
Honey Bee Infecting Lake Sinai Viruses


Abstract
:1. Introduction
2. Results
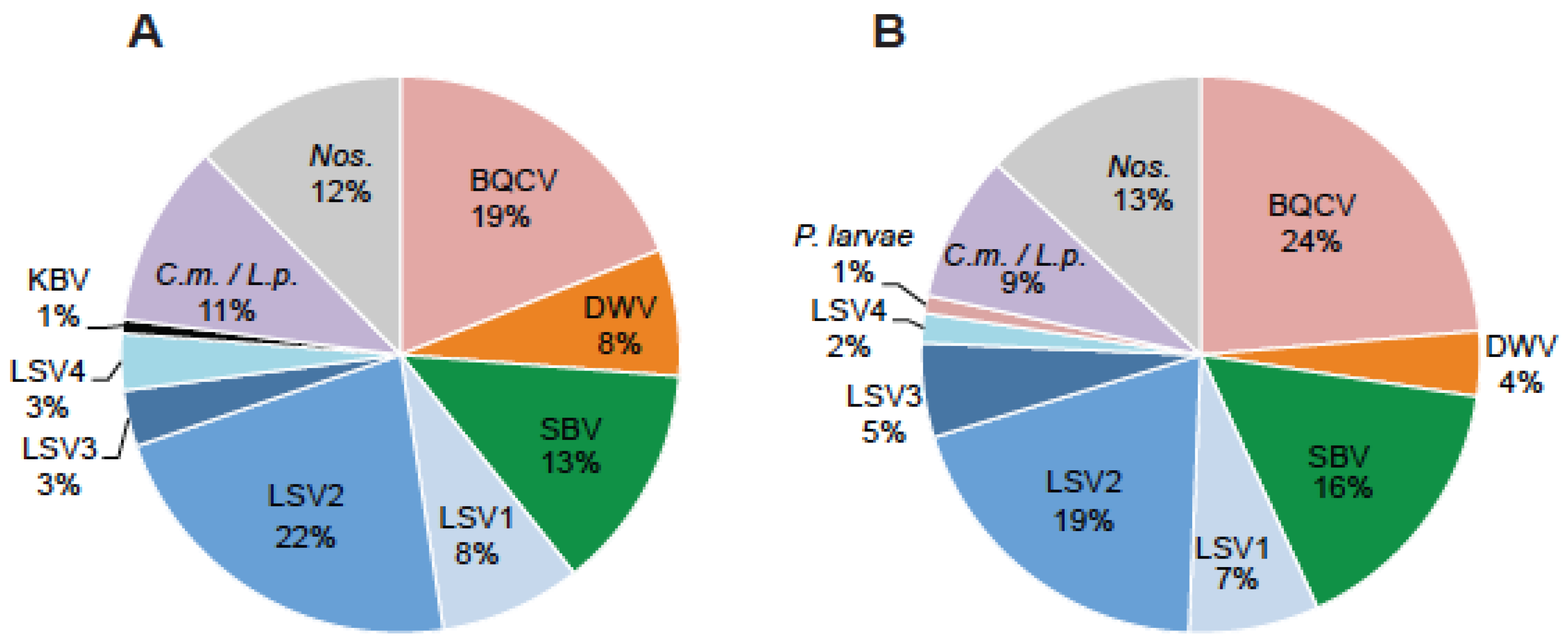
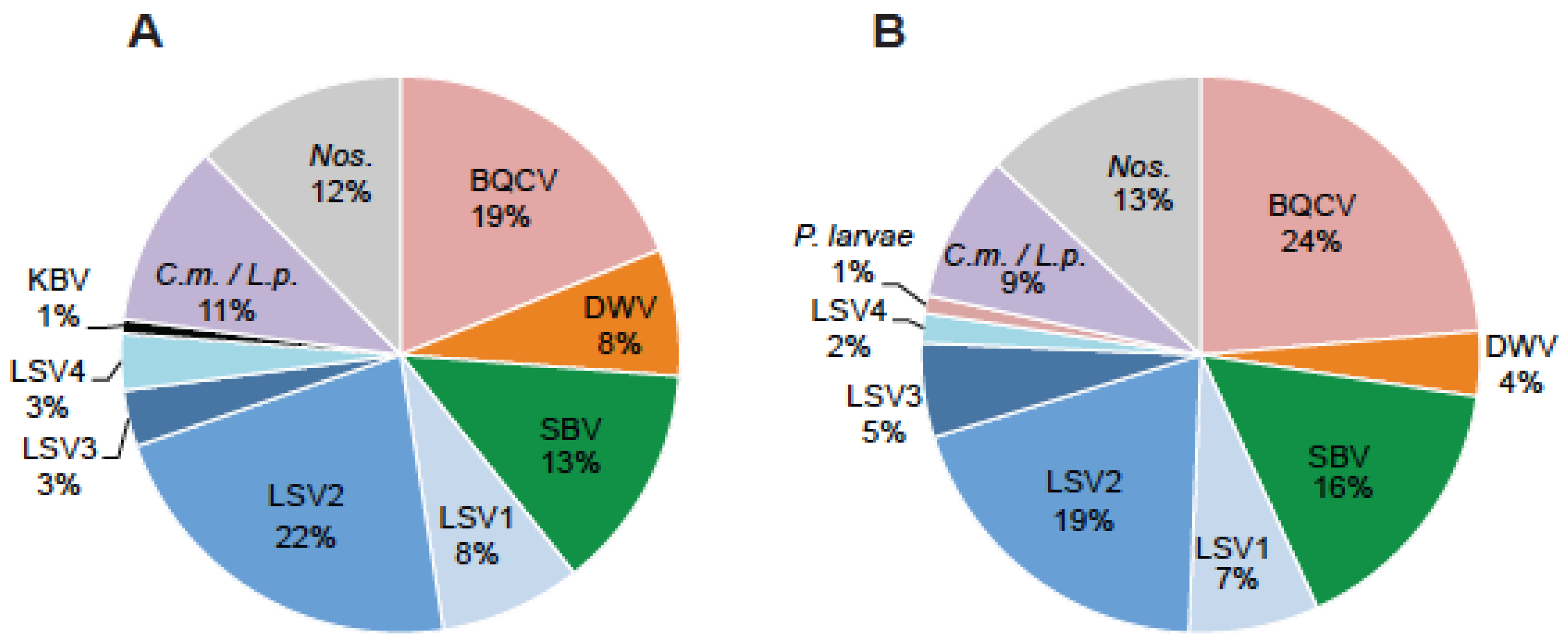
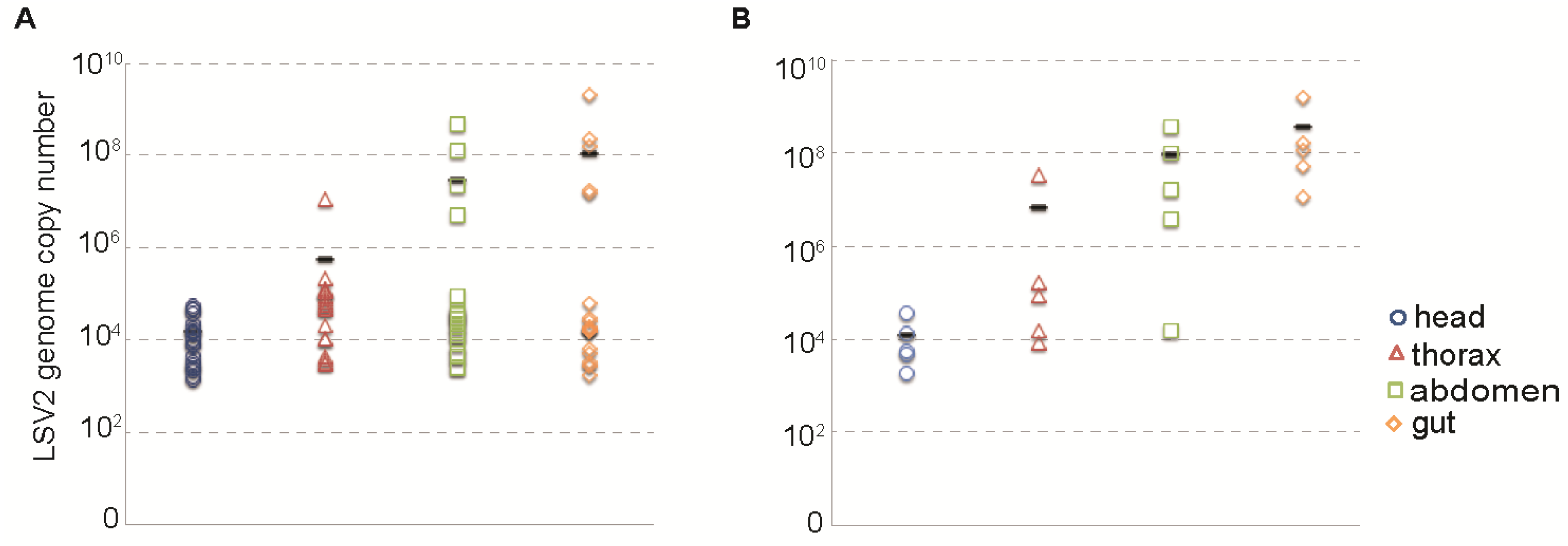
2.1. Lake Sinai Viruses Are Readily Detected in Honey Bee Samples
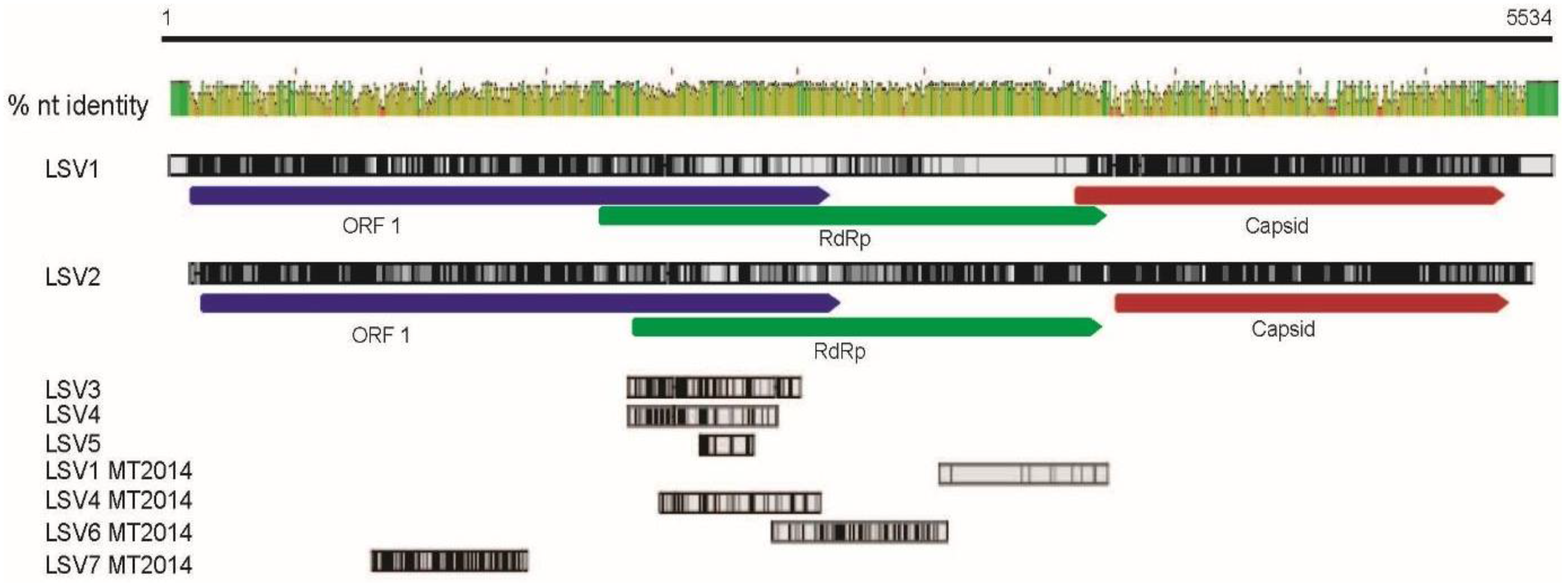
2.2. Lake Sinai Virus Genome Sequences Extended

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Accession Number | Read Length | New Seq. Length |
|---|---|---|---|
| LSV1 3′end | KR022003 | 611 nt | 396 nt |
| LSV2 5′end | KR022002 | 280 nt | 54 nt |
| LSV2 3′end | KR022004 | 645 nt | 501 nt |
| LSV1 2014 MT | KR021356 | 659 nt | n/a |
| LSV4 2014MT | KP892556 | 576 nt | 129 nt |
| LSV6 2014MT | KR021357 | 684 nt | 684 nt |
| LSV7 2015 MT | KR021355 | 604 nt | 604 nt |
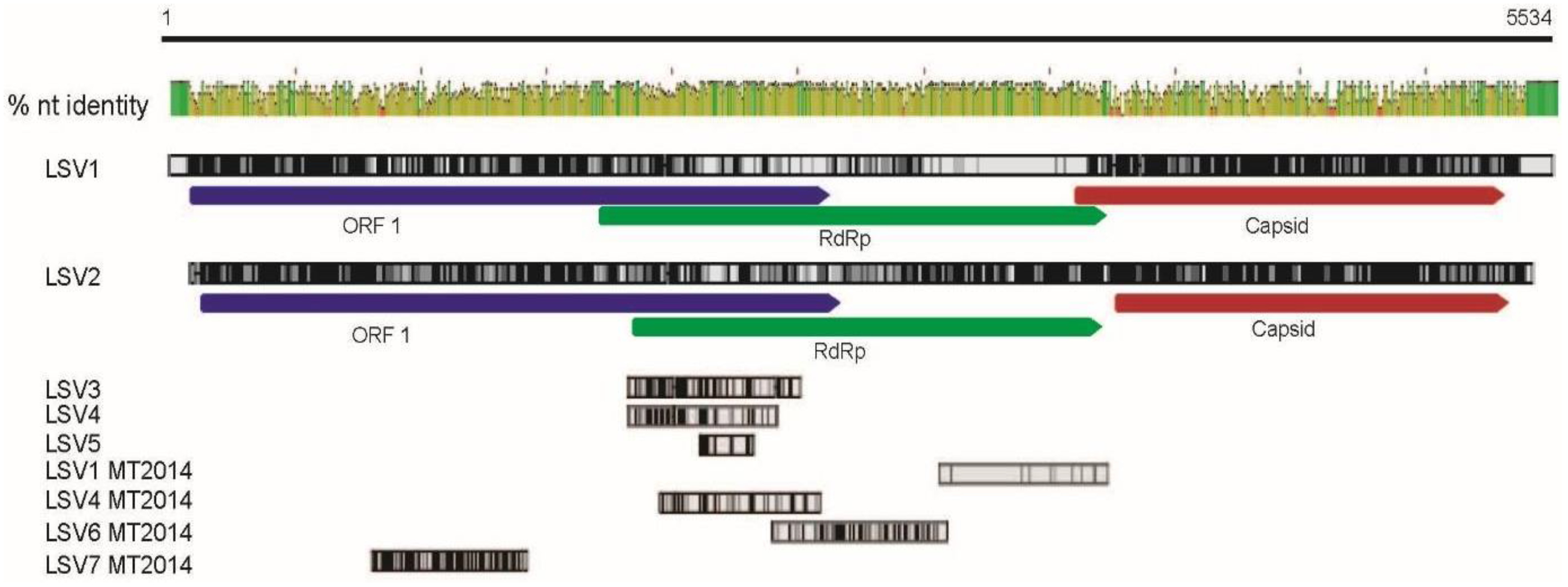
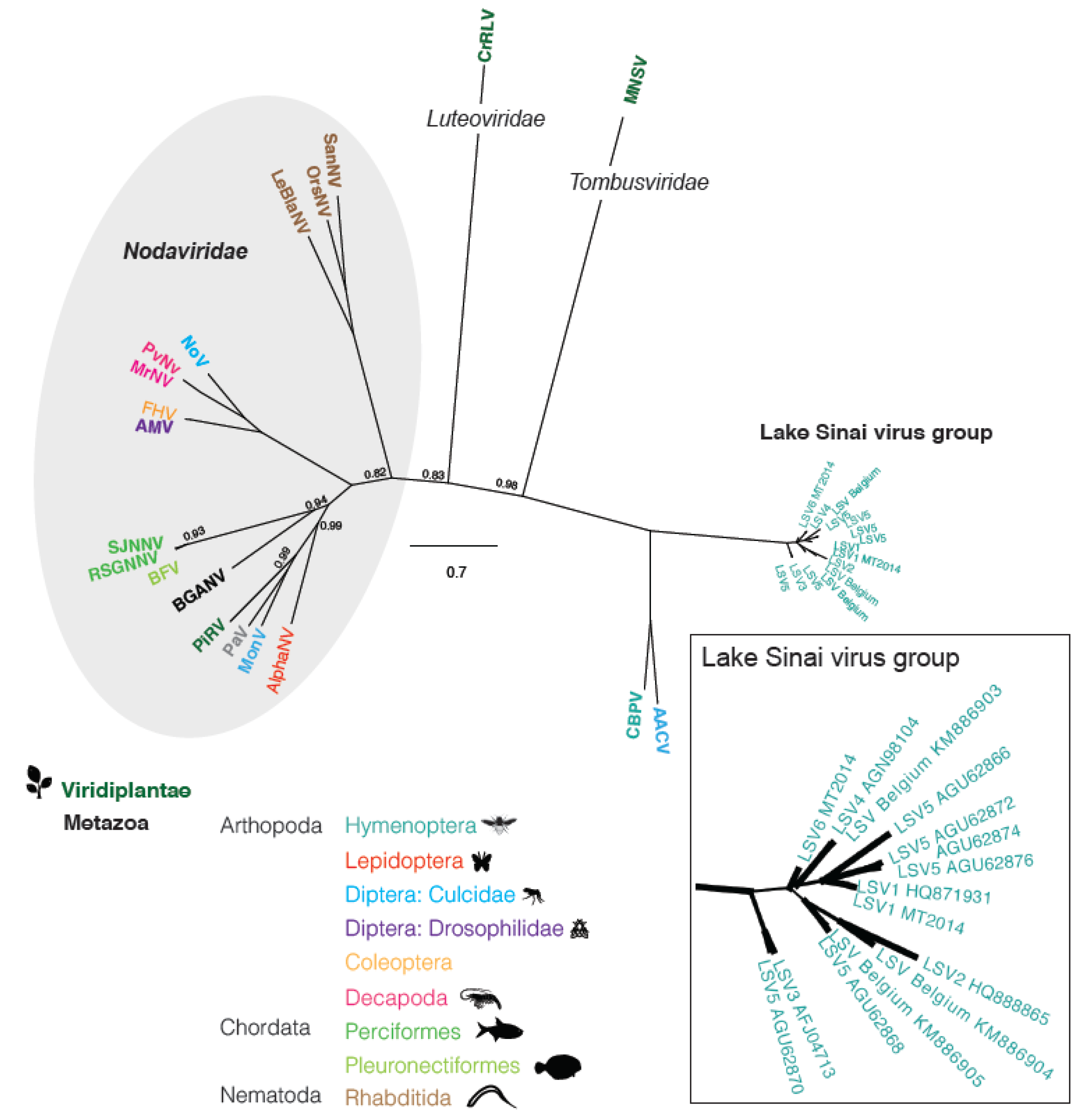
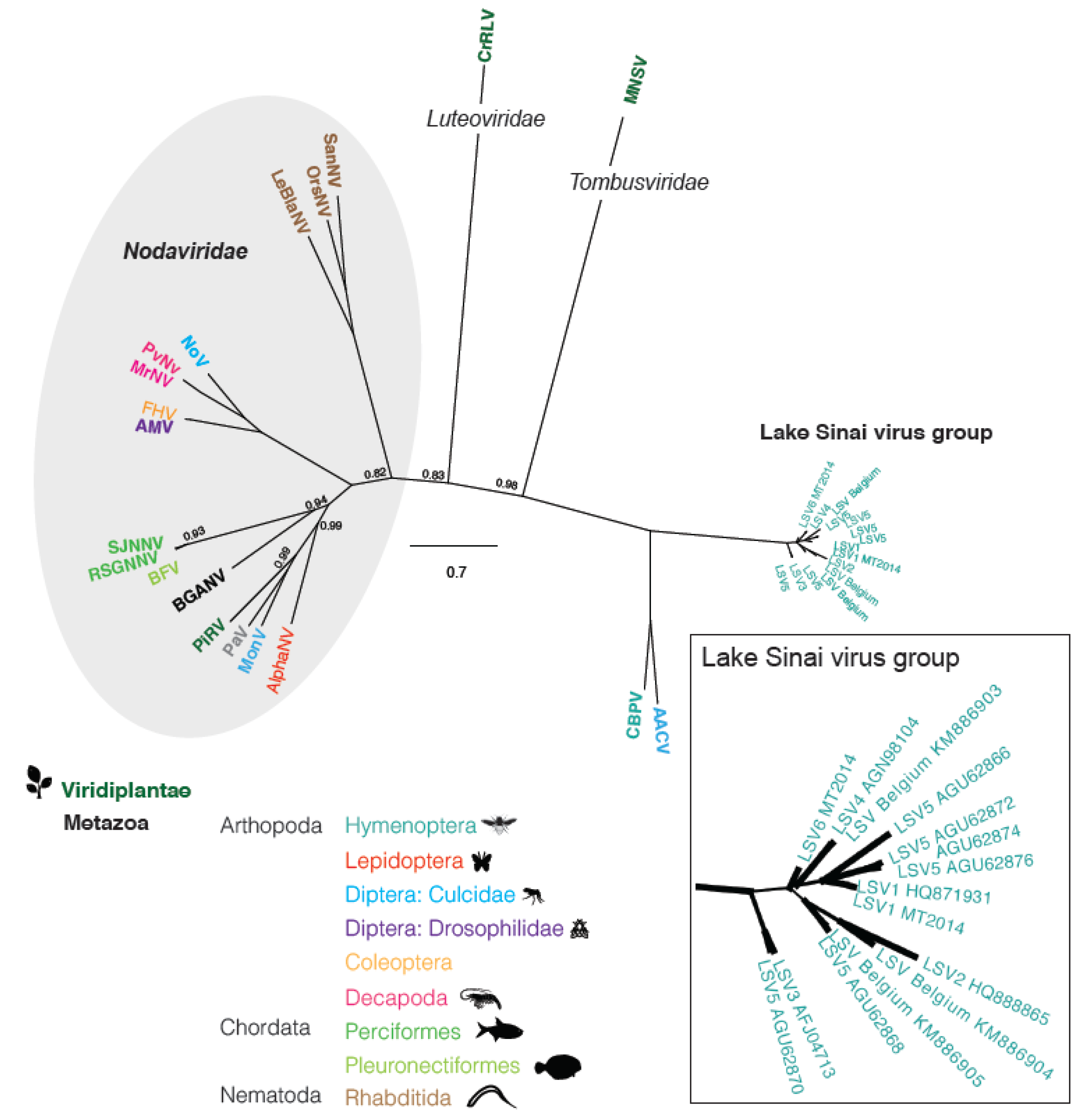
2.3 Phylogenetic Analysis
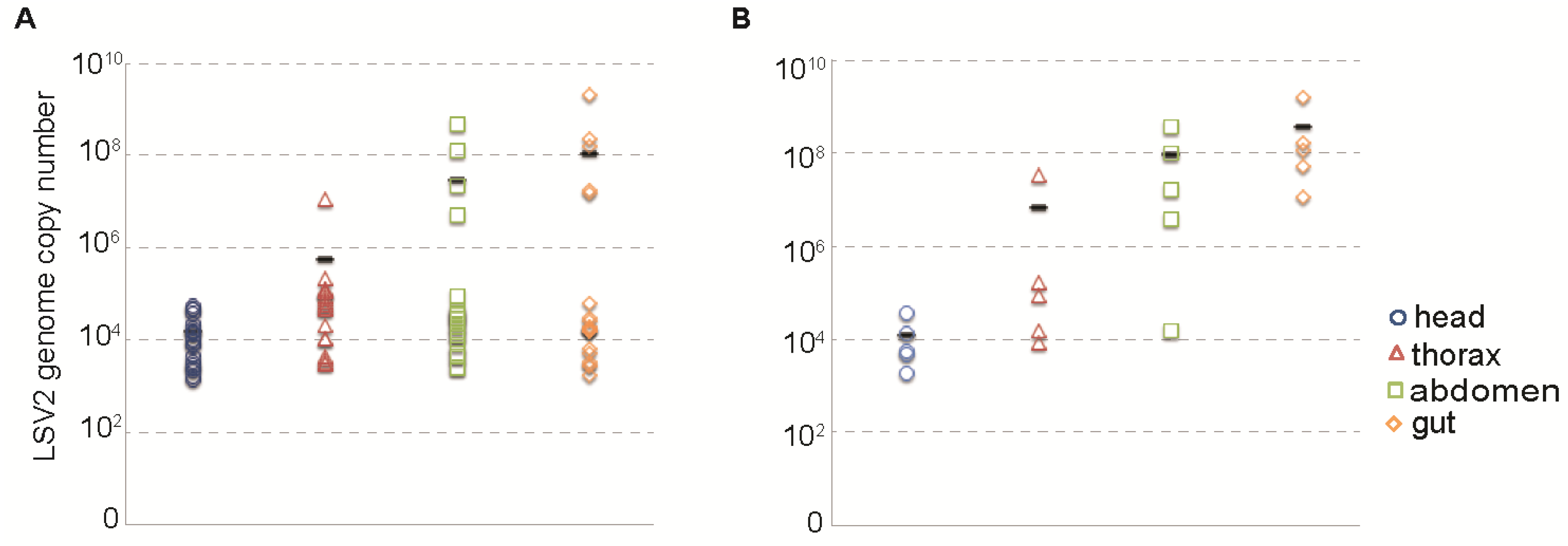
2.4. Relative Distribution of Lake Sinai Virus 2
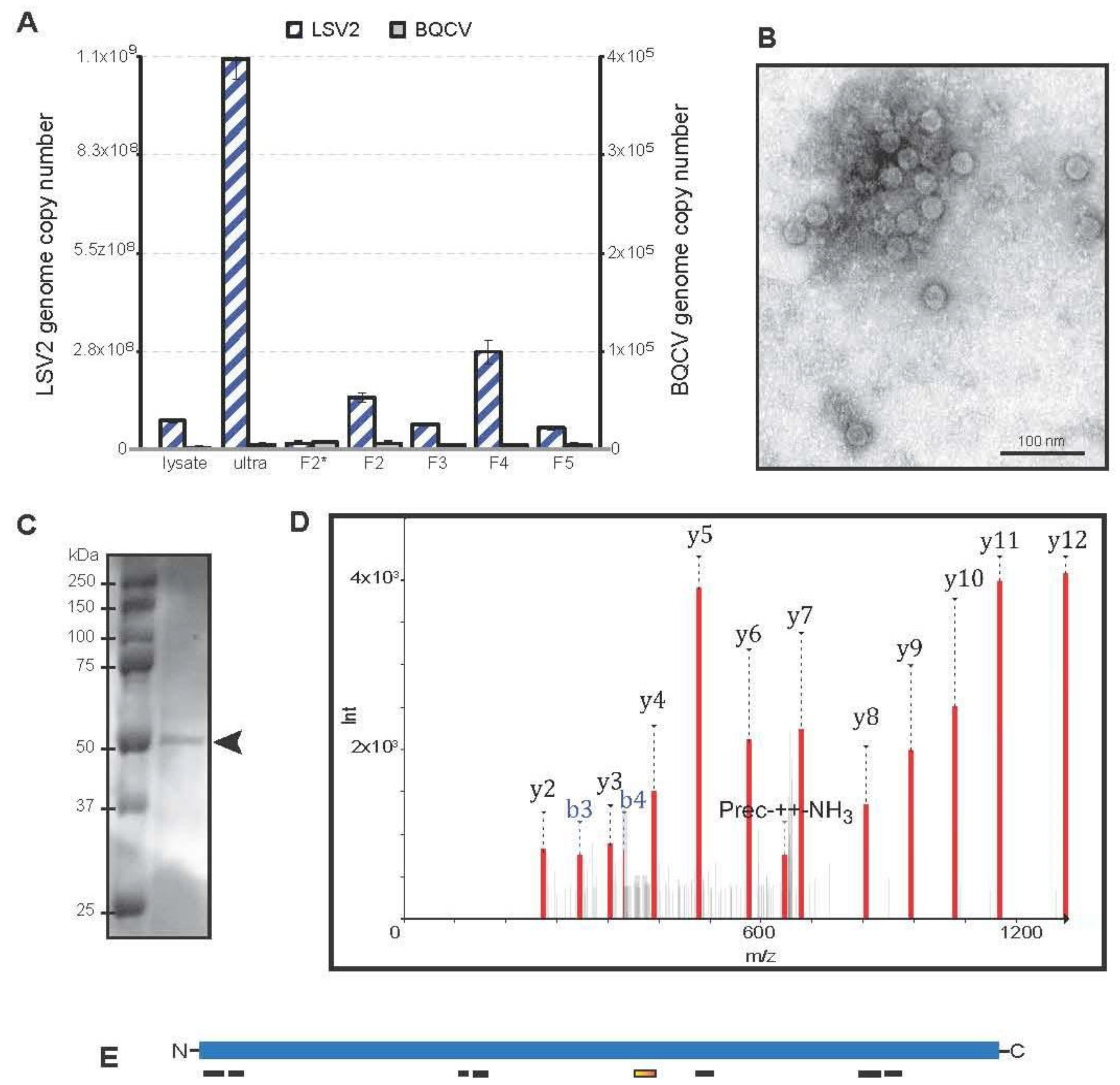
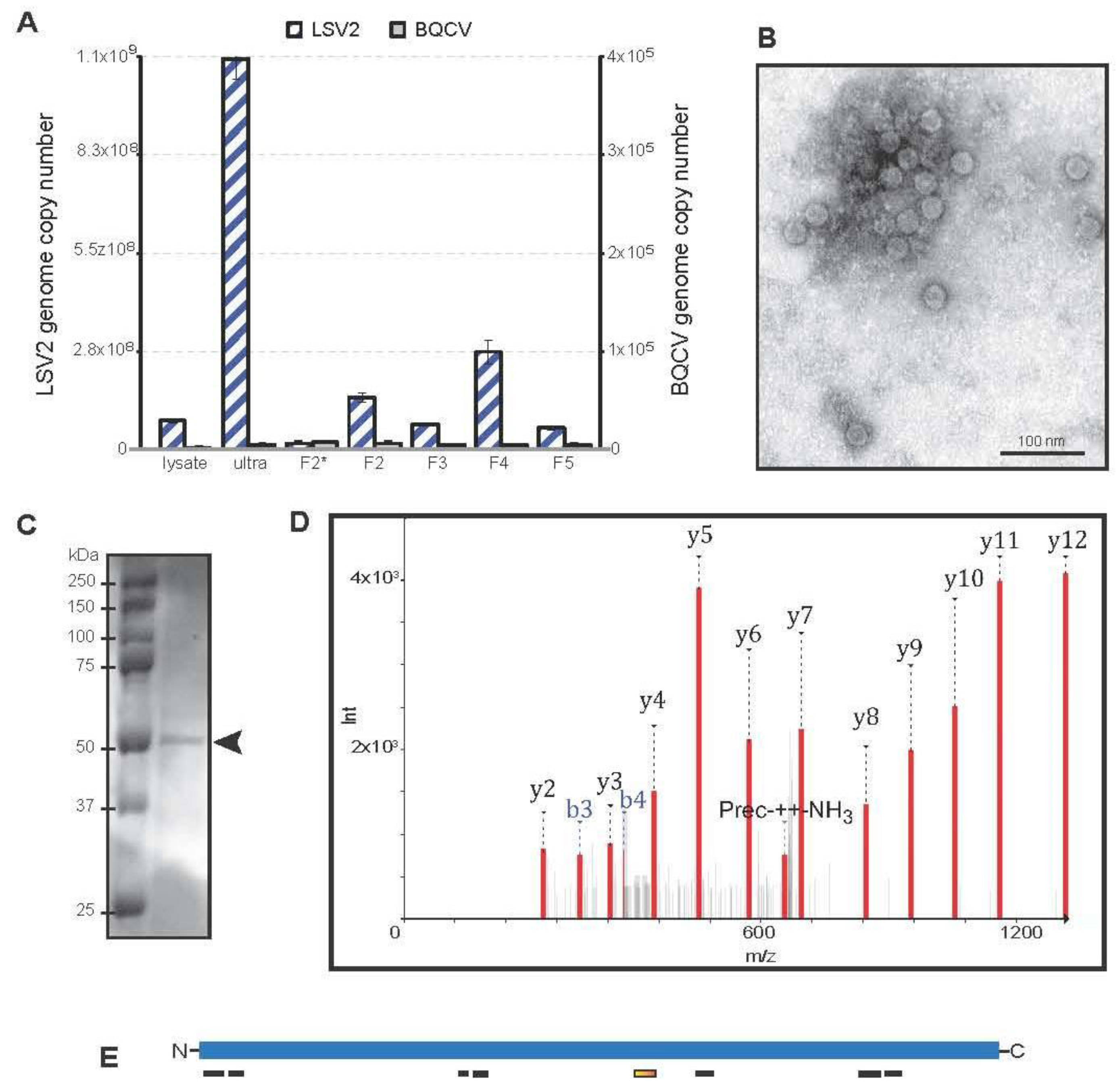
2.5. Lake Sinai Virus 2 Purification and Characterization
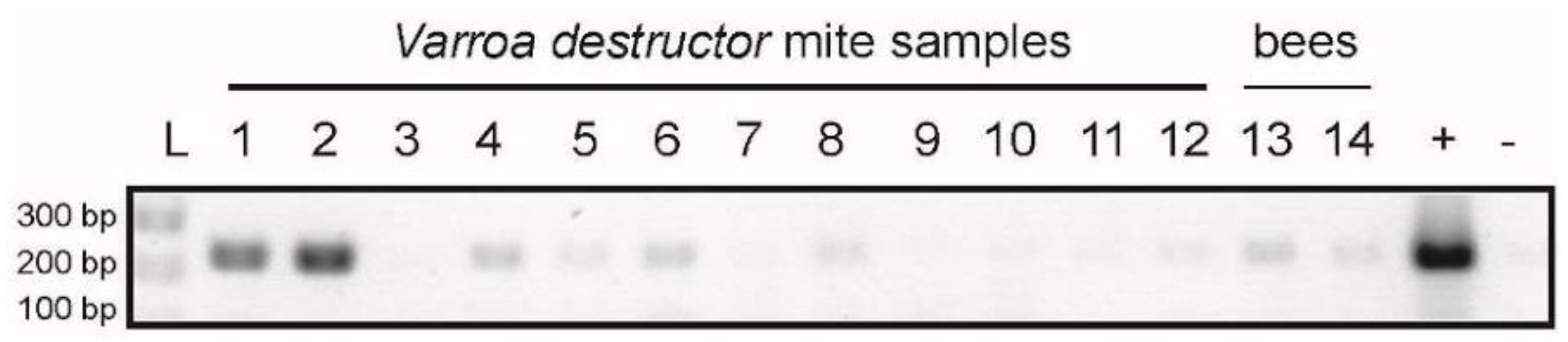
2.6. LSV1 and LSV2 Detected in the Varroa Destructor Mite
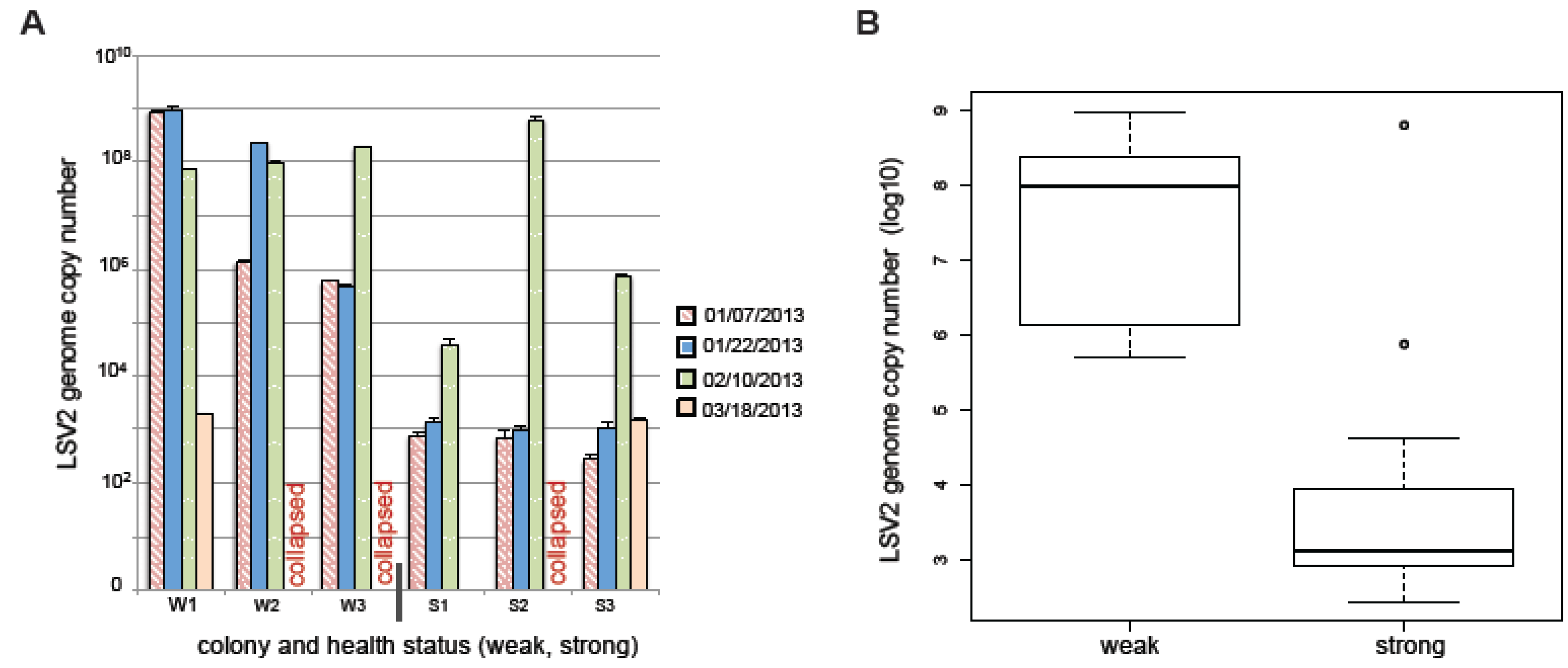
2.7. LSV and Colony Health
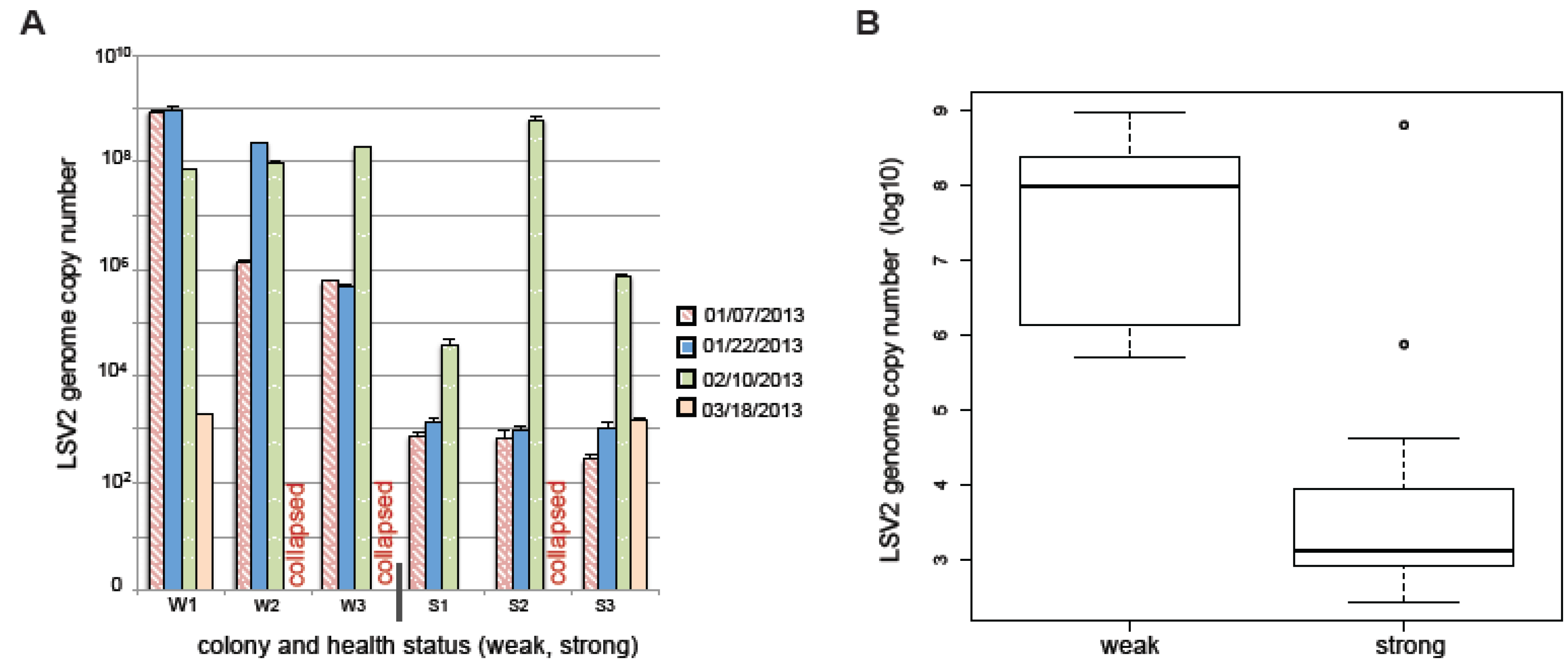
3. Discussion
4. Methods
4.1. Honey Bee and Mite Samples
4.2. RNA Isolation
4.3. Rapid Amplification of cDNA Ends (RACE)
4.4. Reverse Transcription/cDNA Synthesis
4.5. Negative Strand-Specific RT-PCR
4.6. Polymerase Chain Reaction (PCR)
4.7. Quantitative PCR (qPCR)
4.8. Honey Bee Pathogen Screening by PCR and qPCR
4.9. Virus Isolation and Analysis
4.10. Transmission Electron Microscopy (TEM)
4.11. SDS-PAGE
4.12. Phylogenetic Analysis
4.13. Mass Spectrometry of LSV Capsid Protein
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gallai, N.; Salles, J.-M.; Settele, J.; Vaissière, B.E. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef]
- Calderone, N.W. Insect pollinated crops, insect pollinators and US agriculture: Trend analysis of aggregate data for the period 1992–2009. PLoS ONE 2012, 7, e37235. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, R.; Pisa, L.; Andonov, S.; Brodschneider, R.; Charriere, J.-D.; Chlebo, R.; Coffey, M.F.; Crailsheim, K.; Dahle, B.; Gajda, A.; et al. Managed honey bee colony losses in Canada, China, Europe, Israel and Turkey, for the winters of 2008-9 and 2009-10. J. Apic. Res. 2012, 51, 91–114. [Google Scholar] [CrossRef]
- Clermont, A.; Eickermann, M.; Kraus, F.; Georges, C.; Hoffmann, L.; Beyer, M. A survey on some factors potentially affecting losses of managed honey bee colonies in Luxembourg over the winters 2010/2011 and 2011/2012. J. Apic. Res. 2014, 53, 43–56. [Google Scholar] [CrossRef]
- McMenamin, A.J.; Genersch, E. Honey bee colony losses and associated viruses. Curr. Opin. Insect Sci. 2015, 8, 121–129. [Google Scholar] [CrossRef]
- Van Engelsdorp, D.; Hayes, J.; Underwood, R.M.; Pettis, J. A Survey of Honey Bee Colony Losses in the US, Fall 2007 to Spring 2008. PLoS ONE 2008, 3, e4071. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, N.A.; Rennich, K.; Wilson, M.E.; Caron, D.M.; Lengerich, E.J.; Pettis, J.S.; Rose, R.; Skinner, J.A.; Tarpy, D.R.; Wilkes, J.T.; et al. A national survey of managed honey bee 2012–2013 annual colony losses in the USA: Results from the Bee Informed Partnership. J. Apic. Res. 2014, 53, 1–18. [Google Scholar] [CrossRef]
- Spleen, A.M.; Lengerich, E.J.; Rennich, K.; Caron, D.; Rose, R.; Pettis, J.S.; Henson, M.; Wilkes, J.T.; Wilson, M.; Stitzinger, J.; et al. A national survey of managed honey bee 2011–12 winter colony losses in the United States: Results from the Bee Informed Partnership. J. Apic. Res. 2013, 52, 44–53. [Google Scholar] [CrossRef]
- VanEngelsdorp, D.; Caron, D.; Hayes, J.; Underwood, R.; Henson, M.; Rennich, K.; Spleen, A.; Andree, M.; Snyder, R.; Lee, K.; et al. A national survey of managed honey bee 2010-11 winter colony losses in the USA: Results from the Bee Informed Partnership. J. Apic. Res. 2012, 51, 115–124. [Google Scholar] [CrossRef]
- VanEngelsdorp, D.; Evans, J.D.; Saegerman, C.; Mullin, C.; Haubruge, E.; Nguyen, B.K.; Frazier, M.; Frazier, J.; Cox-Foster, D.; Chen, Y.; et al. Colony collapse disorder: A descriptive study. PLoS ONE 2009, 4, e6481. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.D.; Evans, J.D.; Pettis, J. Colony losses, managed colony population decline, and Colony Collapse Disorder in the United States. J. Apic. Res. 2010, 49, 134–136. [Google Scholar] [CrossRef]
- Pettis, J.S.; Delaplane, K.S. Coordinated responses to honey bee decline in the USA. Apidologie 2010, 41, 256–263. [Google Scholar] [CrossRef]
- Nazzi, F.; Pennacchio, F. Disentangling multiple interactions in the hive ecosystem. Trends Parasitol. 2014, 30, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.S.; Tarpy, D.R.; Chen, Y.; Jeffreys, L.; Lopez, D.; Pettis, J.S.; vanEngelsdorp, D.; Evans, J.D. Pathogen webs in collapsing honey bee colonies. PLoS ONE 2012, 7, e43562. [Google Scholar] [CrossRef] [PubMed]
- Cox-Foster, D.L.; Conlan, S.; Holmes, E.C.; Palacios, G.; Evans, J.D.; Moran, N.A.; Quan, P.-L.; Briese, T.; Hornig, M.; Geiser, D.M.; et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 2007, 318, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Evans, J.D.; Robinson, G.E.; Berenbaum, M.R. Changes in transcript abundance relating to colony collapse disorder in honey bees (Apis mellifera). Proc. Natl. Acad. Sci. USA 2009, 106, 14790–14795. [Google Scholar] [CrossRef] [PubMed]
- Tentcheva, D.; Gauthier, L.; Zappulla, N.; Dainat, B.; Cousserans, F.; Colin, M.E.; Bergoin, M. Prevalence and seasonal variations of six bee viruses in Apis mellifera L. and Varroa destructor mite populations in France. Appl. Environ. Microbiol. 2004, 70, 7185–7191. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E.; von der Ohe, W.; Kaatz, H.; Schroeder, A.; Otten, C.; Büchler, R.; Berg, S.; Ritter, W.; Mühlen, W.; Gisder, S.; et al. The German bee monitoring project: A long term study to understand periodically high winter losses of honey bee colonies. Apidologie 2010, 41, 332–352. [Google Scholar] [CrossRef]
- Chen, Y.P.; Pettis, J.S.; Corona, M.; Chen, W.P.; Li, C.J.; Spivak, M.; Visscher, P.K.; Zhao, Y.; DeGrandi-Hoffman, G.; Boncristiani, H.; et al. Israeli Acute Paralysis Virvus: Epidemiology, Pathogenesis and Implications for Honey Bee Health. PLoS Pathog. 2014, 10, e1004261. [Google Scholar] [CrossRef] [PubMed]
- Runckel, C.; Flenniken, M.L.; Engel, J.C.; Ruby, J.G.; Ganem, D.; Andino, R.; DeRisi, J.L. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS ONE 2011, 6, e20656. [Google Scholar] [CrossRef] [PubMed]
- vanEngelsdorp, D.; Lengerich, E.; Spleen, A.; Dainat, B.; Cresswell, J.; Baylis, K.; Nguyen, B.K.; Soroker, V.; Underwood, R.; Human, H.; et al. Standard epidemiological methods to understand and improve Apis mellifera health. J. Apic. Res. 2013, 52. [Google Scholar] [CrossRef]
- International Committee on the Taxonomy of Viruses (ICTV). Available online: http://www.ictvonline.org/virusTaxonomy.asp?msl_id=28 (accessed on 15 June 2015).
- Granberg, F.; Vicente-Rubiano, M.; Rubio-Guerri, C.; Karlsson, O.E.; Kukielka, D.; Belák, S.; Sánchez-Vizcaíno, J.M. Metagenomic detection of viral pathogens in spanish honeybees: Co-infection by aphid lethal paralysis, Israel acute paralysis and Lake Sinai viruses. PLoS ONE 2013, 8, e57459. [Google Scholar] [CrossRef] [PubMed]
- Ravoet, J.; Maharramov, J.; Meeus, I.; de Smet, L.; Wenseleers, T.; Smagghe, G.; de Graaf, D.C. Comprehensive Bee Pathogen Screening in Belgium Reveals Crithidia mellificae as a New Contributory Factor to Winter Mortality. PLoS ONE 2013, 8, e72443. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Zirkel, F.; Kurth, A.; van Cleef, K.W.R.; Drosten, C.; van Rij, R.P.; Junglen, S. A unique Nodavirus with novel features: Mosinovirus expresses two subgenomic RNAs, a capsid gene of unknown origin, and a suppressor of the antiviral RNAi pathway. J. Virol. 2014, 88, 13447–13459. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Siede, R. Honey Bee Viruses. In Advances in Virus Research; Elsevier: Atlanta, GA, USA, 2007; Volume 70, pp. 33–80. [Google Scholar]
- King, A.; Adams, M.J.; Lefkowitz, E.J.; Carstens, E.B. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: San Diego, CA, USA, 2011. [Google Scholar]
- Brutscher, L.M.; Daughenbaugh, K.F.; Flenniken, M.L. Antiviral Defense Mechanisms in Honey Bees. Curr. Opin. Insect Sci. 2015, 10, 71–82. [Google Scholar] [CrossRef]
- Ribière, M.; Olivier, V.; Blanchard, P. Chronic bee paralysis: A disease and a virus like no other? J. Invertebr. Pathol. 2010, 103 (Suppl. 1), S120–S131. [Google Scholar] [CrossRef] [PubMed]
- Ravoet, J.; de Smet, L.; Wenseleers, T.; de Graaf, D.C. Genome sequence heterogeneity of Lake Sinai Virus found in honey bees and Orf1/RdRP-based polymorphisms in a single host. Virus Res. 2015, 201, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Tozkar, C.O.; Kence, M.; Huang, Q.; Evans, J.D. Metatranscriptomic analyses of honey bee colonies. Front. Genet. 2015, 6, e100. [Google Scholar] [CrossRef] [PubMed]
- Ravoet, J.; de Smet, L.; Meeus, I.; Smagghe, G.; Wenseleers, T.; de Graaf, D.C. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 2014, 122, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Expanding networks of RNA virus evolution. BMC Biol. 2012, 10, e54. [Google Scholar] [CrossRef] [PubMed]
- Runckel, C.; DeRisi, J.; Flenniken, M.L. A draft genome of the honey bee trypanosomatid parasite Crithidia mellificae. PLoS ONE 2014, 9, e95057. [Google Scholar] [CrossRef]
- Schwarz, R.S.; Bauchan, G.R.; Murphy, C.A.; Ravoet, J.; de Graaf, D.C.; Evans, J.D. Characterization of Two Species of Trypanosomatidae from the Honey Bee Apis mellifera: Crithidia mellificae Langridge and McGhee, and Lotmaria passim n. gen., n. sp. J. Eukaryot. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Schwarz, R.S. Bees brought to their knees: Microbes affecting honey bee health. Trends Microbiol. 2011, 19, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Chung, B.Y.W.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glücksman, E.; Wang, H.; Brown, T.D.K.; et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS ONE 2013, 8, e80720. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Delaplane, K.S.; van der Steen, J. Standard methods for estimating strength parameters of Apis mellifera colonies. J. Apic. Res. 2013, 52. [Google Scholar] [CrossRef]
- Sagili, R.R.; Burgett, D.M. Evaluating Honey Bee Colonies for Pollination: A Guide for Commercial Growers and Beekeepers. Available online: http://digitalarchives.wa.gov/WA.Media/do/49E233CA126700D7CCB9906279FFAF59.pdf (accessed on 16 June 2015).
- Koonin, E.V.; Wolf, Y.I.; Nagasaki, K.; Dolja, V.V. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat. Rev. Microbiol. 2008, 11, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Yang, X.; Cox-Foster, D.; Cui, L. The role of varroa mites in infections of Kashmir bee virus (KBV) and deformed wing virus (DWV) in honey bees. Virology 2005, 342, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Di Prisco, G.; Pennacchio, F.; Caprio, E.; Boncristiani, H.F.; Evans, J.D.; Chen, Y. Varroa destructor is an effective vector of Israeli acute paralysis virus in the honeybee, Apis mellifera. J. Gen. Virol. 2011, 92, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Ongus, J.R.; Peters, D.; Bonmatin, J.-M.; Bengsch, E.; Vlak, J.M.; van Oers, M.M. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 2004, 85, 3747–3755. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pettis, J.; Evans, J.; Kramer, M. Transmission of Kashmir bee virus by the ectoparasitic mite Varroa destructor. Apidologie 2004, 35, 441–448. [Google Scholar] [CrossRef]
- Boncristiani, H.F.; di Prisco, G.; Pettis, J.S.; Hamilton, M.; Chen, Y.P. Molecular approaches to the analysis of deformed wing virus replication and pathogenesis in the honey bee, Apis mellifera. Virol. J. 2009, 6, e221. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Bailey, L.; Ball, B.V. Standard methods for virus research in Apis mellifera. J. Apic. Res. 2013, 52. [Google Scholar] [CrossRef]
- Pirk, C.; de Miranda, J.R.; Kramer, M. Statistical guidelines for Apis mellifera research. J. Apic. Res. 2013, 52, 1–24. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian phylogenetic inference using DNA sequences: A Markov Chain Monte Carlo Method. Mol. Biol. Evol. 1997, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.O. Phylogenetic systematics turns over a new leaf. Trends Ecol. Evol. 2001, 16, 30–37. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.-I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Barsnes, H.; Berven, F.S.; Sickmann, A.; Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Breiter, D.; Beck, F.; Rahnenführer, J.; Martens, L.; Zahedi, R.P. D-score: A search engine independent MD-score. Proteomics 2013, 13, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Barsnes, H.; Vaudel, M.; Colaert, N.; Helsens, K.; Sickmann, A.; Berven, F.S.; Martens, L. compomics-utilities: An open-source Java library for computational proteomics. BMC Bioinform. 2011, 12, e70. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Côté, R.; Reisinger, F.; Barsnes, H.; Foster, J.M.; Rameseder, J.; Hermjakob, H.; Martens, L. The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 2010, 38, D736–D765. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daughenbaugh, K.F.; Martin, M.; Brutscher, L.M.; Cavigli, I.; Garcia, E.; Lavin, M.; Flenniken, M.L. Honey Bee Infecting Lake Sinai Viruses. Viruses 2015, 7, 3285-3309. https://doi.org/10.3390/v7062772
Daughenbaugh KF, Martin M, Brutscher LM, Cavigli I, Garcia E, Lavin M, Flenniken ML. Honey Bee Infecting Lake Sinai Viruses. Viruses. 2015; 7(6):3285-3309. https://doi.org/10.3390/v7062772
Chicago/Turabian StyleDaughenbaugh, Katie F., Madison Martin, Laura M. Brutscher, Ian Cavigli, Emma Garcia, Matt Lavin, and Michelle L. Flenniken. 2015. "Honey Bee Infecting Lake Sinai Viruses" Viruses 7, no. 6: 3285-3309. https://doi.org/10.3390/v7062772
APA StyleDaughenbaugh, K. F., Martin, M., Brutscher, L. M., Cavigli, I., Garcia, E., Lavin, M., & Flenniken, M. L. (2015). Honey Bee Infecting Lake Sinai Viruses. Viruses, 7(6), 3285-3309. https://doi.org/10.3390/v7062772