
HSV-1 gM and the gK/pUL20 Complex Are Important for the Localization of gD and gH/L to Viral Assembly Sites


Abstract
:1. Introduction
2. Experimental Section
2.1. Cell Lines and Viruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COL426 | TGCGGTCTGCGTCACGGGGCTCCTCGTCCTGGCCTCTGTGTAATAGTGAGAATTCCCTGCTTTTACGCCACGAGGATGACGACGATAAGTAGGG | Forward primer for replacing codons 53–55 of UL10 with three stop codons |
| COL427 | ACCCCGGCATAAGAGCTCGCCGTGGCGTAAAAGCAGGGAATTCTCACTATTACACAGAGGCCAGGACGAGGACAACCAATTAACCAATTCTGATTAG | Reverse primer for replacing codons 53–55 of UL10 with three stop codons |
| COL468 | GGTACGCCCCACCGGCACCAACAACGACACCGCCCTCGTGTGATAGTAAGCTTACCAGACCCTATTGTTTCTGAGGATGACGACGATAAGTAGGG | Forward primer for replacing codons 54–56 of UL53 with three stop codons |
| COL469 | GGGGGGTGCGTCGGGGCCCCCAGAAACAATAGGGTCTGGTAAGCTTACTATCACACGAGGGCGGTGTCGTTGTCAACCAATTAACCAATTCTGATTAG | Reverse primer for replacing codons 54–56 of UL53 with three stop codons |
| COL464 | CCTTCCTCTGGTGGATCGAGATCTGGTCGACGAGGCCGCCTGATAGTAAGCTTAGGGAGAACTGCCACTGGAGAGGATGACGACGATAAGTAGGG | Forward primer for replacing codons 20–23 of UL20 with three stop codons |
| COL465 | GAGGACAATGAAAACTGTTCCTCCAGTGGCAGTTCTCCCTAAGCTTACTATCAGGCGGCCTCGTCGACCAGATCAACCAATTAACCAATTCTGATTAG | Revere primer for replacing codons 20–23 of UL20 with three stop codons |
2.2. Antibody Internalization Assays
| Antigen | Name | Species | Isotype | Application (Dilution) | Source |
|---|---|---|---|---|---|
| Actin | AC-40 | Mouse | IgG2a | WB (1:5000) | Sigma |
| gB | CB24 | Mouse | IgG2b | WB (1:60) | [30] |
| gD | LP2 | Mouse | IgG2a | IF (1:3) | [31] |
| gH | BBH1 | Mouse | IgG2a | WB (1:10) | Abcam |
| gM | gM-B | Rabbit | - | WB (1:1000) | From: H Browne |
| pUL20 | - | Rabbit | - | WB (1:1000) | From: H Browne |
| pUL37 | - | Rabbit | - | WB (1:5000) | From: T Mettenleiter |
| VP5 | Ab6508 | Mouse | IgG2b | IF (1:200); WB (1:1000) | Abcam |
| VP16 | LP1 | Mouse | IgG1 | WB (1:2000) | Abcam |
| VP22 | AGV30 | Rabbit | - | WB (1:2000) | [33] |
2.3. Single Step Growth Curves
2.4. Determination of Plaque Size
2.5. Sucrose Cushion Virus Purification
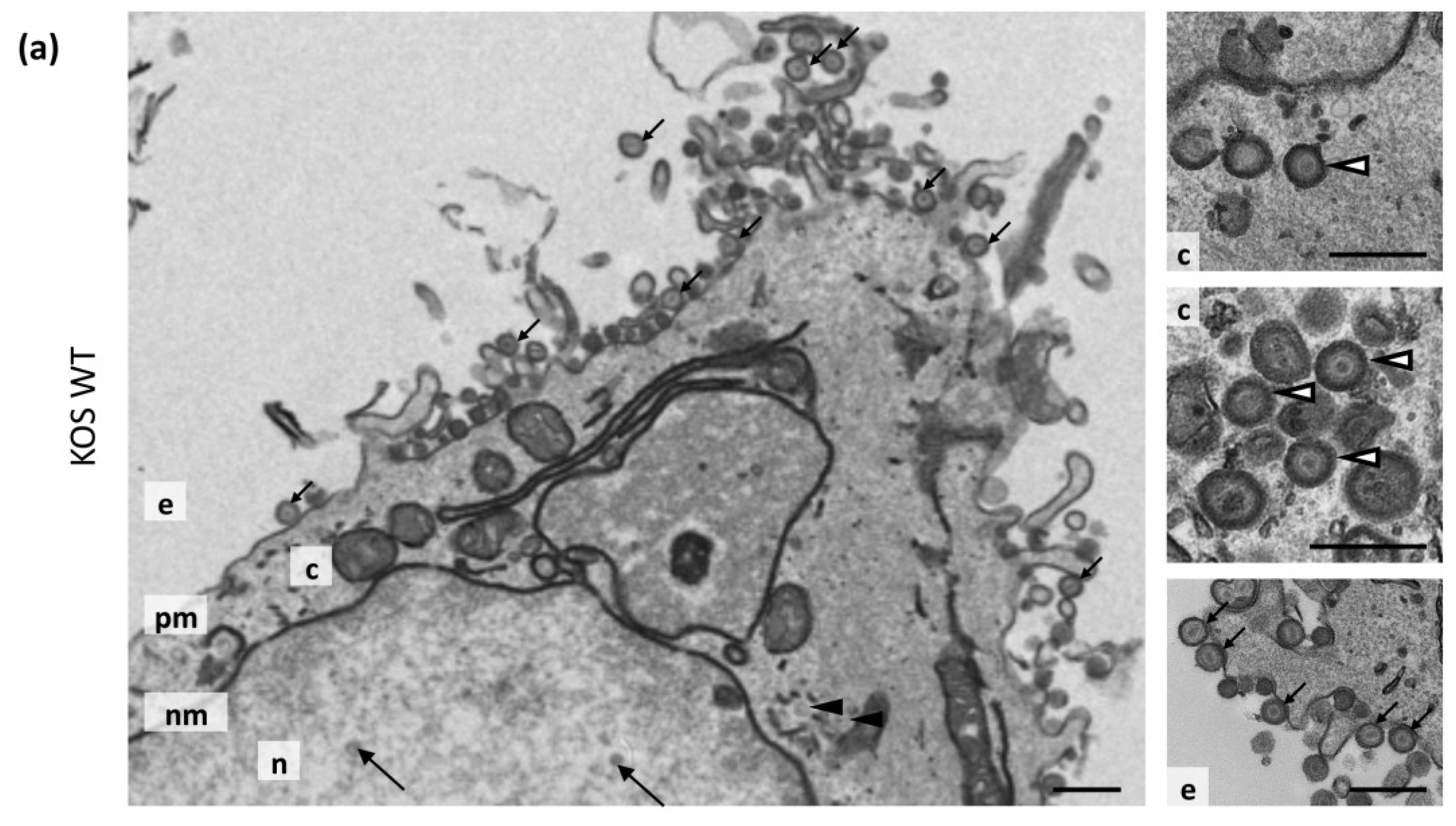
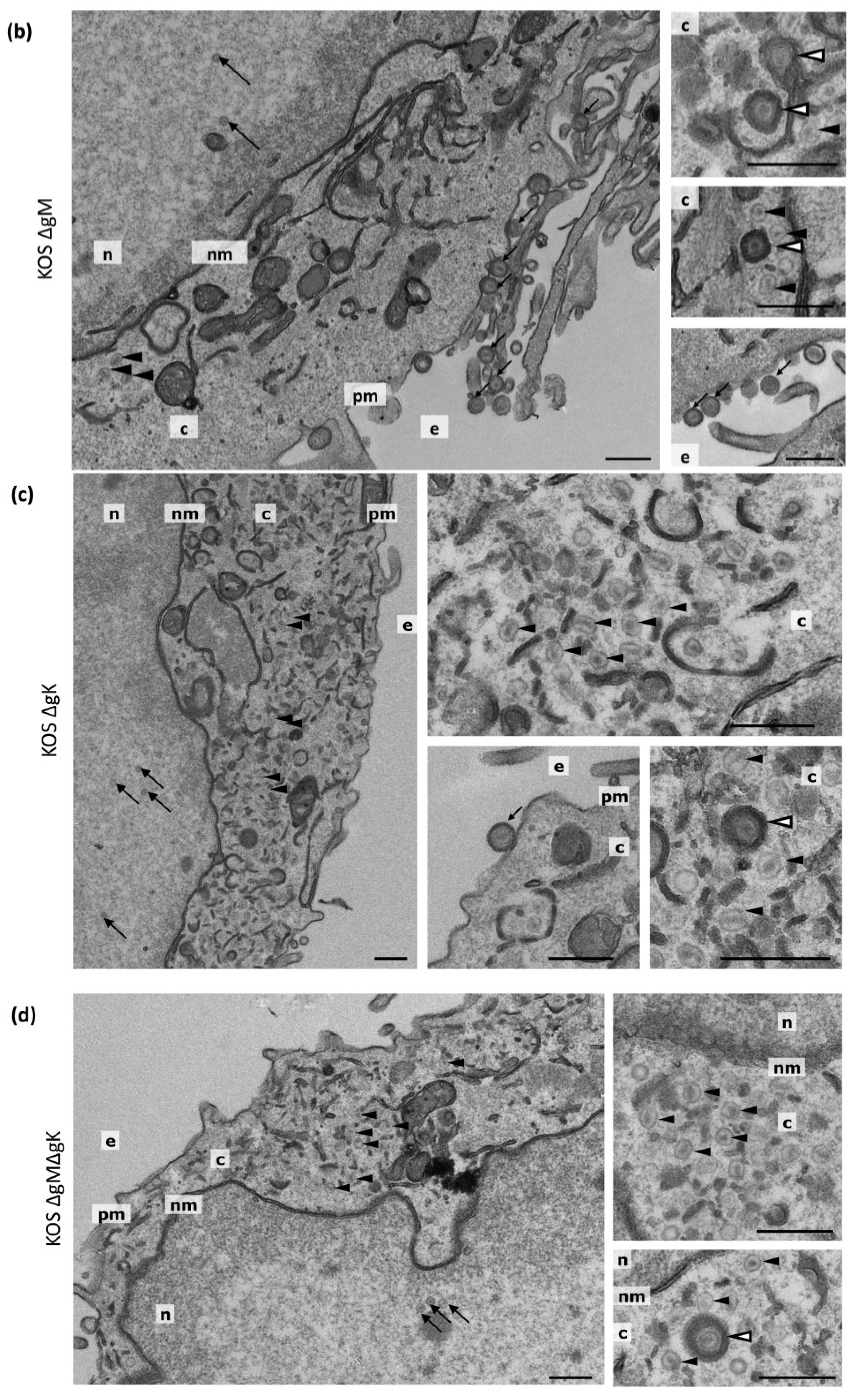
2.6. Transmission Electron Microscopy (TEM) and Quantification
3. Results
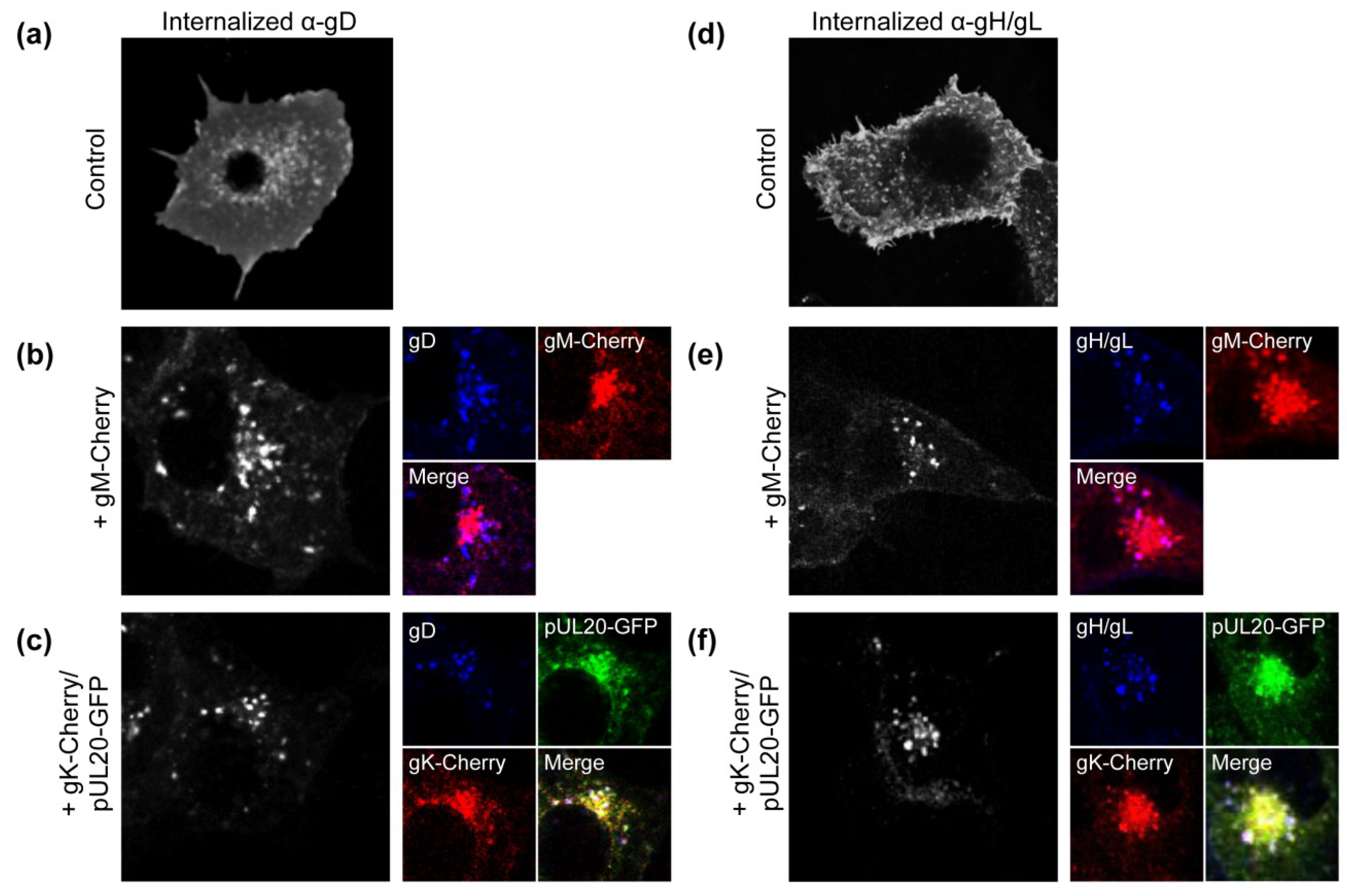
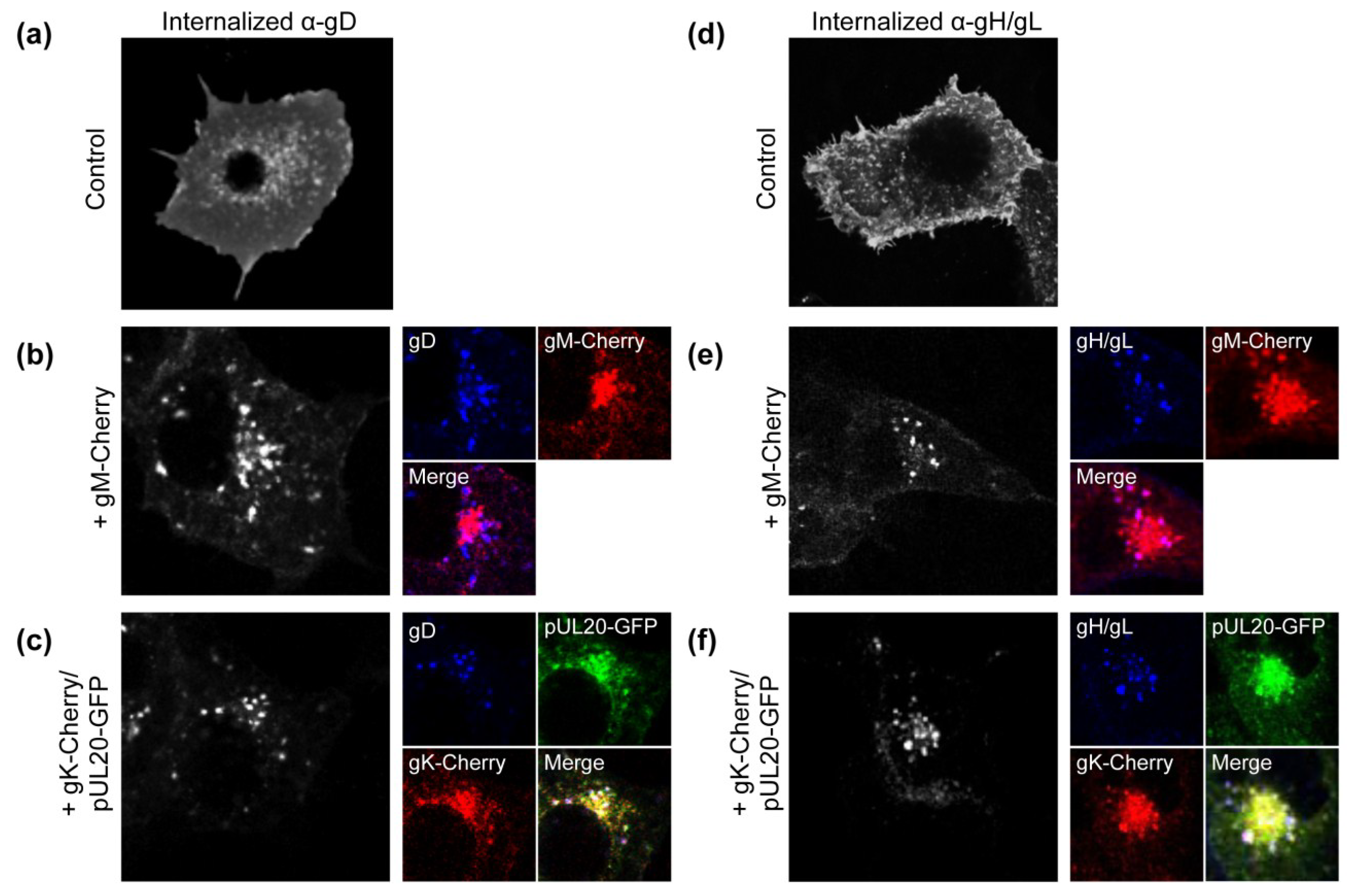
3.1. gK/pUL20 Cause the Internalization of gD and gH/gL in Transfected Cells
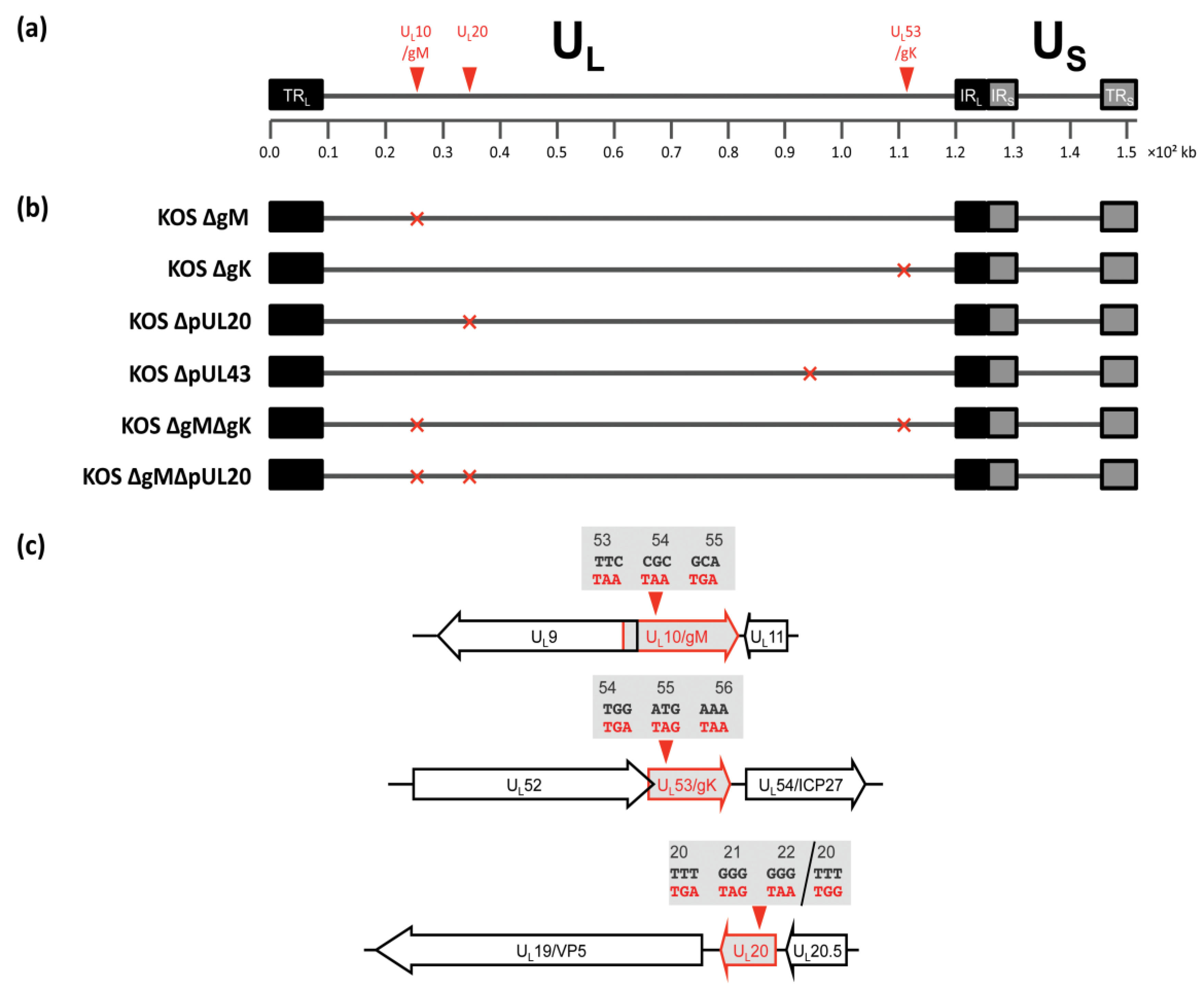
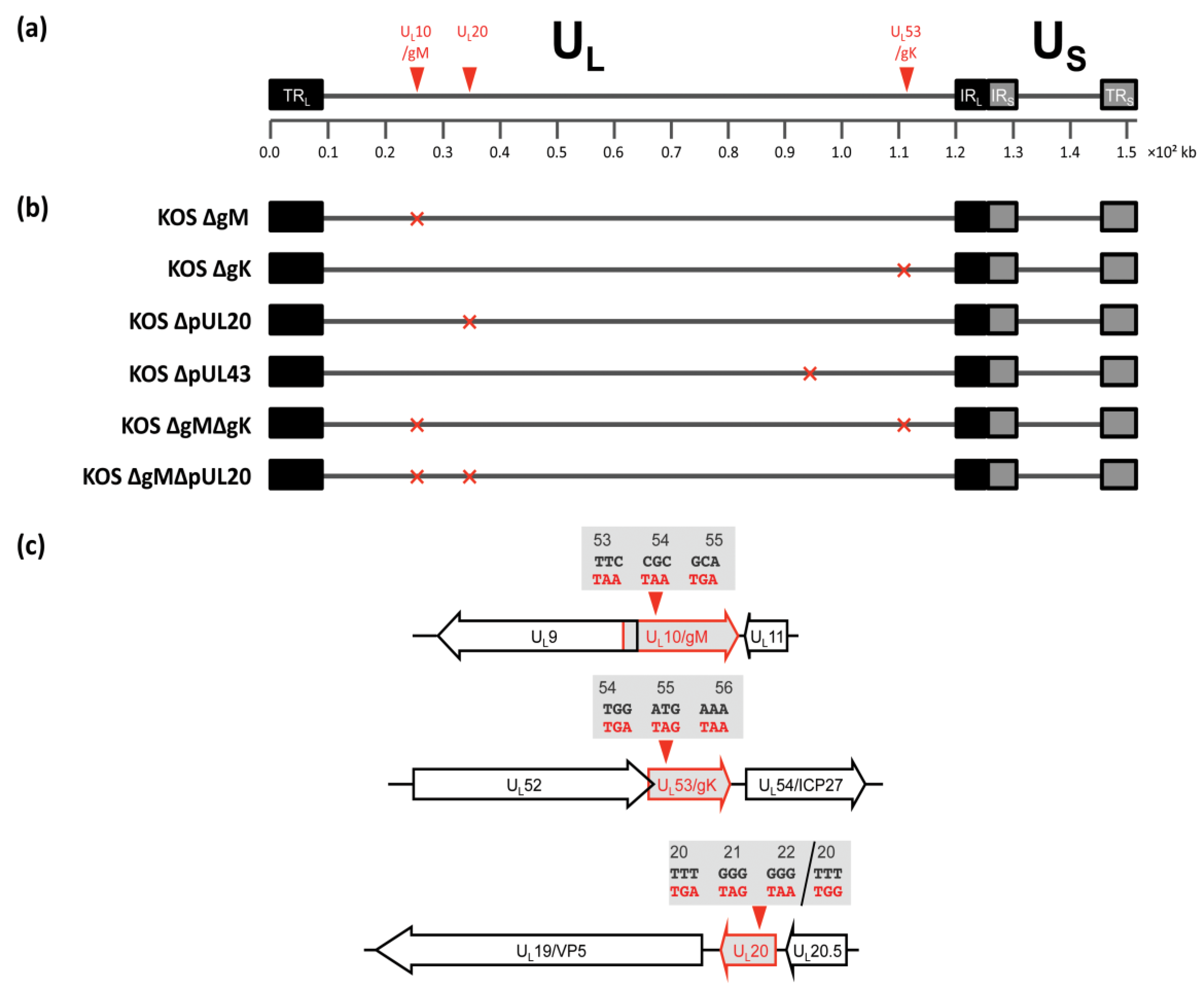
3.2. Construction of Recombinant Viruses Lacking gK/pUL20 and gM
| Virus | Virus Titre (PFU/mL) | |
|---|---|---|
| Non-Complementing Cell Line (Vero) * | Complementing Cell Lines (VK302/Fd20-1) ** | |
| KOS ΔgK | 1.52 × 104 | 2.63 × 106 |
| KOS ΔpUL20 | 3.40 × 103 | 9.70 × 105 |
| KOS ΔgMΔgK | 2.20 × 103 | 1.08 × 106 |
| KOS ΔgMΔpUL20 | 2.70 × 102 | 1.53 × 105 |
3.3. Growth Kinetics of Viruses Lacking gK/pUL20 and gM
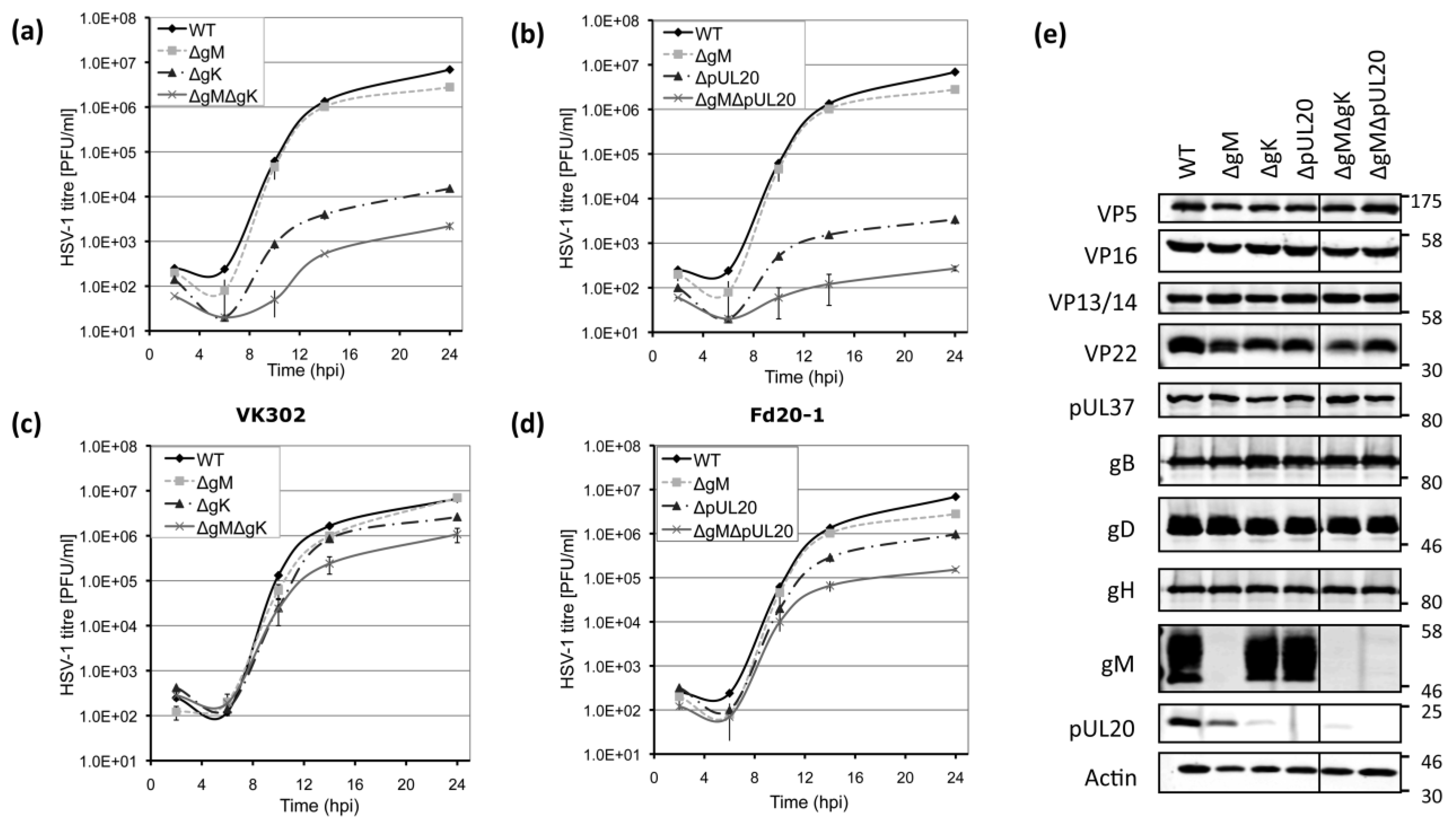
3.4. Viral Protein Expression of Recombinant Viruses Lacking gK/pUL20 and gM
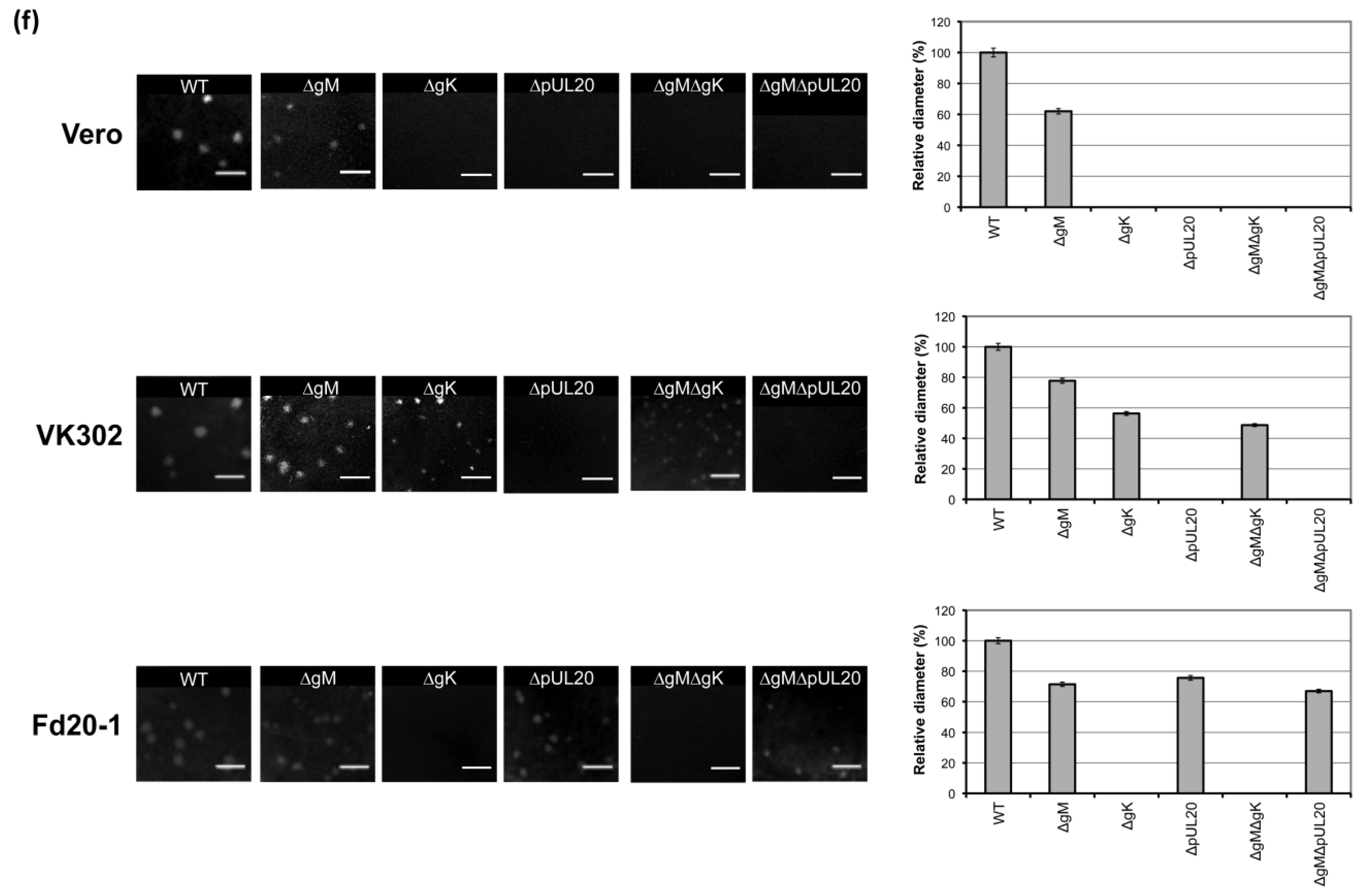
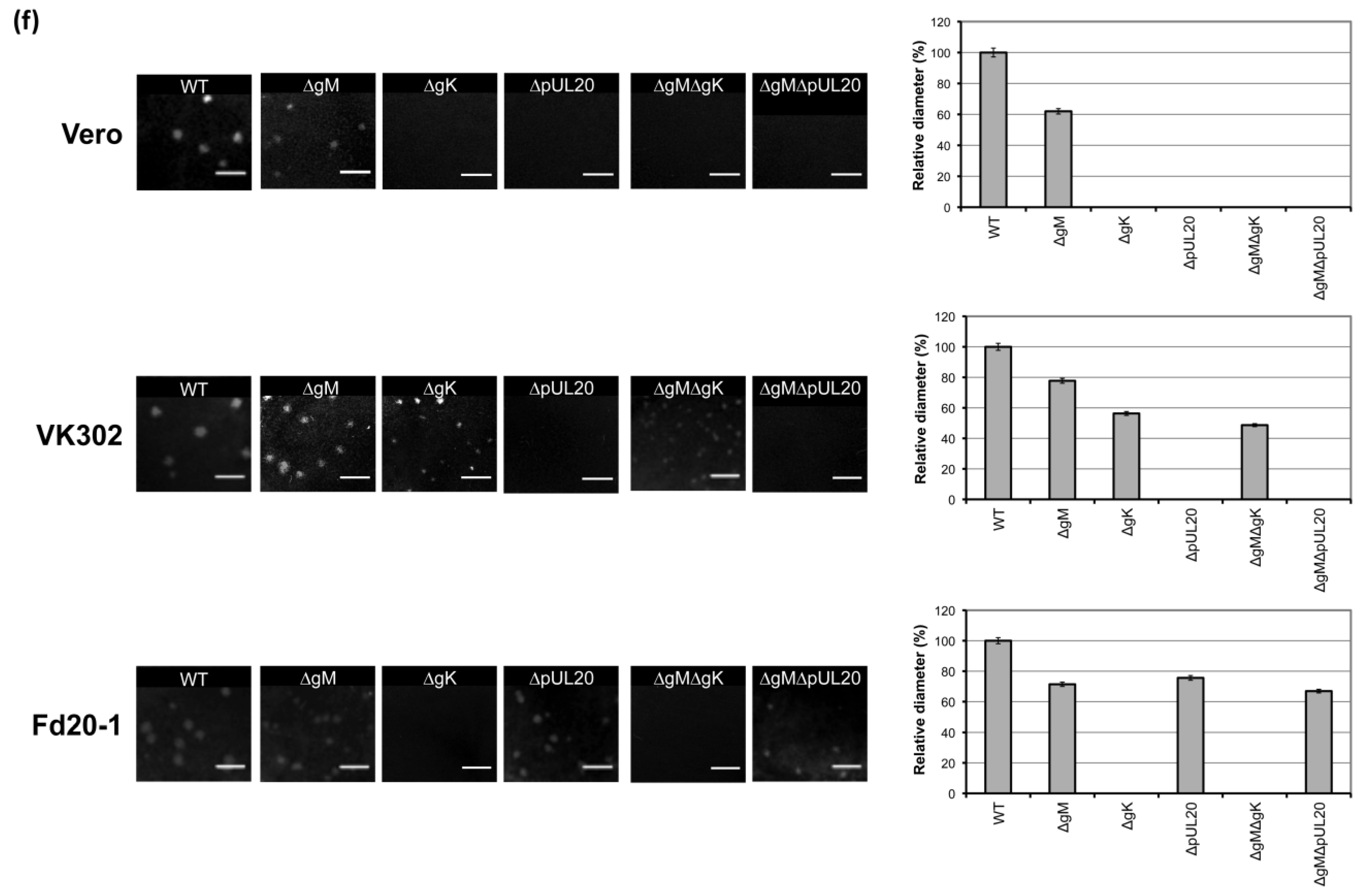
3.5. Plaque Size Analysis of Recombinant Viruses Lacking gK/pUL20 and gM
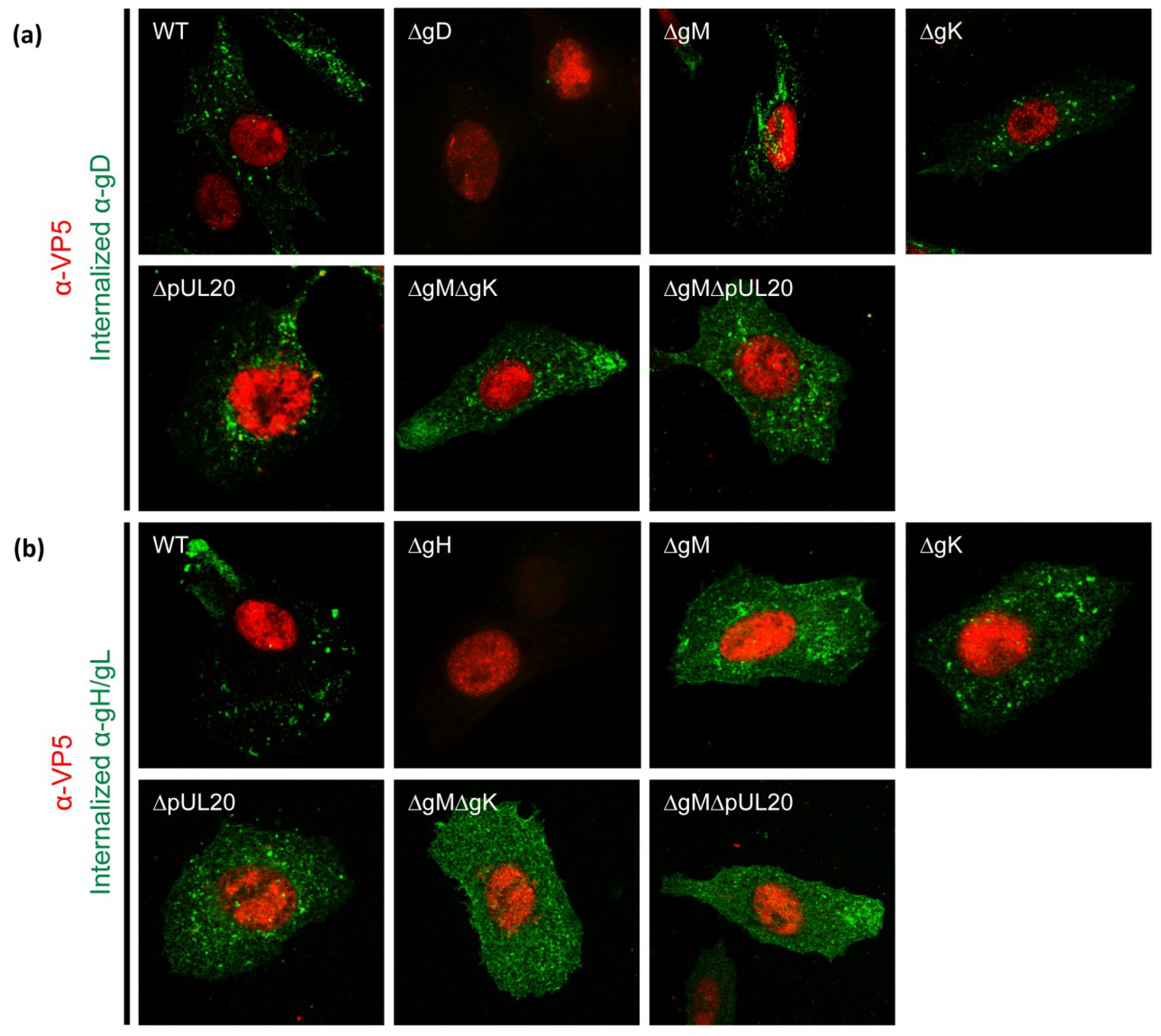
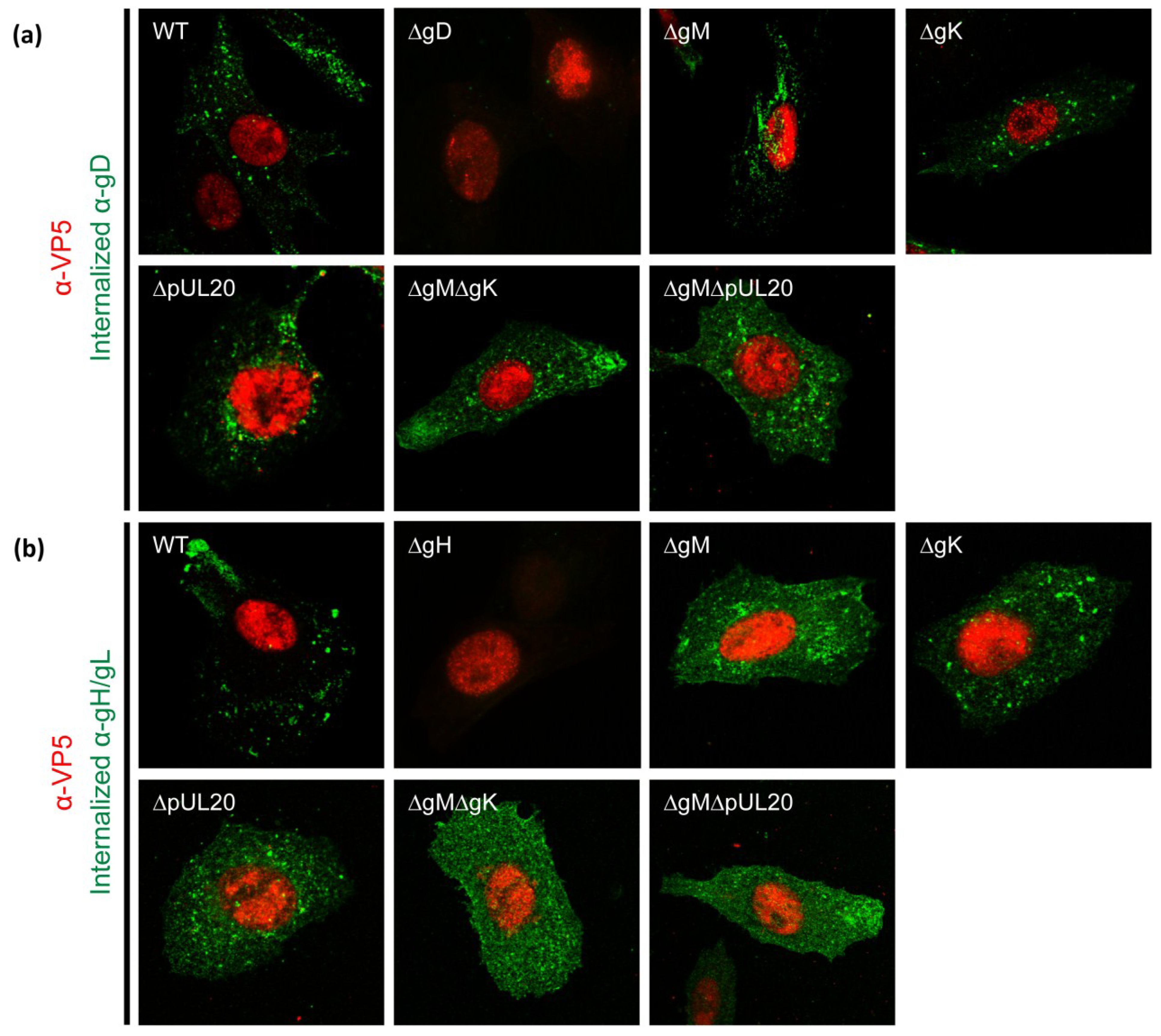
3.6. Internalization of gD and gH/gL in Infected Cells
3.7. Transmission Electron Microscopy
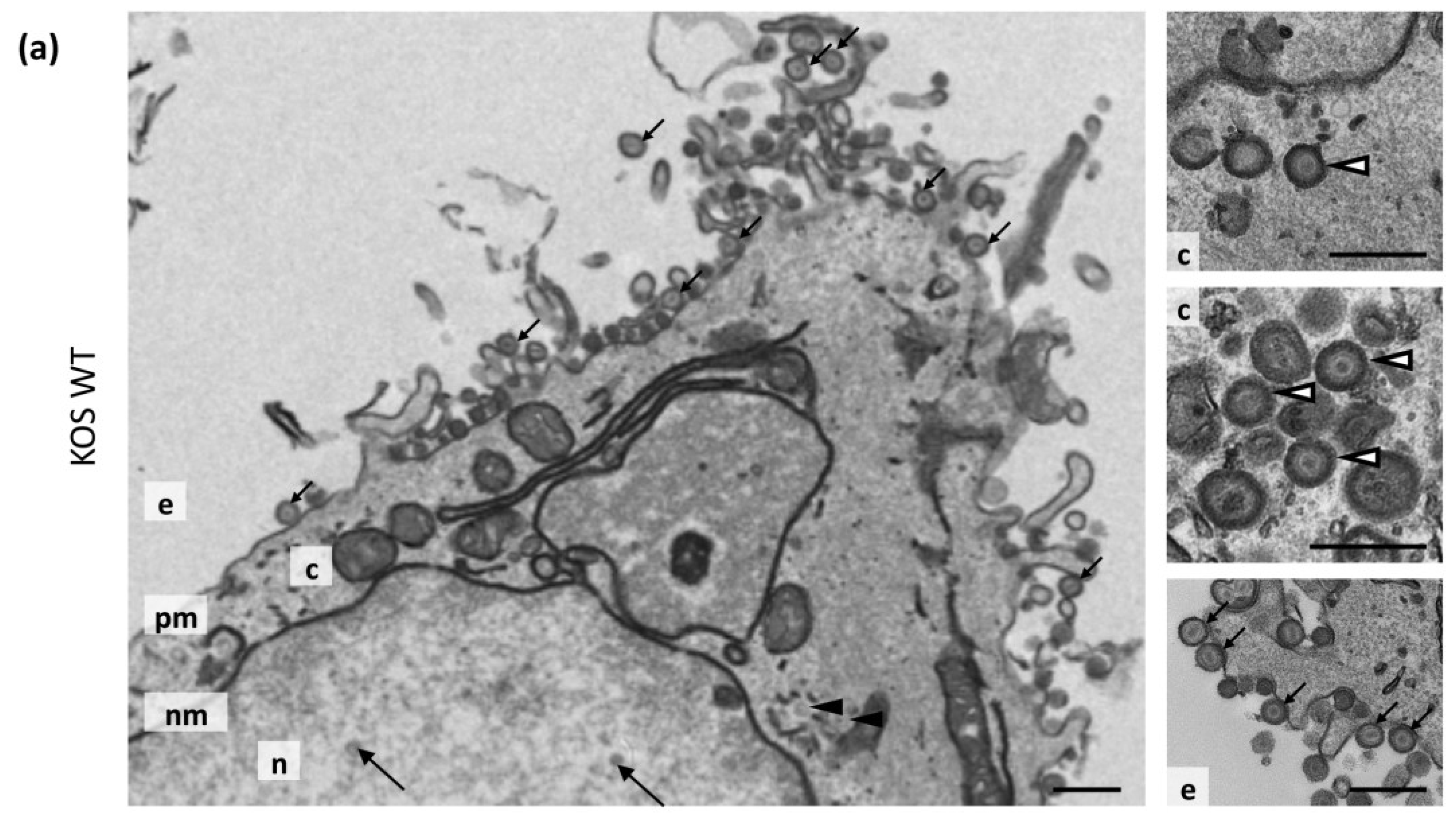
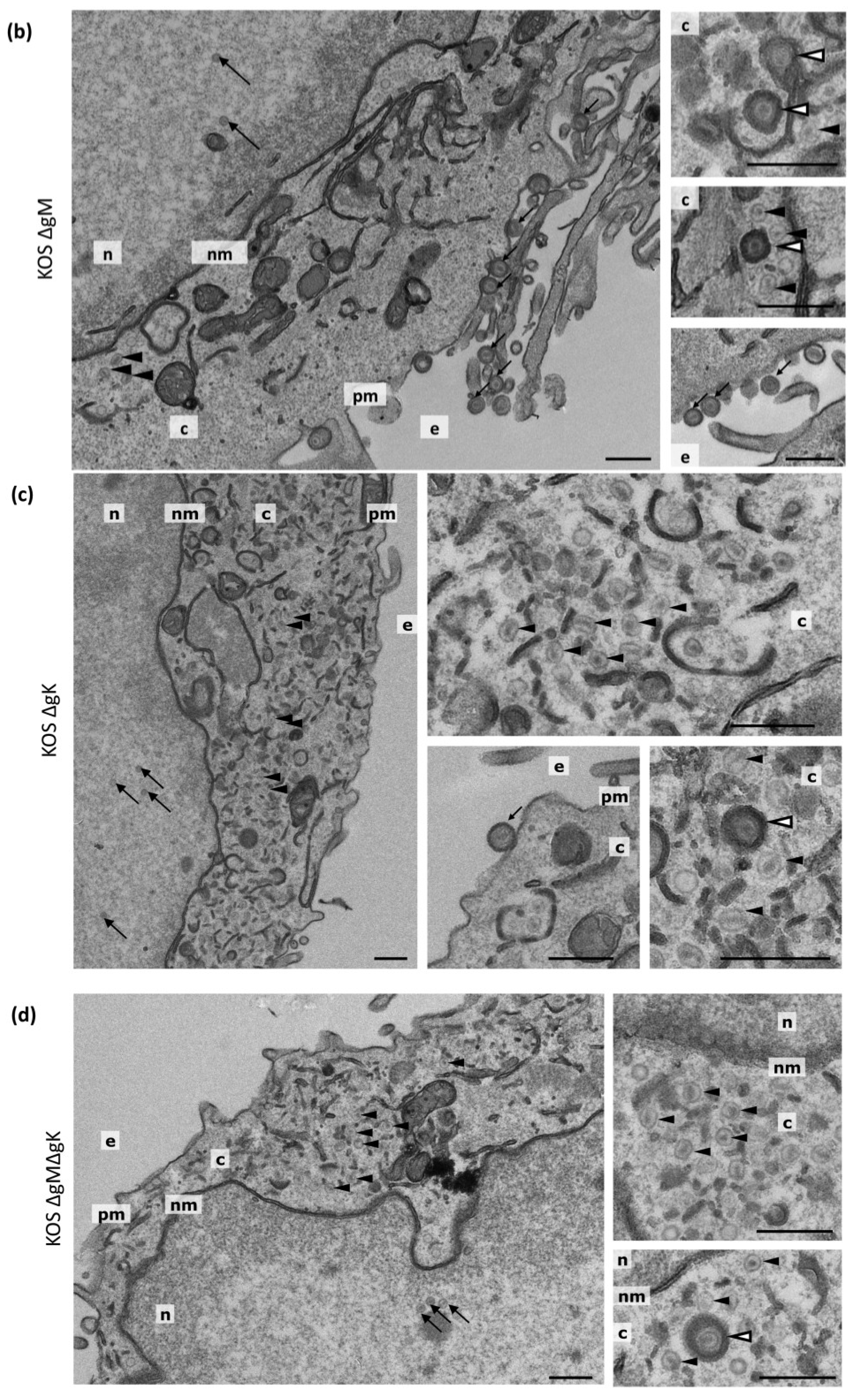
| Virus | Nuclear Capsids | Cytoplasmic Non-Enveloped Capsids | Cytoplasmic Enveloped Virions | Extracellular Virions | Total |
|---|---|---|---|---|---|
| KOS WT | 510 (41.9%) | 31 (2.5%) | 117 (9.6%) | 560 (46.0%) | 1218 |
| KOS ΔgM | 309 (44.5%) | 40 (5.8%) | 60 (8.6%) | 285 (41.4%) | 694 |
| KOS ΔgK | 244 (47.4%) | 258 (50.1%) | 9 (1.7%) | 4 (0.8%) | 515 |
| KOS ΔgMΔgK | 186 (34.8%) | 347 (64.8%) | 2 (0.4%) | 0 (0.0%) | 535 |
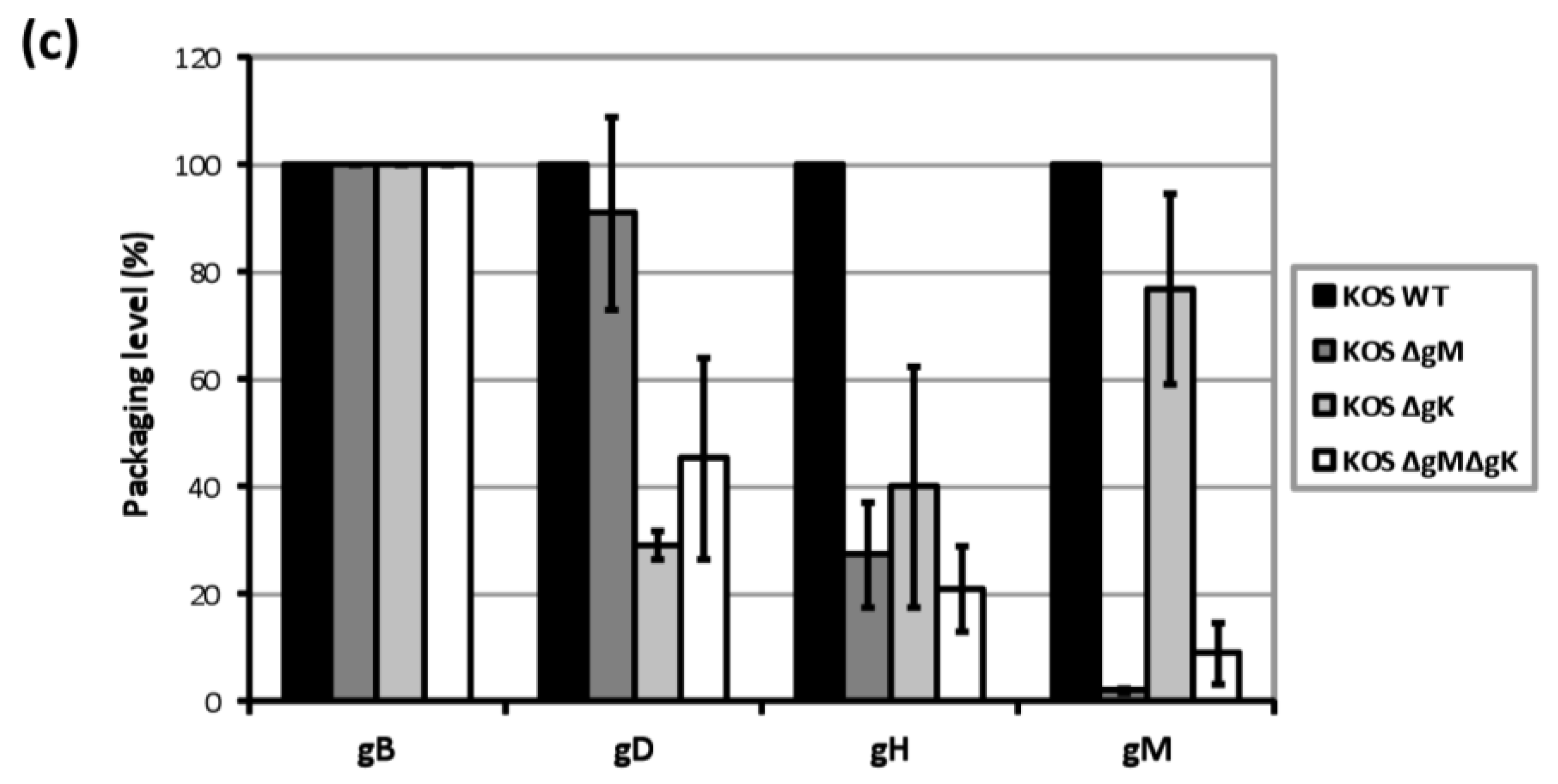
3.8. Analysis of gD and gH/gL Incorporation into Progeny Virions from Recombinant Viruses
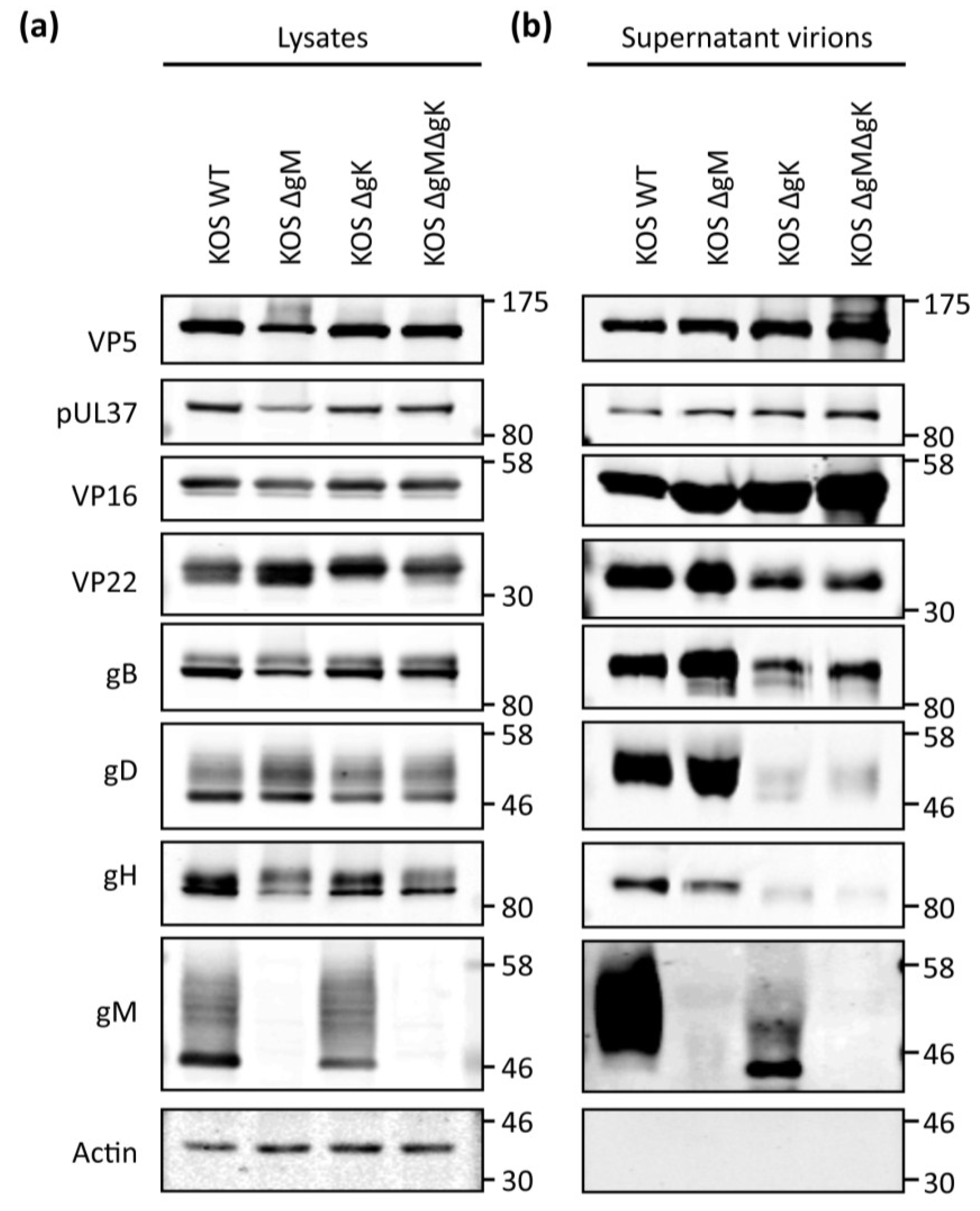

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Traub, L.M. Tickets to ride: Selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 2009, 10, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippe, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes virus fusion and entry: A story with many characters. Viruses 2012, 4, 800–832. [Google Scholar] [CrossRef] [PubMed]
- Alconada, A.; Bauer, U.; Sodeik, B.; Hoflack, B. Intracellular traffic of herpes simplex virus glycoprotein gE: Characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 1999, 73, 377–387. [Google Scholar] [PubMed]
- Beitia Ortiz de Zarate, I.; Cantero-Aguilar, L.; Longo, M.; Berlioz-Torrent, C.; Rozenberg, F. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J. Virol. 2007, 81, 13889–13903. [Google Scholar]
- Crump, C.M.; Bruun, B.; Bell, S.; Pomeranz, L.E.; Minson, T.; Browne, H.M. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 2004, 85, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Bell, S.; Zenner, H.L.; Lau, S.Y.; Crump, C.M. Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J. Gen. Virol. 2012, 93, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, E.; Lombardi, G.; Gianni, T.; Capri, M.; Campadelli-Fiume, G. Coexpression of UL20p and gK inhibits cell-cell fusion mediated by herpes simplex virus glycoproteins gD, gH-gL, and wild-type gB or an endocytosis-defective gB mutant and downmodulates their cell surface expression. J. Virol. 2004, 78, 8015–8025. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Altenschmidt, J.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Identification and characterization of the pseudorabies virus UL43 protein. Virology 2005, 334, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Melancon, J.M.; Olivier, T.L.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 2004, 78, 13262–13277. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Chouljenko, V.N.; Kousoulas, K.G. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 2008, 82, 6310–6323. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Kousoulas, K.G. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 1999, 73, 8457–8468. [Google Scholar] [PubMed]
- Hutchinson, L.; Johnson, D.C. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 1995, 69, 5401–5413. [Google Scholar] [PubMed]
- Hutchinson, L.; Roop-Beauchamp, C.; Johnson, D.C. Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J. Virol. 1995, 69, 4556–4563. [Google Scholar] [PubMed]
- Baines, J.D.; Ward, P.L.; Campadelli-Fiume, G.; Roizman, B. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 1991, 65, 6414–6424. [Google Scholar] [PubMed]
- Bond, V.C.; Person, S. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 1984, 132, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Debroy, C.; Pederson, N.; Person, S. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 1985, 145, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Alvarez, X.; Kousoulas, K.G. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): The UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 2003, 77, 499–510. [Google Scholar] [PubMed]
- MacLean, C.A.; Efstathiou, S.; Elliott, M.L.; Jamieson, F.E.; McGeoch, D.J. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J. Gen. Virol. 1991, 72, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Pogue-Geile, K.L.; Lee, G.T.; Shapira, S.K.; Spear, P.G. Fine mapping of mutations in the fusion-inducing MP strain of herpes simplex virus type 1. Virology 1984, 136, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Holland, T.C. Determination of the transmembrane topology of herpes simplex virus type 1 glycoprotein K. J. Biol. Chem. 1997, 272, 33305–33311. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, R.; Holland, T.C. In vitro characterization of the HSV-1 UL53 gene product. Virology 1992, 186, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Melancon, J.M.; Foster, T.P.; Kousoulas, K.G. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 2004, 78, 7329–7343. [Google Scholar] [CrossRef] [PubMed]
- Stingley, S.W.; Ramirez, J.J.; Aguilar, S.A.; Simmen, K.; Sandri-Goldin, R.M.; Ghazal, P.; Wagner, E.K. Global analysis of herpes simplex virus type 1 transcription using an oligonucleotide-based DNA microarray. J. Virol. 2000, 74, 9916–9927. [Google Scholar] [CrossRef]
- Ward, P.L.; Campadelli-Fiume, G.; Avitabile, E.; Roizman, B. Localization and putative function of the UL20 membrane protein in cells infected with herpes simplex virus 1. J. Virol. 1994, 68, 7406–7417. [Google Scholar] [PubMed]
- Gierasch, W.W.; Zimmerman, D.L.; Ward, S.L.; Vanheyningen, T.K.; Romine, J.D.; Leib, D.A. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods 2006, 135, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [PubMed]
- Babic, N.; Rodger, G.; Arthur, J.; Minson, A.C. A study of primary neuronal infection by mutants of herpes simplex virus type 1 lacking dispensable and non-dispensable glycoproteins. J. Gen. Virol. 1999, 80, 2403–2409. [Google Scholar] [PubMed]
- Browne, H.; Bell, S.; Minson, T.; Wilson, D.W. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: Evidence for reenvelopment during egress. J. Virol. 1996, 70, 4311–4316. [Google Scholar] [PubMed]
- Zenner, H.L.; Yoshimura, S.; Barr, F.A.; Crump, C.M. Analysis of Rab GTPase-activating proteins indicates that Rab1a/b and Rab43 are important for herpes simplex virus 1 secondary envelopment. J. Virol. 2011, 85, 8012–8021. [Google Scholar] [CrossRef] [PubMed]
- Minson, A.C.; Hodgman, T.C.; Digard, P.; Hancock, D.C.; Bell, S.E.; Buckmaster, E.A. An Analysis of the Biological Properties of Monoclonal Antibodies against Glycoprotein D of Herpes Simplex Virus and Identification of Amino Acid Substitutions that Confer Resistance to Neutralization. J. Gen. Virol. 1986, 67, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Buckmaster, E.A.; Gompels, U.; Minson, A. Characterisation and physical mapping of an HSV-1 glycoprotein of approximately 115 X 10(3) molecular weight. Virology 1984, 139, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.D.; Roizman, B. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 1991, 65, 938–944. [Google Scholar] [PubMed]
- Browne, H.; Bell, S.; Minson, T. Analysis of the requirement for glycoprotein m in herpes simplex virus type 1 morphogenesis. J. Virol. 2004, 78, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Melancon, J.M.; Baines, J.D.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 2004, 78, 5347–5357. [Google Scholar] [CrossRef] [PubMed]
- Jayachandra, S.; Baghian, A.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 1997, 71, 5012–5024. [Google Scholar] [PubMed]
- Leege, T.; Fuchs, W.; Granzow, H.; Kopp, M.; Klupp, B.G.; Mettenleiter, T.C. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 2009, 83, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Melancon, J.M.; Luna, R.E.; Foster, T.P.; Kousoulas, K.G. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 2005, 79, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Chouljenko, V.N.; Walker, J.D.; Kousoulas, K.G. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J. Virol. 2013, 87, 8029–8037. [Google Scholar] [CrossRef] [PubMed]
- MacLean, C.A.; Robertson, L.M.; Jamieson, F.E. Characterization of the UL10 gene product of herpes simplex virus type 1 and investigation of its role in vivo. J. Gen. Virol. 1993, 74, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.H.; Harley, C.A.; Mukhopadhyay, A.; Wilson, D.W. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 2005, 86, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Maringer, K.; Stylianou, J.; Elliott, G. A network of protein interactions around the herpes simplex virus tegument protein VP22. J. Virol. 2012, 86, 12971–12982. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Hare, P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 1998, 72, 6448–6455. [Google Scholar] [PubMed]
- Blaho, J.A.; Mitchell, C.; Roizman, B. An amino acid sequence shared by the herpes simplex virus 1 alpha regulatory proteins 0, 4, 22, and 27 predicts the nucleotidylylation of the UL21, UL31, UL47, and UL49 gene products. J. Biol. Chem. 1994, 269, 17401–17410. [Google Scholar] [PubMed]
- Elliott, G.; O’Reilly, D.; O’Hare, P. Identification of phosphorylation sites within the herpes simplex virus tegument protein VP22. J. Virol. 1999, 73, 6203–6206. [Google Scholar] [PubMed]
- Meredith, D.M.; Lindsay, J.A.; Halliburton, I.W.; Whittaker, G.R. Post-translational modification of the tegument proteins (VP13 and VP14) of herpes simplex virus type 1 by glycosylation and phosphorylation. J. Gen. Virol. 1991, 72, 2771–2775. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Reilly, D.; O’Hare, P. Phosphorylation of the herpes simplex virus type 1 tegument protein VP22. Virology 1996, 226, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, J.; Maringer, K.; Cook, R.; Bernard, E.; Elliott, G. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. J. Virol. 2009, 83, 5204–5218. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, A.; Johnson, D.C. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J. Virol. 2006, 80, 3167–3179. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.; Mo, C.; Cheng, J.J.; Sommer, M.; Zerboni, L.; Stamatis, S.; Arvin, A.M. Functions of the C-terminal domain of varicella-zoster virus glycoprotein E in viral replication in vitro and skin and T-cell tropism in vivo. J. Virol. 2004, 78, 12406–12415. [Google Scholar] [CrossRef] [PubMed]
- Tirabassi, R.S.; Enquist, L.W. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J. Virol. 1998, 72, 4571–4579. [Google Scholar] [PubMed]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, S.-Y.K.; Crump, C.M. HSV-1 gM and the gK/pUL20 Complex Are Important for the Localization of gD and gH/L to Viral Assembly Sites. Viruses 2015, 7, 915-938. https://doi.org/10.3390/v7030915
Lau S-YK, Crump CM. HSV-1 gM and the gK/pUL20 Complex Are Important for the Localization of gD and gH/L to Viral Assembly Sites. Viruses. 2015; 7(3):915-938. https://doi.org/10.3390/v7030915
Chicago/Turabian StyleLau, Sheung-Yee Kathy, and Colin M. Crump. 2015. "HSV-1 gM and the gK/pUL20 Complex Are Important for the Localization of gD and gH/L to Viral Assembly Sites" Viruses 7, no. 3: 915-938. https://doi.org/10.3390/v7030915
APA StyleLau, S.-Y. K., & Crump, C. M. (2015). HSV-1 gM and the gK/pUL20 Complex Are Important for the Localization of gD and gH/L to Viral Assembly Sites. Viruses, 7(3), 915-938. https://doi.org/10.3390/v7030915