Abstract
Co-evolution of viruses and their hosts has reached a fragile and dynamic equilibrium that allows viral persistence, replication and transmission. In response, infected hosts have developed strategies of defense that counteract the deleterious effects of viral infections. In particular, single-strand DNA editing by Apolipoprotein B Editing Catalytic subunits proteins 3 (APOBEC3s) is a well-conserved mechanism of mammalian innate immunity that mutates and inactivates viral genomes. In this review, we describe the mechanisms of APOBEC3 editing during viral replication, the viral strategies that prevent APOBEC3 activity and the consequences of APOBEC3 modulation on viral fitness and host genome integrity. Understanding the mechanisms involved reveals new prospects for therapeutic intervention.
1. APOBEC3s Edit Single-Stranded DNA
The APOBEC3 enzymes are deaminases that edit single-stranded DNA (ssDNA) sequences by transforming deoxycytidine into deoxyuridine [1,2,3]. APOBEC3s are involved in the mechanisms of innate defense against exogenous viruses and endogenous retroelements [3]. The human genome codes for seven APOBEC3 genes clustered in tandem on chromosome 22 (namely A3A, A3B, A3C, A3DE, A3F, A3G, and A3H) and surrounded by the CBX6 and CBX7 genes. All APOBEC3 genes encode a single- or a double-zinc-coordinating-domain protein. Each zinc-domain belongs to one of the three distinct phylogenic clusters termed Z1, Z2 and Z3. The seven APOBEC3 genes arose via gene duplications and fusions of a key mammalian ancestor with a CBX6-Z1-Z2-Z3-CBX7 locus organization. Aside from mice and pigs, duplications of APOBEC3 genes have occurred independently in different lineages: humans and chimpanzees (n = 7), horses (n = 6), cats (n = 4), and sheep and cattle (n = 3) [4,5]. Read-through transcription, alternative splicing and internal transcription initiation may further extend the diversity of APOBEC3 proteins.
APOBEC3s are interferon-inducible genes [6] that are highly expressed in immune cells despite being present in almost all cell types [7,8]. The sub-cellular localization differs between the APOBEC3s isoforms: A3DE/A3F/A3G are excluded from chromatin throughout mitosis and become cytoplasmic during interphase, A3B is nuclear and A3A/A3C/A3H are cell-wide during interphase [9].
APOBEC3s exert an antiviral effect either dependently or independently of their deaminase activity. The deaminase activity involves the removal of the exocyclic amine group from deoxycytidine to form deoxyuridine. This process can generate different types of substitutions. First, DNA replication through deoxyuridine leads to the insertion of a deoxyadenosine, therefore causing a C to T transition. Alternatively, Rev1 translesion synthesis DNA polymerase can insert a C in front of an abasic site that is produced through uracil excision by uracil-DNA glycosylase (UNG2) leading to a C-to-G transversion [10]. In addition to inducing deleterious mutations in the viral genome, deamination of deoxycytidine can also initiate degradation of uracilated viral DNA via a UNG2-dependent pathway [11,12]. On the other hand, deaminase-independent inhibition requires binding of APOBEC3s to single-stranded DNA or RNA viral sequences at various steps of the replication cycle [13,14,15,16,17,18,19,20,21,22,23].
2. APOBEC3 Edition during Viral Replication Cycles
The mechanism of APOBEC3s inactivation is dependent on the type of virus and its mode of replication.
2.1. Retroviruses
Retroviruses are plus-strand single-stranded RNA viruses replicating via a DNA intermediate generated in the cytoplasm by reverse transcription. Human retroviruses notably include HIV (human immunodeficiency virus) and HTLV (human T-lymphotropic virus).
2.1.1. HIV-1
Historically, the first member of the APOBEC3 family was discovered in a groundbreaking study on HIV-1 [24]. A3G has indeed been shown to inhibit HIV infection and to be repressed by the viral Vif protein. Later on, a similar function was also attributed to other APOBEC3 proteins, namely A3DE, A3F and A3H [25,26,27]. Figure 1 illustrates the different mechanisms of HIV-1 inhibition by APOBEC3s. After binding of the HIV virion to the host cell membrane, the viral single-stranded RNA (ssRNA) genome is released into the cytoplasm and converted into double-stranded DNA (dsDNA) by reverse transcription. This dsDNA is then inserted into the host genome as an integrated provirus.
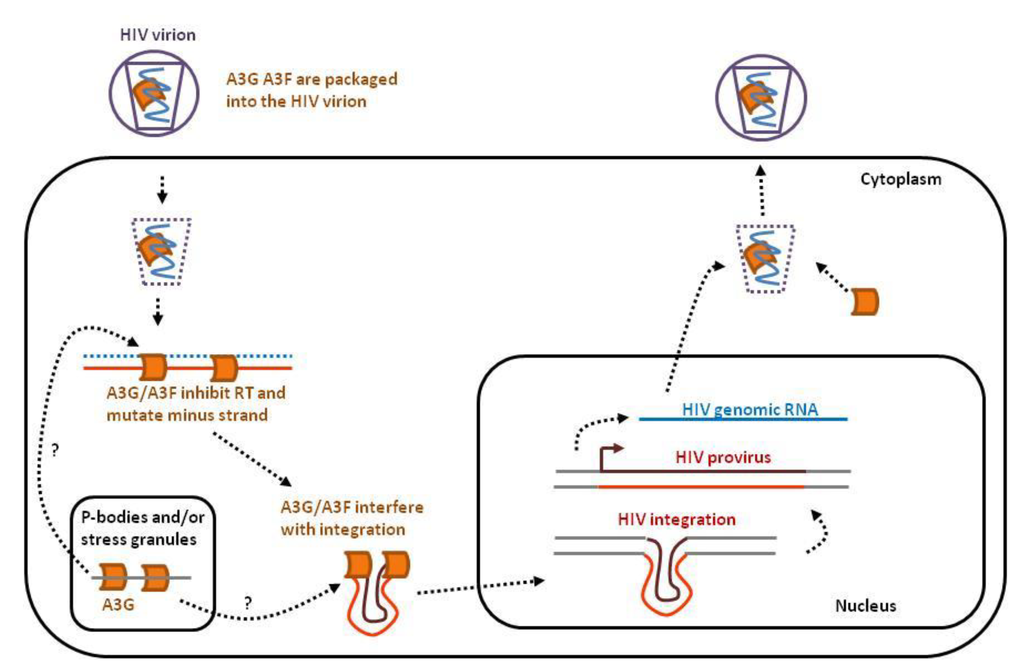
Figure 1.
APOBEC3s interfere with several key steps of the HIV infectious cycle. After binding of the HIV virion to the cell membrane, the single-stranded RNA genome (in blue) is released into the cytoplasm together with APOBEC3G and 3F (orange). APOBEC3 proteins expressed by the host cell concentrate in P-bodies and stress granules. A3G and A3F inhibit reverse transcription, mutate viral DNA and perturb proviral integration into the host genome. In the absence of HIV Vif, A3G and A3F will be incorporated into the budding virions.
A3DE, A3F, A3G and A3H are expressed by CD4+ T cells upon HIV infection, are packaged into virions and lead to proviral DNA mutations [27]. A3G and A3F notably concentrate in cytoplasmic microdomains (non-membrane structures) called mRNA-processing bodies or P-bodies [28,29,30]. P-bodies are sites of RNA storage, translational repression and decay [31]. A3G exerts its anti-HIV effect mainly via its deaminase function inducing abundant and deleterious mutations within the HIV provirus, whereas A3F acts more preferentially through its deaminase-independent activity [32]. This deaminase-independent effect involves inhibition of reverse transcription priming and extension [14,15,16,17] and interference with proviral integration [21,22,23].
APOBEC3-induced mutations are almost always G-to-A transitions of the plus-strand genetic code. Moreover, the mutation load is not homogeneous along the HIV provirus but presents two highly polarized gradients, each peaking just 5′ to the central polypurine tract (cPPT) and 5′ to the LTR (long terminal repeat) proximal polypurine tract (3′PPT) [33]. As illustrated in Figure 2, this mutational signature is due to the mechanism of HIV reverse transcription. Binding of the human tRNALys3 to the primer binding sequence (PBS) initiates the minus strand DNA synthesis by the virus-encoded reverse transcriptase protein (RT). The RT-associated ribonuclease H activity (RNAse H) selectively degrades the RNA strand of the RNA:DNA hybrid leaving the nascent minus-strand DNA free to hybridize with the complementary sequence at the 3′ end of the viral genomic ssRNA. After minus strand transfer, the viral RNA is reverse-transcribed into DNA. Whilst DNA synthesis proceeds, the RNAse H function cleaves the RNA strand of the RNA:DNA. Two specific purine-rich sequences (polypurine tracts cPPT and 3′PPT) that are resistant to RNAse H remain annealed with the nascent minus strand DNA. The reverse transcriptase uses the PPTs as primers to synthesize the plus-strand DNA. Finally, another strand transfer allows the production of the 5′ end of the plus-strand DNA (reviewed in [34]). From this complex multistep process, it appears that only the minus strand can be single-stranded (light red in Figure 2). Thus, G to A mutations observed on the plus strand (dark red in Figure 2) originate from C-to-T mutations on the minus strand. The gradient of mutational load actually correlates with the time that the minus strand remains single-chain [33].
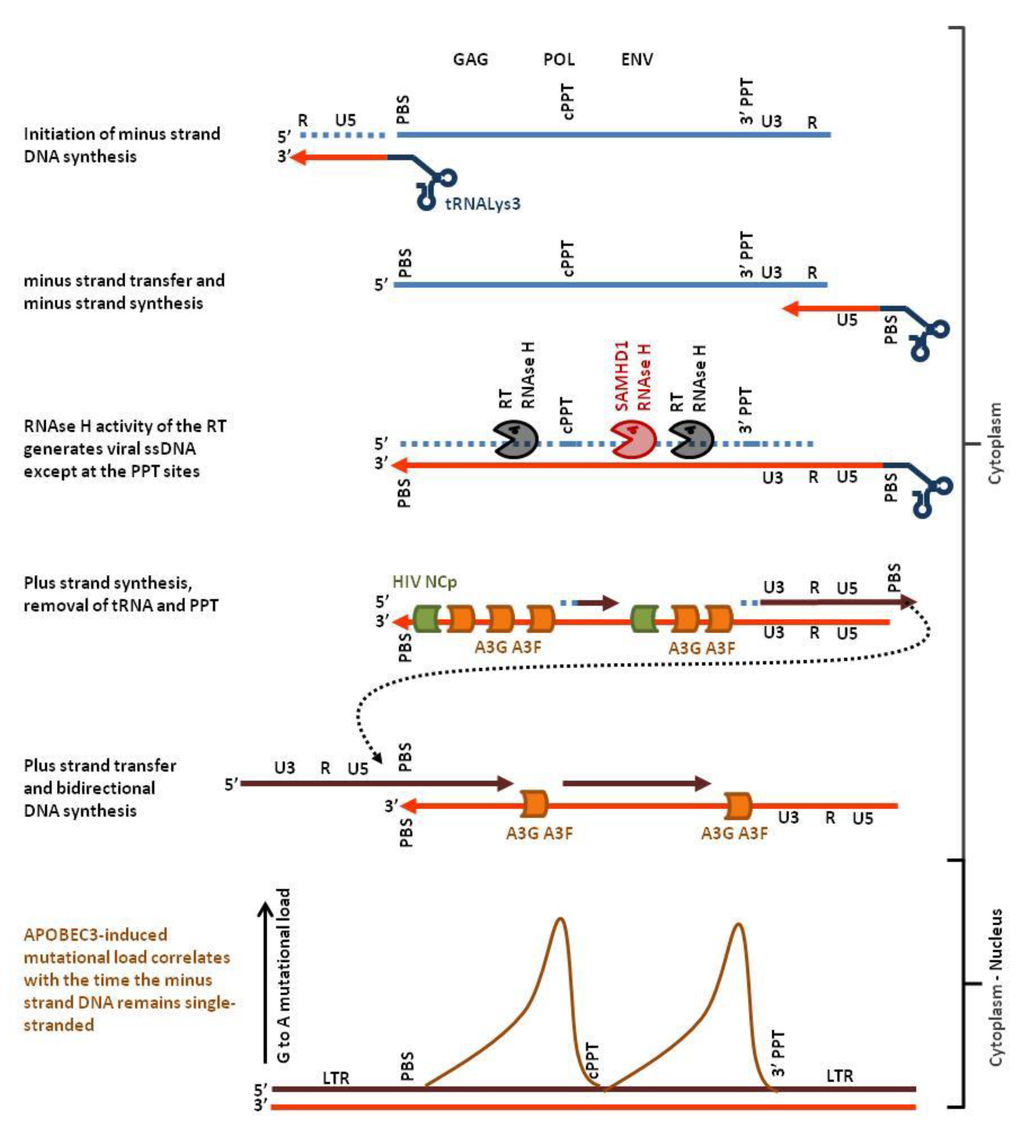
Figure 2.
Hotspots of APOBEC3 editing in the HIV genome. Host cell tRNALys3 (dark blue) hybridizes to the primer binding sequence (PBS) of the single stranded plus-strand RNA genome (light blue) and initiates minus strand DNA synthesis (light red). After strand transfer, reverse transcription proceeds up to the PBS yielding minus-strand DNA. RNAse H then hydrolyses the RNA (dotted light blue) of the RNA:DNA hybrid leaving the minus-strand DNA single-stranded. APOBEC3 G and F (orange) have now access the ssDNA genome, deaminate deoxycytidine and inhibit plus strand DNA synthesis (dark red). RNAse H activity of SAMHD1 promotes exposure of the minus-strand DNA (red pacman) whereas HIV nucleocapsid (green) limits APOBEC3-edition. Deoxycytidine deamination of the minus strand generates G-to-A mutations on the plus strand. Since plus-strand DNA synthesis starts from the PolyPurine Tracts (cPPT, 3′PPT), ssDNA located distant to these sites will be accessible to APOBEC3 edition over a longer period of time. Therefore, the APOBEC3-related mutational load will also be higher (brown curve, schematic representation of the data from [33]).
To counteract inactivation, HIV-1 encodes the Vif protein that inhibits APOBEC3s. Vif prevents A3G, A3F and A3H from being packaged into the virion by recruitment to a cullin5-elonginB/C-Rbx2-CBFβ E3 ubiquitin ligase complex, resulting in their polyubiquitination and subsequent proteasomal degradation [35,36,37]. Other mechanisms can limit APOBEC3 access to the single-chain minus-strand DNA generated during reverse transcription. By stabilizing the viral core, the glycosylated Gag protein of the murine leukemia virus renders the reverse transcription complex resistant to APOBEC3 and to other cytosolic viral sensors [38]. The HIV nucleocapsid protein (NCp) is able to bind ssDNA in a sequence aspecific manner and prevents A3A from mutating genomic DNA during transient strand separation [39]. Degradation of the RNA strand from the RNA:DNA hybrid by the RNAse H activity of the reverse transcriptase contributes to expose the minus strand as a single-chain nucleic acid. Interestingly, the host factor SAMHD1 (sterile alpha motif and histidine-aspartic acid domain containing protein 1) restricts HIV via its RNAse H function, activity that may facilitate the access of the APOBEC3s to the transiently single-stranded minus strand [40,41].
2.1.2. HTLV-1
Another human retrovirus, human T-lymphotropic virus 1 (HTLV-1), is also a target of A3G [42,43]. As in HIV-1 infection, A3G induces G-to-A transitions on the plus strand via deamination of deoxycytidines on the minus strand. HTLV-1 proviruses contain A3G-related base substitutions, including non-sense mutations [43]. Because HTLV-1 proviral loads mainly result from clonal expansion of infected cells, non-sense mutations are stabilized and amplified by mitosis, provided that viral factors stimulating proliferation are functional [44,45,46,47]. Although HTLV-1 does not seem to encode for a Vif-like protein, the frequencies of G-to-A changes in HTLV-1 proviruses are low, likely due to the mode of replication of HTLV-1 by clonal expansion [43,48]. This phenotype has also been associated with the ability of the viral nucleocapsid to limit A3G encapsidation [49].
2.1.3. HERVs
Human endogenous retroviruses (HERV) are transposable elements which were evolutionary integrated into human lineage after infection of germline cells. HERVs are abundant in the human genome (about 8%) and exert important regulatory functions such as control of cellular gene transcription [50]. HERVs contain canonical retroviral gag, pol and env genes surrounded by two LTRs. Nevertheless, most HERVs are defective for replication because of inactivating mutations or deletions [51]. These mutations are likely associated with A3G activity because of a particular signature with a mutated C present in a 5′GC context instead of 5′TC for other APOBEC3s [52,53,54]. Interestingly, A3G is still able to inhibit a reconstituted functional form of HERV-K in cell culture [52].
2.1.4. Simian Foamy Virus
SFV (simian foamy virus) is a retrovirus that is widespread among non-human primates and can be transmitted to humans [55]. A3F and A3G target SFV genome in vitro, leading to G-to-A transitions on the plus strand [56]. SFV genomes found in humans also display G-to-A mutations [57,58,59]. SFV codes for the accessory protein Bet, limiting APOBEC3 action [60,61,62,63].
2.2. Retroelements
About half of the human genome is constituted by repetitive elements. Among them, non-LTR retroelements LINE-1 (long interspersed nuclear element-1), SINE (short interspersed nuclear elements) and Alu are capable of retrotransposition, i.e., inserting a copy of themselves elsewhere in the genome. Since retrotranpositions can be harmful for genome integrity, these events are tightly controlled. In fact, only a small proportion of endogenous retroelements remains active in the germline cells because APOBEC3s protect the host genome from unscheduled retrotransposition (Figure 3). LINE-1 retrotransposition is initiated by transcription of a full-length LINE-1 RNA and translation of ORF1p and ORF2p. These two proteins associate with LINE-1 RNA to form the LINE-1 RiboNucleoProtein (L1 RNP) complex. Upon translocation of L1 RNP into the nucleus, LINE-1 is reverse transcribed and integrated into a new site of the host genome. A3C restricts LINE-1 retrotransposition in a deaminase-independent manner by redirecting and degrading the L1 RNP complex in P-bodies [20]. Within the nucleus, A3C also impairs LINE-1 minus strand DNA synthesis [20]. A3A prevents LINE-1 retrotransposition by deaminating the LINE-1 minus strand DNA [64]. Consistently, RNAse H treatment increases deamination of the LINE-1 minus strand [64].
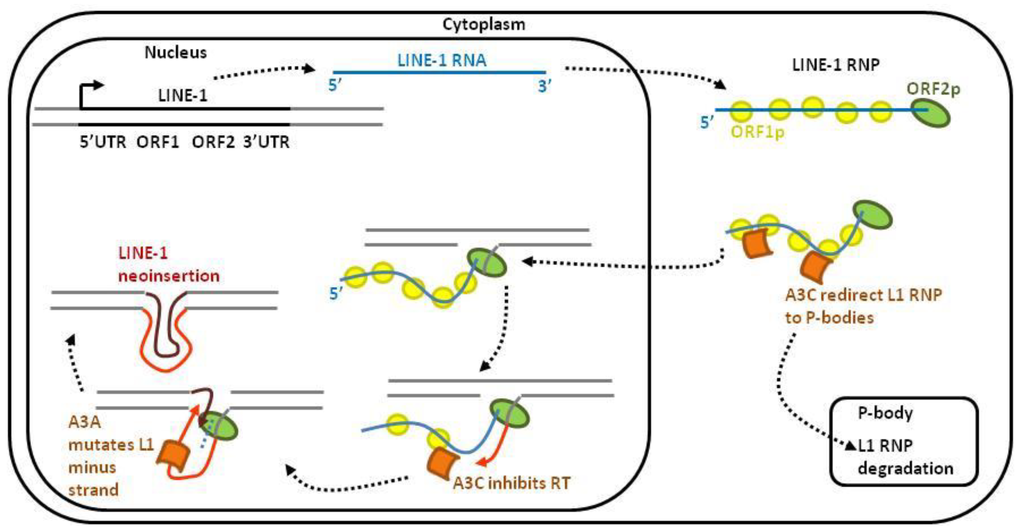
Figure 3.
LINE-1 retrotransposons are targeted by APOBEC3s. After transcription, the LINE-1 mRNA is transported into the cytoplasm. After translation, the ORF1- and ORF2-encoded proteins associate with the LINE-1 RNA and form a ribonucleoprotein (RNP) complex. The LINE-1 RNP enters the nucleus, where the ORF2p endonuclease domain cleaves the chromosomal DNA. After cleavage, the 3′-hydroxyl is used by the LINE-1 reverse transcriptase to synthesize a cDNA of LINE-1. This target-site-primed reverse transcription typically results in the insertion of a 5′-truncated LINE-1 element into a new genomic location. Different APOBEC3s-dependent mechanisms control LINE-1 retrotransposition: (1) in the cytoplasm, A3C interacts with and redirects the L1-RNP into P-bodies for degradation; (2) in the nucleus, A3C inhibits reverse transcriptase processing while A3A mutates the minus strand LINE-1 DNA.
2.3. Hepadnaviruses
Since their genome is partially single-strand, hepadnaviruses, such as human hepatitis B virus (HBV), are susceptible to APOBEC3 editing. Except A3DE, all APOBEC3s are able to edit the HBV genome in vitro, A3A being the most efficient [65,66]. APOBEC3 editing of HBV DNA has also been validated in vivo [67,68]. Since both minus and plus strands are susceptible to APOBEC3 editing, the mutational signature is more complex than in retroviruses [65,66,67]. HBV viral particles contain a partially double-stranded circular DNA genome (relaxed circular DNA or rcDNA; Figure 4). After uncoating of the viral particle, the rcDNA migrates into the nucleus, where minus-strand DNA synthesis is completed to generate the covalently closed circular double-stranded DNA genome (cccDNA).
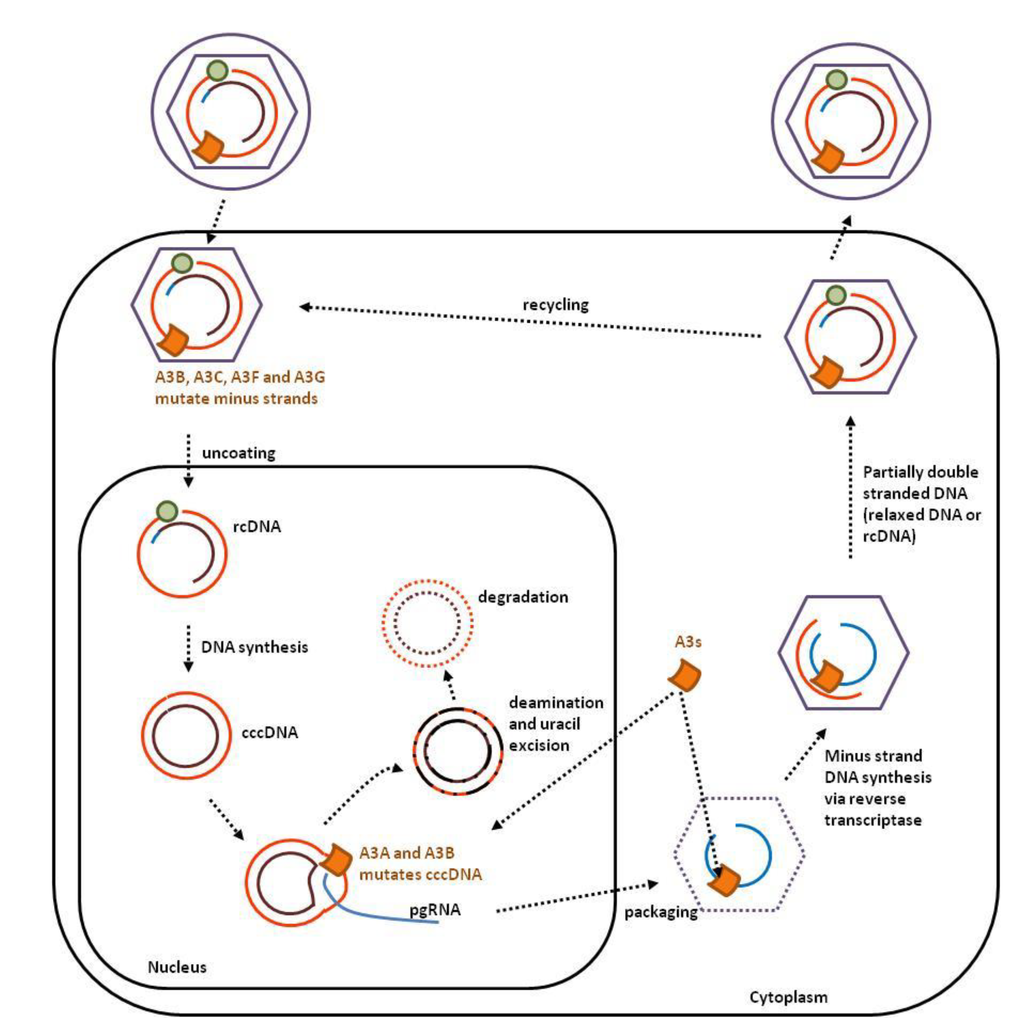
Figure 4.
APOBEC3s interfere with several steps of the HBV replication cycle. The HBV viral particle contains a partially double-stranded DNA genome (relaxed circular DNA or rcDNA) that can be edited by A3G and A3F. Unlike HIV, HBV does not appear to encode Vif-like protein. Upon transfer into the nucleus, the plus strand of the rcDNA is replicated to form the covalently closed circular DNA genome (cccDNA). A3A and A3B deaminate the cccDNA genome leading to uracil excision and subsequent degradation.
In the nucleus, A3A and A3B deaminate HBV cccDNA (Figure 4). Since APOBEC3s require a ssDNA substrate, it is predicted that cccDNA melts during transcription. APOBEC3 deamination of deoxycytidine introduces deleterious mutations in the viral genome and initiates its catabolism via the uracil DNA glycosylase dependent pathway [12].
After transcription of the cccDNA, the pregenomic RNA (pgRNA) translocates into the cytoplasm and is reverse-transcribed into circular partially double-stranded DNA. This mechanism involves priming by the viral P protein, a strand transfer directed by DR1 annealing and degradation of the RNA template by the RNAse H activity of the reverse transcriptase (Figure 5, dotted light blue). The 5′ end of the pgRNA anneals with DR2, directs a second strand transfer and primes plus-strand DNA synthesis, yielding rcDNA. The minus strand DNA is deaminated proportionally to its exposure to APOBEC3s (Figure 5, orange curve) [68]. Since different subcellular compartments are involved (cytoplasm, nucleus, extracellular viral particles), multiple nuclear and cytoplasmic APOBEC3s (i.e., A3A, A3B, A3C, A3F and A3G) edit the HBV genome [65].
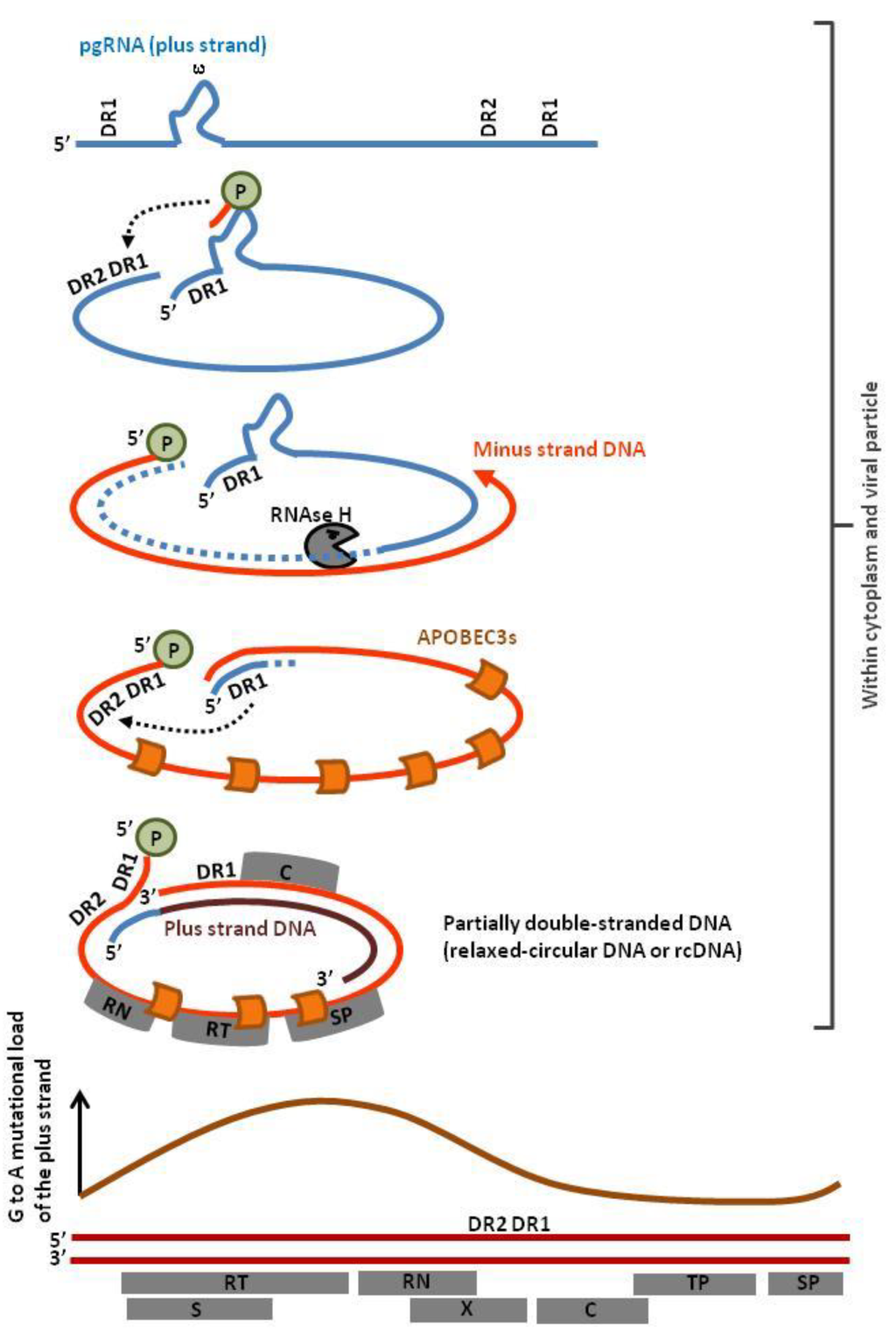
Figure 5.
Profile of APOBEC3 editing of the HBV genome. The viral P protein initiates reverse transcription at the stem loop structure ε. The pregenomic RNA (pgRNA) contains two direct repeat sequences (DR1) at the 5′ and 3′ end of the viral genome, allowing strand transfer to the 5′ end of the viral genome. While synthesis of the minus-strand DNA proceeds, the RNAse H activity of the reverse transcriptase degrades the pgRNA except at the 5′ end. After a second strand transfer, the undigested pgRNA anneals with the direct repeat sequence DR2 and primes plus-strand gDNA synthesis, yielding relaxed circular DNA (rcDNA). Mutational load correlates with the time of exposure of ssDNA (orange curve, schematic representation of data extracted from reference [68]). Abbreviations within grey boxes read as follow: RT, Reverse Transcriptase; RN, RNAse; TP, Terminal Protein; SP, Spacer Domain; S, short surface gene; X, X gene, C, Core gene.
Compared to HIV, additional APOBEC3 proteins (A3A, A3B and A3C) target the HBV genome in the nucleus. Incorporation of HIV into chromatin instead of an episome for HBV may protect the provirus from APOBEC3s editing by a mechanism involving Tribbles 3 proteins [69].
2.4. Herpesviruses
Herpesviruses such as herpes simplex virus-1 (HSV-1) and Epstein-Barr virus (EBV) have a linear double-stranded DNA genome that is edited by APOBEC3 on both strands [70]. After infection, the HSV-1 capsid is transported to the nuclear pores and delivers the double-stranded linear DNA into the nucleus. After circularization of the viral genome, bidirectional DNA synthesis is initiated at the origins of replication [71,72]. This process requires DNA denaturation by the origin binding protein (UL9). The helicase/primase (UL5/UL8/UL52) and single-stranded DNA binding proteins (ICP8 coded by the UL29 gene) then associate with the origin of replication and recruit the DNA polymerase/UL42 complex (Figure 6). During DNA synthesis and transcription, nuclear APOBEC3s have access to single-stranded viral DNA. APOBEC3-edition of HSV-1 and EBV genomes is higher in the minus strand (G to A as opposed to C to T) [70]. It is hypothesized that, due to discontinued replication, the lagging strand exposes more viral ssDNA than the leading strand. HSV-1 and EBV encode orthologs of uracil-DNA glycosylases (UDG) that excise uridine at the replication fork. The HSV-1 UDG (UL2) binds to UL30, associates with the viral replisome and directs replication-coupled BER (base excision repair) to ensure genome integrity [73]. The viral UDG might therefore protect against APOBEC3 editing.
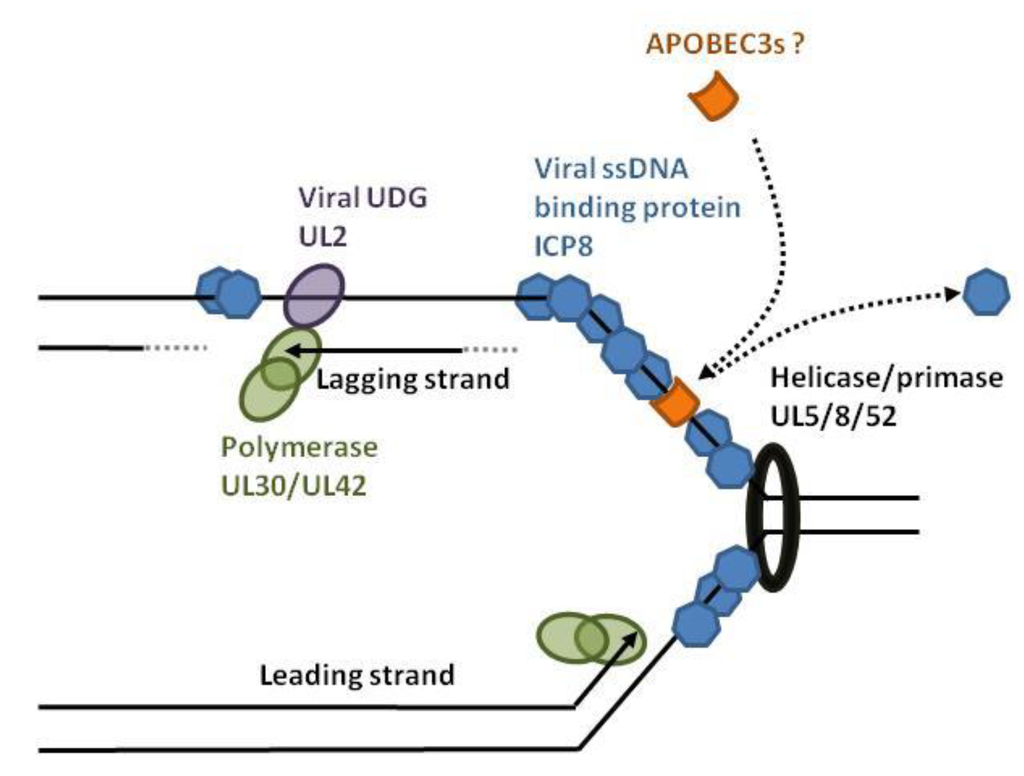
Figure 6.
HSV-1 replication fork and hypothetical model of APOBEC3 editing. In the nucleus, replication of HSV-1 is initiated by the origin binding protein (UL9) that melts double-stranded DNA. The helicase/primase complex (UL5/UL8/UL52) unwinds and anneals RNA primers, allowing DNA replication by the UL30/UL42 complex. The viral protein ICP8 covers the transiently exposed single-stranded DNA and competes with the APOBEC3 binding. The viral UL2 is a uracil-DNA glycosylase (UDG) that favors replication-coupled base excision DNA repair.
2.5. Papillomavirus
Human papillomaviruses (HPVs) are circular double-stranded DNA viruses. A3A, A3C and A3H are able to deaminate both strands of the 8Kb viral genome [74,75,76]. APOBEC3-edited HPV DNA is found in benign and precancerous cervical lesions [74]. Replication of the HPV genome occurs in the nucleus and is primarily based on the host replication machinery. The HPV protein E1 recruits ssDNA-binding protein RPA (replication protein A) during replication to cover the transiently exposed viral ssDNA [77].
2.6. TT Virus
Transfusion-transmitted virus (TTV) is a non-enveloped virus causing a persistent and asymptomatic infection. Having a circular single-stranded DNA genome, TTV is a prototypical substrate of APOBEC3s and shows APOBEC3-related mutations [78].
Together, these data show that viruses are targeted by particular isoforms of APOBEC3 depending on their modes of replication (Table 1) and have developed strategies to dampen ssDNA edition. A3G and A3F are restricted to the cytoplasm whereas A3A, A3B and A3C preferentially act in the nucleus. Importantly, the mutational load is proportional to the duration of single-stranded DNA exposure to APOBEC3s.

Table 1.
Summary of the anti-viral activity of the different APOBEC3 isoforms. * It has been recently shown that A3A can also edit RNA transcripts [79].
| Sub-cellular localization | Substrate edited | Retro viruses | Retro elements | Hepadna viruses | Herpes viruses | ||||
|---|---|---|---|---|---|---|---|---|---|
| HIV-1 | HTLV-1 | HERVs | SFV | ||||||
| A3A | cell wide | single stranded DNA, RNA * | + | + | + | ||||
| A3B | nuclear | single stranded DNA | + | ||||||
| A3C | cell wide | single stranded DNA | + | + | + | ||||
| A3DE | cytoplasmic | single stranded DNA | + | ||||||
| A3F | cytoplasmic | single stranded DNA | + | + | + | ||||
| A3G | cytoplasmic | single stranded DNA | + | + | + | + | + | ||
| A3H | cell wide | single stranded DNA | + | + | + | ||||
3. Therapeutic Strategies by Perturbation of the Viral Mutation Rate
Viral quasi-species refer to a population of distinct but closely related viral genomes that differ only by a limited number of mutations. The distribution of variants is dominated by a master sequence that displays the highest fitness within a given environment (Figure 7A). High mutation rates during viral replication are the driving force for quasi-species generation. Lethal mutations or inappropriate adaptation to the environmental conditions (e.g., anti-viral therapy, immune pressure) will clear unfit genomes. When conditions change, the fittest quasi-species may differ from the master sequence. Providing that the distribution contains an adequate variant, a new population will grow (Figure 7B). The rate of mutation and the selection pressure will dictate the wideness of the distribution. If the environmental changes are too drastic or the quasi-species distribution too narrow, the viral population will be unable to recover [80].
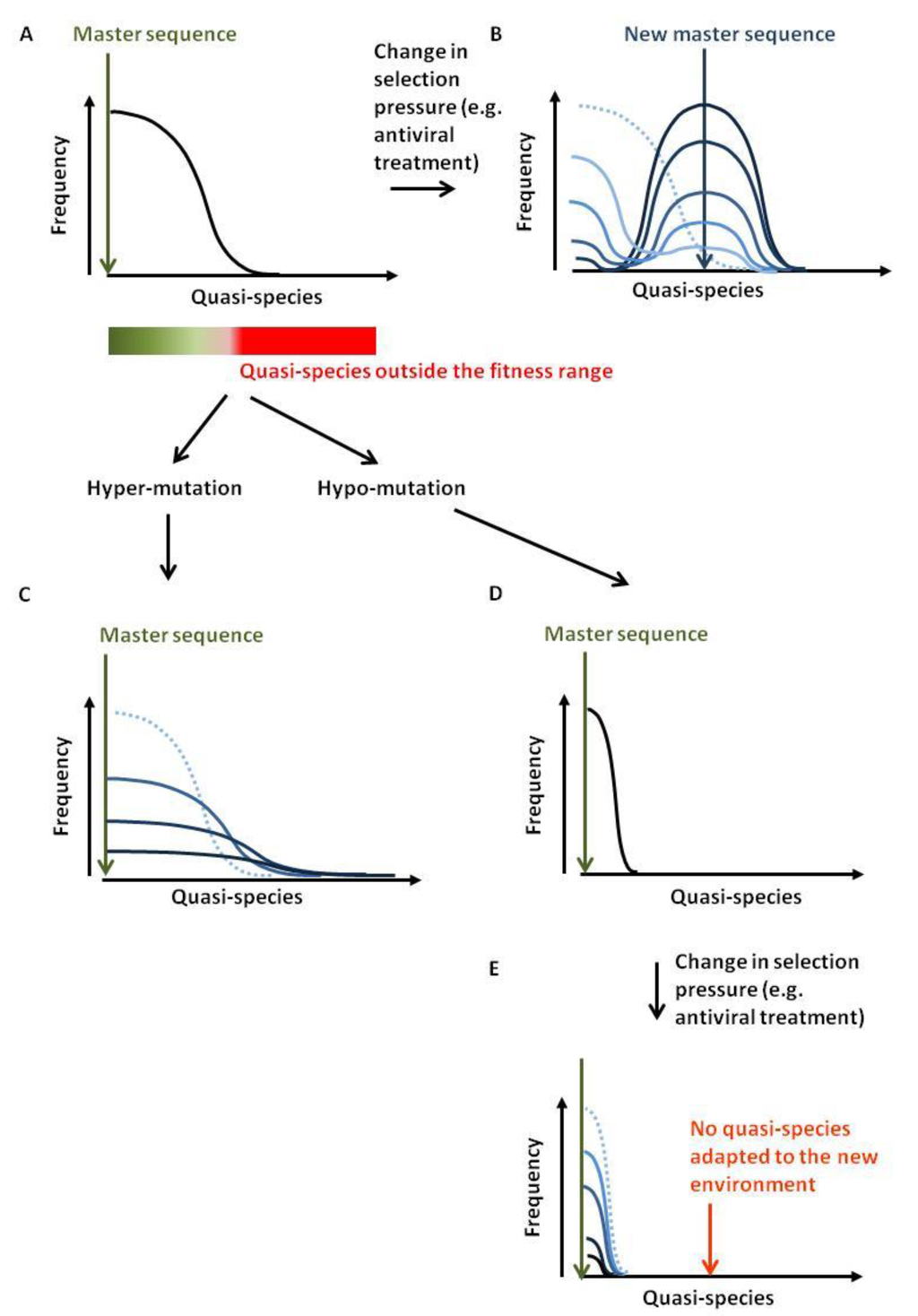
Figure 7.
Antiviral strategy by hypo- or hyper-mutation. (A) Viral quasi-species refer to as a population of distinct but closely related viral genomes that only differ by a limited number of mutations. The frequency of these quasi-species spreads around a master sequence. The boundary of this population is dictated by the selection forces acting against the viral diversification. At equilibrium, quasi-species generated outside the fitness range will not persist. (B) If selection criteria are modified, the fittest sequence will change. If the original distribution contained this sequence, the population will first shrink and then re-grow around a new master sequence (from light to dark blue corresponding to the time evolution). (C) Excess of APOBEC3-directed mutations will affect fitness of the newly created quasi-species up to complete disappearance. (D,E) Hypo-mutation will restrict the range of quasi-species and limit adaptability to new environmental conditions.
Emergence of quasi-species is thus a major issue that limits antiviral therapy. Viral populations can indeed accommodate environmental changes due to improved immunity (vaccination) or pharmacological inhibition. It is possible to affect quasi-species adaptability by modulating the frequencies of mutation [81,82].
The first approach, referred to as lethal mutagenesis or the hyper-mutation strategy, aims to introduce an excess of mutations in the viral genomes. If the mutational load per viral genome is too high, a substantial proportion of the new viruses will be defective or inadequately adapted to their environment. Introducing mutations in viruses would therefore decrease viral load (Figure 7C). In principle, exogenous induction of APOBEC3 expression could achieve this goal. This strategy has recently been exemplified for HBV, where forced expression of A3A and A3B induced HBV cccDNA hypermutation with no detectable effect on genomic DNA [12]. Nevertheless, this approach raises serious safety issues because APOBEC3 mutations could also drive cancer development [83,84,85]. Indeed, A3A and A3B over-expression in yeast creates mutational clusters and genomic rearrangements similar to those observed in human cancers, the mutational burden being magnified by DNA strand breaks [86,87,88,89]. Because the processing of double-strand break repair transiently exposes single-stranded nucleic acids, DNA repair could provide a substrate for nuclear deaminases. What would, for example, happen if an HBV-infected liver cell is being forced to express deaminases and at the same time has to repair DNA strand breaks generated by reactive oxygen species produced during alcohol catabolism [90]?
Therefore, it would be safer to promote hyper-mutation by targeting the viral factors that inhibit APOBEC3s. In that respect, Vif inhibitors are being developed [91,92]. Inhibition of viral ssDNA-binding proteins (like HSV-1 ICP8) might lead to increased access for endogenously expressed APOBEC3s to the viral ssDNA (Figure 6). Promotion of RNAse H activity during retrotranscription might facilitate the binding of the APOBEC3s to the viral ssDNA (Figure 2). Because reverse transcription is thought to start within the virion, promotion of APOBEC3s loading in to the viral particle will increase editing (MLV glyco-Gag shields the reverse transcription complex from APOBEC3 and cytosolic sensors [38]). Alternatively, it would be possible to target viral DNA repair mechanisms (e.g., via inhibitors against the viral UDG UL2 of HSV-1, Figure 6). In these cases, safety issues are related to the emergence of sub-lethal APOBEC3-mutations and promotion of drug resistant quasi-species.
The reverse strategy would be to reduce mutation rate by inhibiting APOBEC3, thereby narrowing the quasi-species spectrum and limiting viral adaptability to new environmental conditions (Figure 7D,E). APOBEC3s inhibitors are currently being developed and evaluated [93,94]. This approach, which paradoxically targets a well-conserved mechanism of mammalian innate immunity, would preserve host genome integrity. Potential risks of this therapy pertain to adequate control of endogenous retroelements and opportunistic infections.
4. Conclusions
Single-strand DNA editing by APOBEC3 proteins is a very powerful mechanism of mammalian innate immunity that mutates and inactivates viral genomes. The outcome of infection is the result of a finely tuned balance between onset of mutations, generation of quasi-species and APOBEC inhibition by viral factors. Understanding the mechanisms involved reveals new prospects for therapeutic strategies that interfere with APOBEC3 deamination of cytosine residues in nascent viral DNA.
Acknowledgments
This work was supported by the “Fonds National de la Recherche Scientifique” (FNRS), the Télévie, the Interuniversity Attraction Poles (IAP) Program “Virus-host interplay at the early phases of infection” BELVIR initiated by the Belgian Science Policy Office, the Belgian Foundation against Cancer (FBC), the Sixth Research Framework Programme of the European Union (project “The role of infections in cancer” INCA LSHC-CT-2005-018704), the “Neoangio” excellence program and the “Partenariat Public Privé”, PPP INCA, of the “Direction générale des Technologies, de la Recherche et de L’Energie/DG06” of the Walloon government, the “Action de Recherche Concertée Glyvir” (ARC) of the “Communauté française de Belgique”, the “Centre anticancéreux près ULg” (CAC), the “Subside Fédéral de Soutien à la Recherche Synbiofor and Agricultureislife” projects of Gembloux Agrobiotech (GxABT), the “ULg Fonds Spéciaux pour la Recherche” and the “Plan Cancer” of the “Service Public Fédéral”. We thank Sathya Neelature Sriramareddy, Srikanth Perike, Hélène Gazon, Alix de Brogniez, Alexandre Carpentier, Pierre-Yves Barez, Bernard Staumont and Malik Hamaidia for their careful reading and helpful comments.
Author Contributions
L.W. and N.A.G. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Harris, R.S. The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar] [PubMed]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed. Res. Int. 2013, 2013, e683095. [Google Scholar] [CrossRef] [PubMed]
- LaRue, R.S.; Andresdottir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jonsson, S.R.; Landau, N.R.; Lochelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Munk, C.; Willemsen, A.; Bravo, I.G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 2012, 12, e71. [Google Scholar] [CrossRef] [PubMed]
- Mehta, H.V.; Jones, P.H.; Weiss, J.P.; Okeoma, C.M. IFN-alpha and lipopolysaccharide upregulate APOBEC3 mRNA through different signaling pathways. J. Immunol. 2012, 189, 4088–4103. [Google Scholar] [CrossRef] [PubMed]
- Refsland, E.W.; Stenglein, M.D.; Shindo, K.; Albin, J.S.; Brown, W.L.; Harris, R.S. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38, 4274–4284. [Google Scholar] [CrossRef] [PubMed]
- Koning, F.A.; Newman, E.N.; Kim, E.Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef] [PubMed]
- Lackey, L.; Law, E.K.; Brown, W.L.; Harris, R.S. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 2013, 12, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Saetrom, P.; Aas, P.A.; Pettersen, H.S.; Kavli, B.; Slupphaug, G. Error-free versus mutagenic processing of genomic uracil—Relevance to cancer. DNA Repair 2014, 19, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Weil, A.F.; Ghosh, D.; Zhou, Y.; Seiple, L.; McMahon, M.A.; Spivak, A.M.; Siliciano, R.F.; Stivers, J.T. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc. Natl. Acad Sci. USA 2013, 110, E448–E457. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Huthoff, H.; Autore, F.; Gallois-Montbrun, S.; Fraternali, F.; Malim, M.H. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009, 5, e1000330. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Cen, S.; Niu, M.; Saadatmand, J.; Kleiman, L. Inhibition of tRNA(3)(Lys)—Primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 2006, 80, 11710–11722. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Hope, T.J. APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology 2008, 375, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ao, Z.; Chen, L.; Kobinger, G.; Peng, J.; Yao, X. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J. Virol. 2012, 86, 3777–3786. [Google Scholar] [CrossRef] [PubMed]
- Gillick, K.; Pollpeter, D.; Phalora, P.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 2013, 87, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.V.; Klawitter, S.; Held, U.; Berger, A.; Vasudevan, A.A.; Bock, A.; Hofmann, H.; Hanschmann, K.M.; Trosemeier, J.H.; Flory, E.; et al. Human LINE-1 restriction by APOBEC3C is deaminase independent and mediated by an ORF1p interaction that affects LINE reverse transcriptase activity. Nucleic Acids Res. 2014, 42, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Wang, T.; Liu, B.; Tian, C.; Xiao, Z.; Kappes, J.; Yu, X.F. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 2007, 81, 7238–7248. [Google Scholar] [CrossRef] [PubMed]
- Mbisa, J.L.; Barr, R.; Thomas, J.A.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar] [CrossRef] [PubMed]
- Vetter, M.L.; D’Aquila, R.T. Cytoplasmic APOBEC3G restricts incoming Vif-positive human immunodeficiency virus type 1 and increases two-long terminal repeat circle formation in activated T-helper-subtype cells. J. Virol. 2009, 83, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.L.; Doehle, B.P.; Bogerd, H.P.; Cullen, B.R. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004, 23, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Witkowska, H.E.; Hall, S.C.; Santiago, M.; Soros, V.B.; Esnault, C.; Heidmann, T.; Greene, W.C. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 15588–15593. [Google Scholar] [CrossRef] [PubMed]
- Gallois-Montbrun, S.; Kramer, B.; Swanson, C.M.; Byers, H.; Lynham, S.; Ward, M.; Malim, M.H. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007, 81, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Wichroski, M.J.; Robb, G.B.; Rana, T.M. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2006, 2, e41. [Google Scholar] [CrossRef] [PubMed]
- Phalora, P.K.; Sherer, N.M.; Wolinsky, S.M.; Swanson, C.M.; Malim, M.H. HIV-1 replication and APOBEC3 antiviral activity are not regulated by P bodies. J. Virol. 2012, 86, 11712–11724. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Koizumi, Y.; Takeuchi, J.S.; Misawa, N.; Kimura, Y.; Morita, S.; Aihara, K.; Koyanagi, Y.; Iwami, S.; Sato, K. Quantification of deaminase activity-dependent and -independent restriction of HIV-1 replication mediated by APOBEC3F and APOBEC3G through experimental-mathematical investigation. J. Virol. 2014, 88, 5881–5887. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Rusniok, C.; Vartanian, J.P.; Wain-Hobson, S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 2006, 34, 4677–4684. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 Reverse Transcriptase Still Remains a New Drug Target: Structure, Function, Classical Inhibitors, and New Inhibitors with Innovative Mechanisms of Actions. Mol. Biol. Int. 2012, 2012, e586401. [Google Scholar] [CrossRef] [PubMed]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2012, 481, 371–375. [Google Scholar]
- Zhao, K.; Du, J.; Rui, Y.; Zheng, W.; Kang, J.; Hou, J.; Wang, K.; Zhang, W.; Simon, V.A.; Yu, X.F. Evolutionarily conserved pressure for the existence of distinct G2/M cell cycle arrest and A3H inactivation functions in HIV-1 Vif. Cell Cycle 2015, 14, 838–847. [Google Scholar] [PubMed]
- Stavrou, S.; Nitta, T.; Kotla, S.; Ha, D.; Nagashima, K.; Rein, A.R.; Fan, H.; Ross, S.R. Murine leukemia virus glycosylated Gag blocks apolipoprotein B editing complex 3 and cytosolic sensor access to the reverse transcription complex. Proc. Natl. Acad. Sci. USA 2013, 110, 9078–9083. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Hercik, K.; Byeon, I.J.; Ahn, J.; Hill, S.; Hinchee-Rodriguez, K.; Singer, D.; Byeon, C.H.; Charlton, L.M.; Nam, G.; et al. Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res. 2014, 42, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nunez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Mahieux, R.; Suspene, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ma, G.; Nosaka, K.; Tanabe, J.; Satou, Y.; Koito, A.; Wain-Hobson, S.; Vartanian, J.P.; Matsuoka, M. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J. Virol. 2010, 84, 7278–7287. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; Wain-Hobson, S.; Gessain, A.; Plumelle, Y.; Wattel, E. Adult T-cell leukemia/lymphoma on a background of clonally expanding human T-cell leukemia virus type-1-positive cells. Blood 1996, 88, 4646–4650. [Google Scholar] [PubMed]
- Etoh, K.; Tamiya, S.; Yamaguchi, K.; Okayama, A.; Tsubouchi, H.; Ideta, T.; Mueller, N.; Takatsuki, K.; Matsuoka, M. Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res. 1997, 57, 4862–4867. [Google Scholar] [PubMed]
- Mortreux, F.; Gabet, A.S.; Wattel, E. Molecular and cellular aspects of HTLV-1 associated leukemogenesis in vivo. Leukemia 2003, 17, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Malani, N.; Melamed, A.; Gormley, N.; Carter, R.; Bentley, D.; Berry, C.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood 2011, 117, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Hill, S.A.; Princler, G.; Lloyd, P.; Heidecker, G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc. Natl. Acad. Sci. USA 2007, 104, 2915–2920. [Google Scholar] [CrossRef] [PubMed]
- Medstrand, P.; van de Lagemaat, L.N.; Dunn, C.A.; Landry, J.R.; Svenback, D.; Mager, D.L. Impact of transposable elements on the evolution of mammalian gene regulation. Cytogenet. Genome Res. 2005, 110, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Stoye, J.P. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 2012, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Malim, M.H.; Bieniasz, P.D. Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 2008, 82, 8762–8770. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Priet, S.; Ribet, D.; Heidmann, O.; Heidmann, T. Restriction by APOBEC3 proteins of endogenous retroviruses with an extracellular life cycle: Ex vivo effects and in vivo “traces” on the murine IAPE and human HERV-K elements. Retrovirology 2008, 5, e75. [Google Scholar] [CrossRef] [PubMed]
- Anwar, F.; Davenport, M.P.; Ebrahimi, D. Footprint of APOBEC3 on the genome of human retroelements. J. Virol. 2013, 87, 8195–8204. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Gessain, A. Origin, evolution and innate immune control of simian foamy viruses in humans. Curr. Opin. Virol. 2015, 10, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gartner, K.; Wiktorowicz, T.; Park, J.; Mergia, A.; Rethwilm, A.; Scheller, C. Accuracy estimation of foamy virus genome copying. Retrovirology 2009, 6, e32. [Google Scholar] [CrossRef] [PubMed]
- Delebecque, F.; Suspene, R.; Calattini, S.; Casartelli, N.; Saib, A.; Froment, A.; Wain-Hobson, S.; Gessain, A.; Vartanian, J.P.; Schwartz, O. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 2006, 80, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Betsem, E.; Gessain, A. Viral latency in blood and saliva of simian foamy virus-infected humans. PLoS ONE 2013, 8, e77072. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.T.; Small, C.T.; Soliven, K.; Engel, G.A.; Feeroz, M.M.; Wang, X.; Craig, K.L.; Hasan, M.K.; Emerman, M.; Linial, M.L.; et al. A novel bayesian method for detection of APOBEC3-mediated hypermutation and its application to zoonotic transmission of simian foamy viruses. PLoS Comput. Biol. 2014, 10, e1003493. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef] [PubMed]
- Lochelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar] [CrossRef] [PubMed]
- Jaguva Vasudevan, A.A.; Perkovic, M.; Bulliard, Y.; Cichutek, K.; Trono, D.; Haussinger, D.; Munk, C. Prototype foamy virus Bet impairs the dimerization and cytosolic solubility of human APOBEC3G. J. Virol. 2013, 87, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.R.; Narvaiza, I.; Planegger, R.A.; Weitzman, M.D.; Moran, J.V. APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. eLife 2014, 3, e02008. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Guetard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Guetard, D.; Suspene, R.; Rusniok, C.; Wain-Hobson, S.; Vartanian, J.P. Genetic editing of HBV DNA by monodomain human APOBEC3 cytidine deaminases and the recombinant nature of APOBEC3G. PLoS ONE 2009, 4, e4277. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Henry, M.; Marchio, A.; Suspene, R.; Aynaud, M.M.; Guetard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Beggel, B.; Munk, C.; Daumer, M.; Hauck, K.; Haussinger, D.; Lengauer, T.; Erhardt, A. Full genome ultra-deep pyrosequencing associates G-to-A hypermutation of the hepatitis B virus genome with the natural progression of hepatitis B. J. Viral. Hepat. 2013, 20, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Aynaud, M.M.; Suspene, R.; Vidalain, P.O.; Mussil, B.; Guetard, D.; Tangy, F.; Wain-Hobson, S.; Vartanian, J.P. Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J. Biol. Chem. 2012, 287, 39182–39192. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Aynaud, M.M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, I.; Tang, K.W.; Elias, P. Replication and recombination of herpes simplex virus DNA. J. Biol. Chem. 2011, 286, 15619–15624. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Coen, D.M. Herpes simplex viruses: Mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef] [PubMed]
- Bogani, F.; Corredeira, I.; Fernandez, V.; Sattler, U.; Rutvisuttinunt, W.; Defais, M.; Boehmer, P.E. Association between the herpes simplex virus-1 DNA polymerase and uracil DNA glycosylase. J. Biol. Chem. 2010, 285, 27664–27672. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Melendy, T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 2004, 78, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Noguchi, C.; Akiyama, R.; Matsushita, M.; Kunihiro, K.; Tanaka, S.; Abe, H.; Mitsui, F.; Kitamura, S.; Hatakeyama, T.; et al. G to A hypermutation of TT virus. Virus Res. 2010, 149, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Thomas Taggart, R.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 6881. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Harris, R.S. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol. 2009, 4, 605. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Enhancing immunity to HIV through APOBEC. Nat. Biotechnol. 2008, 26, 1089–1090. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2013, 2, e00534. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Dhar, A.; Boissy, R.J.; Hirano, M.; Rubel, A.A.; Rogozin, I.B.; Pavlov, Y.I. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol. Direct. 2012, 7, e47. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Stepchenkova, E.I.; Waisertreiger, I.S.; Noskov, V.N.; Dhar, A.; Eudy, J.D.; Boissy, R.J.; Hirano, M.; Rogozin, I.B.; Pavlov, Y.I. Genome-wide mutation avalanches induced in diploid yeast cells by a base analog or an APOBEC deaminase. PLoS Genet. 2013, 9, e1003736. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Liu, D.; Lv, W.; Wang, X.; Wang, J.; Lv, M.; Huang, W.; Wu, J.; Zhang, H.; Jin, H.; et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J. Virol. 2012, 86, 5497–5507. [Google Scholar] [CrossRef] [PubMed]
- Pery, E.; Sheehy, A.; Nebane, N.M.; Brazier, A.J.; Misra, V.; Rajendran, K.S.; Buhrlage, S.J.; Mankowski, M.K.; Rasmussen, L.; White, E.L.; et al. Identification of a Novel HIV-1 Inhibitor Targeting Vif-dependent Degradation of Human APOBEC3G. J. Biol. Chem. 2015, 290, 10504–10517. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shandilya, S.M.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.E.; Li, M.; Harris, R.S.; Harki, D.A. Small-molecule APOBEC3G DNA cytosine deaminase inhibitors based on a 4-amino-1,2,4-triazole-3-thiol scaffold. ChemMedChem 2013, 8, 112–117. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).