) at 1×, 3 mM MgSO4, and 1× RT/Platinum Taq polymerase. Bovine serum albumin was also added at a final concentration of 0.25 mg/mL. All assays were run on the LightCycler 480 (Roche Applied Science, Indianapolis, ID, USA). Assays that produced an amplicon less than 200 bp were conducted at the following cycling conditions: 50 °C for 15 min (1 cycle); 95 °C for 5 min (1 cycle); 95 °C for 1 s, 50 °C for 20 s and 72 °C for 1 s (45 cycles). A single fluorescence read was taken at the end of each 72 °C step. Those assays with amplicons larger than 200 bp had an extension step of 30 s at 72 °C.
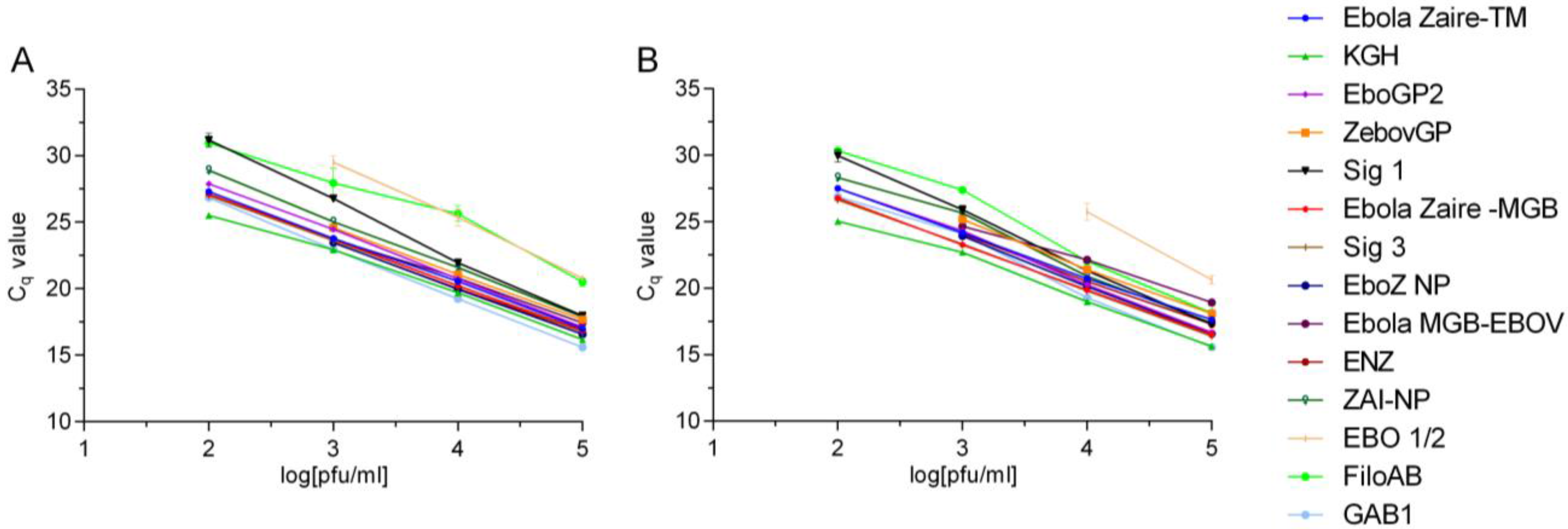
Preliminary LoDs were determined for each assay using 1:10 serial dilutions of EBOV Kikwit and EBOV Makona RNA. A nonlinear regression analysis was conducted over the linear range of the RNA dilution series for each assay to compare differences in the y-intercept and the slope between the Kikwit and Makona viral RNAs. Ebola Zaire-TM and Ebola Zaire-MGB assays were run according to published protocols [
22].
2.4. BioVelocity Analysis for Identifying Diagnostic Signatures
One method for the rapid generation of new diagnostic assays is to use high performance computing to find highly conserved and unique regions of genomes. BioVelocity is a bioinformatics tool based on an innovative algorithm and approach to genomic reference indices. Using a fast and accurate hashing algorithm, BioVelocity is capable of quickly finding conserved and signature sequences from within a given genomic reference or set of raw sequencing reads. This process allows one to find target regions for new assay design within a particular genus, species, or strain in minutes (to be published elsewhere; BioVelocity will also be available in the Illumina App Store in June 2015).
BioVelocity takes advantage of a CRAY-XMT2 supercomputer with four terabytes of RAM to demonstrate dramatic improvements in performance over current technologies with 50× faster speeds, increased functionality, increased throughput, and improved accuracy. The CRAY-XMT2 allows for the use of a brute force index, built out of all possible base pair sequences of various k-mer lengths. This index is used to map against thousands of references and allows for quick alignment of k-mers amongst them simultaneously.
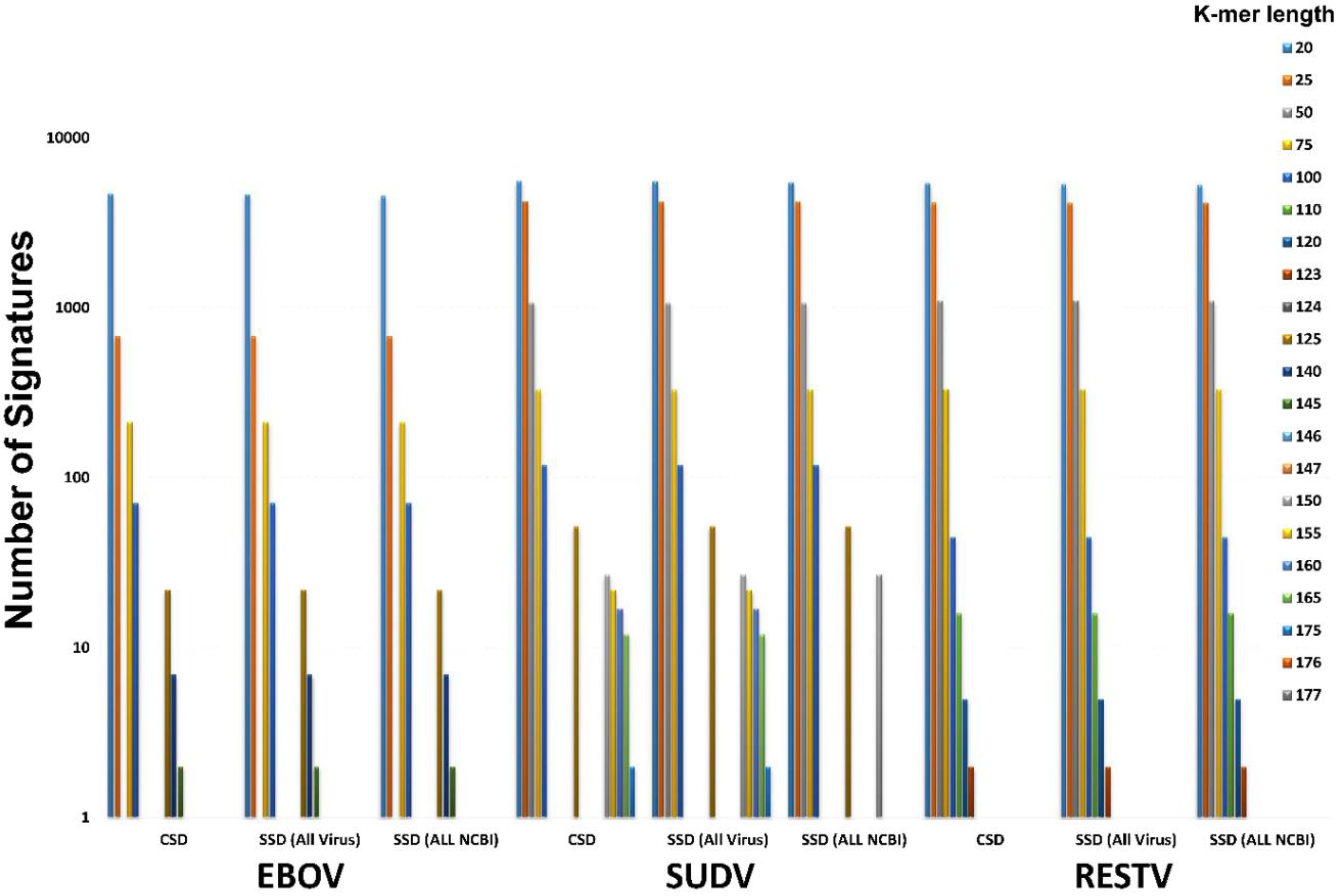
Once a target is selected, in this case EBOV, the conserved regions must be isolated. To determine conserved regions BioVelocity needs two sources of data: a list of full genome references of the desired target organism and a k-merized genome reference or read set of the same target organism. Using BioVelocity, an index was created using all available EBOV whole genome references, 134 in total from NCBI (as of December 2014). Conserved sequence sizes can be manually set based on the necessary application. For example, to find conserved sequences that are 50 base pairs in length, a single full genome reference is split into 50-mers and Conserved Sequence Domain (CSD) detection pipeline is run against the index. BioVelocity scans all 50-mer alignments to identify perfect match in every reference sequence located within the index. If a particular 50-mer perfectly aligned to each of the 134 EBOV references it would be considered conserved and exported into a FASTA file of conserved k-mers.
Signature Sequence Domain (SSD) detection pipeline is designed to compare one organism, either a reference genome or read set, to all other references found within the index to determine unique k-mers. Similar to CSD, SSD uses BioVelocity’s index to quickly align k-mers against thousands of references, but as opposed to CSD, SSD looks for k-mers which have no perfect alignments against any reference genome within the index. After running CSD, BioVelocity collects all conserved k-mers for an organism (e.g., EBOV) and uses this newly acquired k-mer set to determine signature k-mers through the alignment against an index comprised of all of NCBI’s full genome references (excluding the target organism, EBOV). Thus, a set of signature k-mers for the target of interest, EBOV, is generated. These k-mers are prime locations for assays to target due to their conserved status, while also minimizing likelihood of false-positives since they are unique to the target.
The detection pipelines for both CSD and SSD were run for EBOV to determine how many potential signature k-mers are available to target new assay design. The results are shown both against all viruses in NCBI and against all organisms in NCBI (
Table S3). The first column indicates the k-mer size used, the second column indicates the conserved sequences found in EBOV, and the following columns indicate the number of conserved sequences that are also signature sequences based on varying degrees of similarity. For instance, a signature found at 90% means that there are no other sequences that are similar at a 90% or greater level in the index. This is useful for ensuring that target regions can sustain some genomic plasticity before losing their signature status.
2.5. Validation of BioVelocity Signatures
BLAST (NCBI) was used to identify ebolavirus orthologs to each gene (NP, VP35, VP40, GP, VP30, VP24, and L), using EBOV AF086833 as the initial comparator sequence. The BLAST program blastn was used with parameters limiting the search to taxid ebolavirus, and the maximum number of sequences returned set to 250. The e-value maximum was set to 100, and the word size was 11. Because of the diversity among ebolaviruses, the match/mismatch penalties were set to 1, −1, and the gap penalties were set to 0 for existence and 2 for extension. The BLAST results were saved in FASTA format and aligned using ClustalW in BioEdit with some manual editing of the alignments. The precise signature regions were selected after alignment and the ranges of percent identity per species were calculated using BioEdit.
4. Discussion
Rapid and accurate diagnosis of the etiological agent present in a clinical sample is critical for appropriate responses including patient triage and outbreak control measures. Accurate detection also curtails disease transmission by preventing false-positive and false-negative misdiagnoses that may result in catastrophic consequences. Highly sensitive and specific assays also help to monitor response to therapy by quantitation of viral load during treatment and to assess the efficacy of therapeutics currently undergoing clinical trials. The current gold standard for EBOV diagnosis is rRT-PCR, and accurate diagnosis of the presence of EBOV in clinical samples relies on optimal RT-PCR assay kinetics.
Whole genome sequence analysis of the 2014 EBOV viral samples unraveled a major vulnerability of some of the current diagnostic assays due to genetic changes in the RT-PCR target genes [
15]. We therefore sought to perform quantitative comparisons of various existing assays before drawing any conclusion about potential inefficiencies of the current assays or design and performance of new assays. In the current study we took an empirical approach to evaluate many of the RT-PCR assays using the 2014 EBOV viral genomic material as a template in comparison to EBOV Kikwit. This analysis indicated varying efficiencies and sensitivities between the assays using the same RNA template and assay conditions. In fact, two of the currently fielded assays developed prior to the onset of the 2014 outbreak (one has less than 100% signature identity to the 2014 EBOV sequences) performed equally well compared to EBOV Kikwit template. These assays are being successfully used in clinical diagnostics in Western Africa. Most notably, we found minimal, if any, difference in assay performance between the old and new EBOV templates, giving confidence in the results of the diagnostic tests.
We also took an in silico approach to assess the effectiveness of the assays against all the available filovirus sequences in GenBank. This analysis indicated that out of the 24 assays analyzed only three assays showed perfect matches to their target sequences. All the other assays showed potential false-negative results. If the identities of the assay sequences were relaxed to 90/90, many of the assays met the standard. In other words, these assays would be functional depending on the location of the mismatches. Three assays failed even at this relaxed criteria due to false-positive or false-negative results. False-positive results, i.e., an assay meant to detect EBOV that also detects other members of ebolavirus may be acceptable in the case of EVD since the treatment options are the same for EVD caused by any ebolavirus. But there are other instances when false-positive results would result in unnecessary treatments, e.g., misdiagnosis of EVD in the setting of another viral infection with different transmission dynamics, treatment, and prognosis. However, false-negative results, i.e., misdiagnosis or non-diagnosis can have serious negative consequences.
Given the rapid accumulation of mutations in ebolavirus genomes and other RNA viruses, systematic and periodic
in silico evaluation of the current assay signatures against newly added sequences in public databases is crucial for deploying effective diagnostic tests for reemerging pathogens [
18]. Empirical testing of assays may not be feasible for every new isolate due to difficulties in obtaining and testing materials under the requisite containment levels in a rapid time frame.
It is well established that the introduction of EBOV in the current and past human outbreaks was from zoonotic reservoirs [
9,
10]. Hence, real-time biosurveillance of zoonotic sources in known hot spots of EVD may aid in preventing an outbreak in humans before it occurs. Biosurveillance may also allow tracking the changing genetic profiles of the virus itself in the environmental reservoir. Biosurveillance can be accomplished using currently available molecular assays and the genomic changes can be tracked by using unbiased high throughput sequencing of environmental samples using next-generation sequencing.
In this context, capturing the genetic diversity of geographically and temporally diverse hemorrhagic fever viruses is paramount for assessing and maintaining the effectiveness of assays deployed. As shown in the Bio
Velocity analysis performed in this study, lack of sequences representing the genomic diversity may hinder the effective design of diagnostic assays and hence would miss a new genetic variant that emerges as the causal organism of an outbreak. It is imperative that samples from outbreaks are made available in a timely fashion for sequencing [
27] and genomic sequence data generated are shared. Immediate release and availability of genomic sequences in public databases would allow for crowd sourcing of data analysis and rapid development of new assays or improvement of old assays. It is worth noting that crowd sourcing played a key part in rapid whole genome sequencing and post sequencing analysis of the outbreak pathogen, as well as in the development of new diagnostics during the 2011
E. coli O104:H4 outbreak that originated in Germany [
28]. Deep sequencing to decipher quasi-species would also elucidate the genetic trends in population within hosts. Using these data, one can predict the potential impact of genetic drifts on signature erosion and consequent failure of existing assays and develop new signatures if the need arises.
Previous studies documented the impact mismatches can have on real-time PCR efficiency [
24,
29]. Based on these observations and data presented here, we suggest that an
in silico analysis be performed on any new genomic sequence available to assess the effectiveness of the current assays. If the assays fails to meet specified quality metrics, a new assay design is definitely warranted. Contingent on the regulatory status of the existing assay, the timeliness and ability to impact the emerging outbreak may have to be considered. Any redesign to the assay will require, at minimum, additional analytical testing, and a regulatory submission to the FDA for review and approval.
Rational design of new assays targeting highly conserved regions of the genomes containing discriminatory sequences is essential for the effectiveness of diagnostic assays. The majority of the existing species-specific assays were designed based on limited sequence information and were gene-specific due to historical or laboratory specific interests. A rational approach would be to use all and ideally a large set of whole genome sequences to identify conserved segments. One such approach is described here using BioVelocity with subsequent in silico validation of the identified signatures. To provide not only unique but also robust signatures, the index organism genome diversity needs to be properly characterized. Thanks to the recent explosion in whole genome sequencing, this was possible in EBOV and to lesser extent in SUDV and RESTV due to limited number of sequences available. Identifying similar signatures in other species also requires more genome sequences from temporally and geographically diverse isolates that represent the entire genetic diversity of the species.
There is also the desire to use orthogonal approaches for increasing the confidence of the test results. A number of laboratory developed immuno-assays that complement molecular assays are in use in order to increase the confidence of the diagnostic result. Additionally, multiplexing molecular assays for simultaneous analysis of multiple samples and targets is highly desired in any outbreak scenario. This approach not only addresses issues with genomic change but also diagnosis of pathogens that present with similar symptoms. Enabling high throughput analysis in an outbreak scenario will immensely aid in rapid processing of hundreds of samples. We also would like to draw the attention of the research community that some of the sequence-based next generation sequencing platforms with high-throughput, multiplexing and rapid time to result capability will undoubtedly change the future of diagnostics in an outbreak scenario. A recent study based on the Illumina platform addressed such a possibility using a rapid sequencing approach [
30].
Improving current assays to address genomic changes of an emerging threat is of paramount concern demonstrated here and in other studies [
15,
17]; however, the implications of obtaining regulatory approvals and the timely availability of diagnostics during an active outbreak should also be considered. The Emergency Use Authorization (EUA) authority granted by Congress allows the FDA Commissioner to provide medical interventions (medical devices, diagnostics, therapeutics and vaccines) to the American people and Armed forces, to be used when there are no adequate, approved, and available alternatives. While significantly less onerous than the 510 k process, this regulatory path still requires a rigorous level of testing with respect to sensitivity and specificity for the assays as well as documented manufacturing processes to ensure quality of the product. Amending EUA approved diagnostic test applications that address genomic changes requires additional analytical testing, and review of the testing data by the FDA before a new approval is issued. In this context, a recent report by Kugelman
et al. raises the issue on the effectiveness of therapeutics and vaccines due to genetic drift [
17]. While that report focused on therapeutics, similar inferences can be drawn to the diagnostics in use from the current study.
In the current EBOV outbreak, the World Health Organization (WHO) expressed a desire to put in place an emergency quality assessment mechanism. According to the WHO statement [
11], this is a rapid review process for assessing a diagnostic’s quality, safety, and performance similar to the EUA process. Information sought included the recommended specimen types; evidence of test performance, including sensitivity and specificity; suppliers of critical components or raw materials and services, and data on current inventory and manufacturing capacity and quality. The overarching objective is to guide bulk procurement decisions by WHO and other partners that will get the best available tests to Western Africa in a short time frame [
11].
The PSET and BioVelocity pipelines described here can be easily adapted to assess signature erosion and assay performance of any virus or any other organism rapidly and develop new assay signatures and improve molecular assays. Currently, efforts are underway to improve molecular assays of various viruses such as Lassa and Crimean-Congo hemorrhagic fever (CCHF) virus and bacteria such as Burkholderia mallei and B. pseudomallei for which current assays are not optimal with respect to specificity and sensitivity.
It is quite apparent that developing diagnostic assays for RNA viruses is an arms race: rapid genetic changes (especially in the assay target sequences) in viral genomes necessitate changes in the assays in order to detect that genetic variant. These new signatures in turn decay over time forcing new assay designs. It is hoped that some of the considerations outlined in this study will guide future diagnostics development and raise awareness and initiate a discussion on these and related regulatory issues in light of the current EVD crisis in order to be better prepared for a future emerging or reemerging biothreat.
5. Conclusions
In this study we assessed the performance of existing EBOV assays using in vitro and in silico (PCR Signature Erosion Tool) approaches. Because viral evolution is dynamic, the genetic diversity of an outbreak species can expand during the course of an epidemic. Just as viral evolution is adaptive, the development of diagnostic tests must be open-ended to identify newly emerging quasi-species, and to mitigate the risks of lost sensitivity and false-negative results. This is particularly relevant to sequence-based diagnostic tests. In addition, flexibility in the regulatory process is essential. An accelerated FDA approval process should exist to account for inevitable changes to molecular assay signatures. In order to increase the confidence of diagnostic assays, orthogonal point of care diagnostics need to be readily available, with a minimal time to develop and deploy.
The rational deployment and development of sequence-based diagnostic tests depends upon WGS done early, quickly, deeply and repeatedly during an outbreak, and sharing the WGS openly with the scientific community including product developers for characterization and diagnostic design. It is also imperative to perform deep sequencing in order to capture changing genomic profiles present in very minor proportions in the population within a host so that one can predict and adjust the signatures if the possibility of clonal expansion of a minor variant impacting the assay performance exists.
It is critically important to assess the performance of assays in high consequence outbreaks against the species circulating in the outbreak. WGS of the circulating species can have a material impact on the spread of the disease, and on lives. While WGS is most desirable, more limited sequencing of the highly conserved regions may also produce a similar improvement. Sharing assay results with patient symptoms and outcome, via a multi-platform data repository that links these data set is needed. Ease of data entry and automated data communications are key requirements for such a system.
Currently deployed assays, even though designed to target regions of the genome believed to be conserved, have not been tested against new sequences available since their creation and may not have targeted the most highly conserved regions of the genome in the circulating viral population. We applied BioVelocity to identify specific and robust signatures for each ebolavirus and then validated these results in silico. For EBOV, ten unique signatures were identified, none of which overlap with the currently used signatures including the 12 EBOV assays examined in this study indicating a gene-centric approach in the past assay design strategy rather than uniqueness. We have used a novel approach using the power of a large-memory kmer-based technique to rapidly design conserved and unique signatures. BioVelocity could not run on normal cluster architectures with limited memory, or would take orders of magnitude longer if secondary storage were used for the kmer hash tables.
Tools such as BioVelocity enable identification of unique and robust signatures for qPCR targets in minutes; however, to develop robust signatures with low false-negative rates, an adequate number of reference genomes are needed. The required size of the reference genome database is related to the genetic mutation rate and diversity of the population, and the tolerance for false-negative rates.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}