
Advanced Molecular Surveillance of Hepatitis C Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
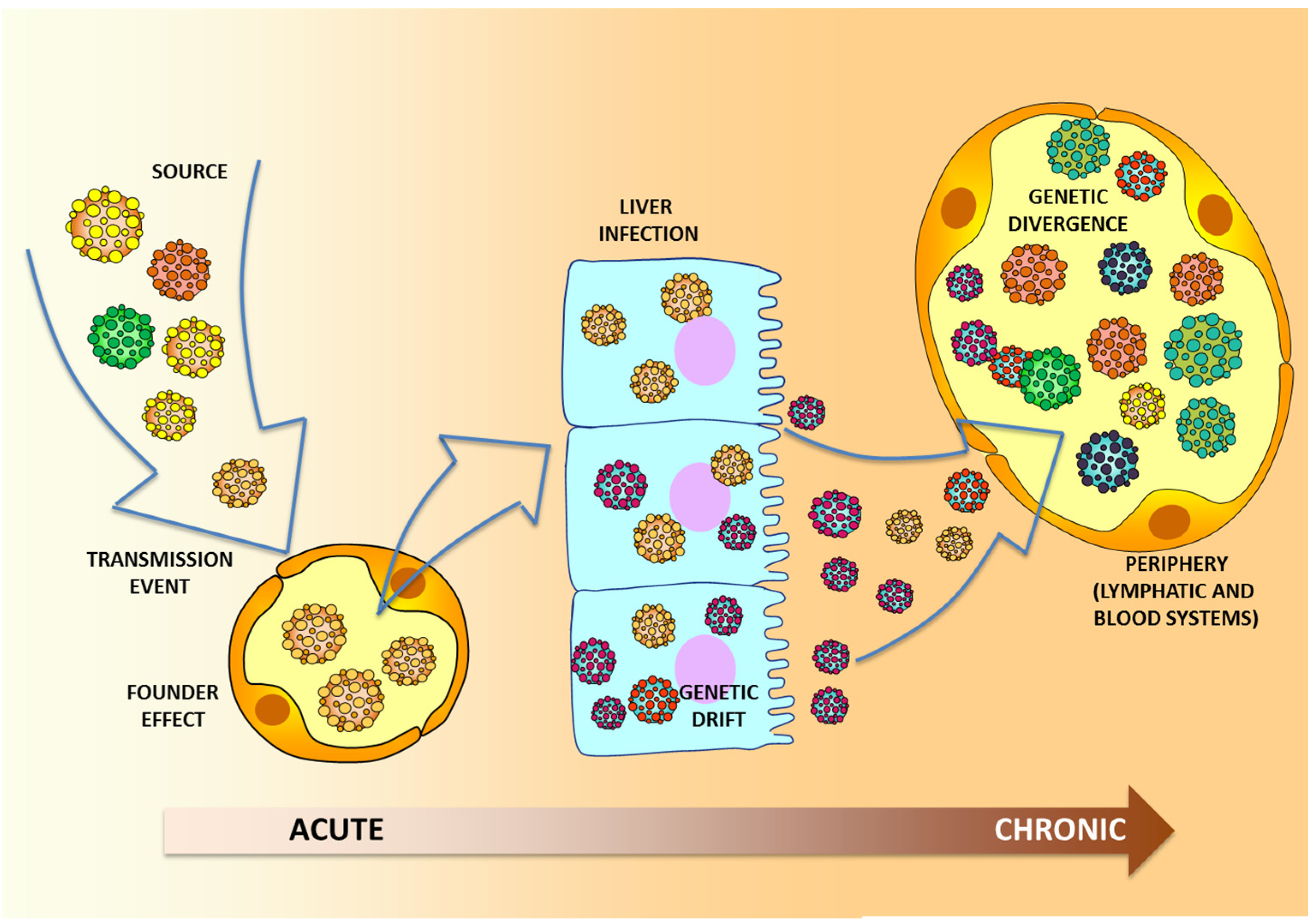{kind=link}
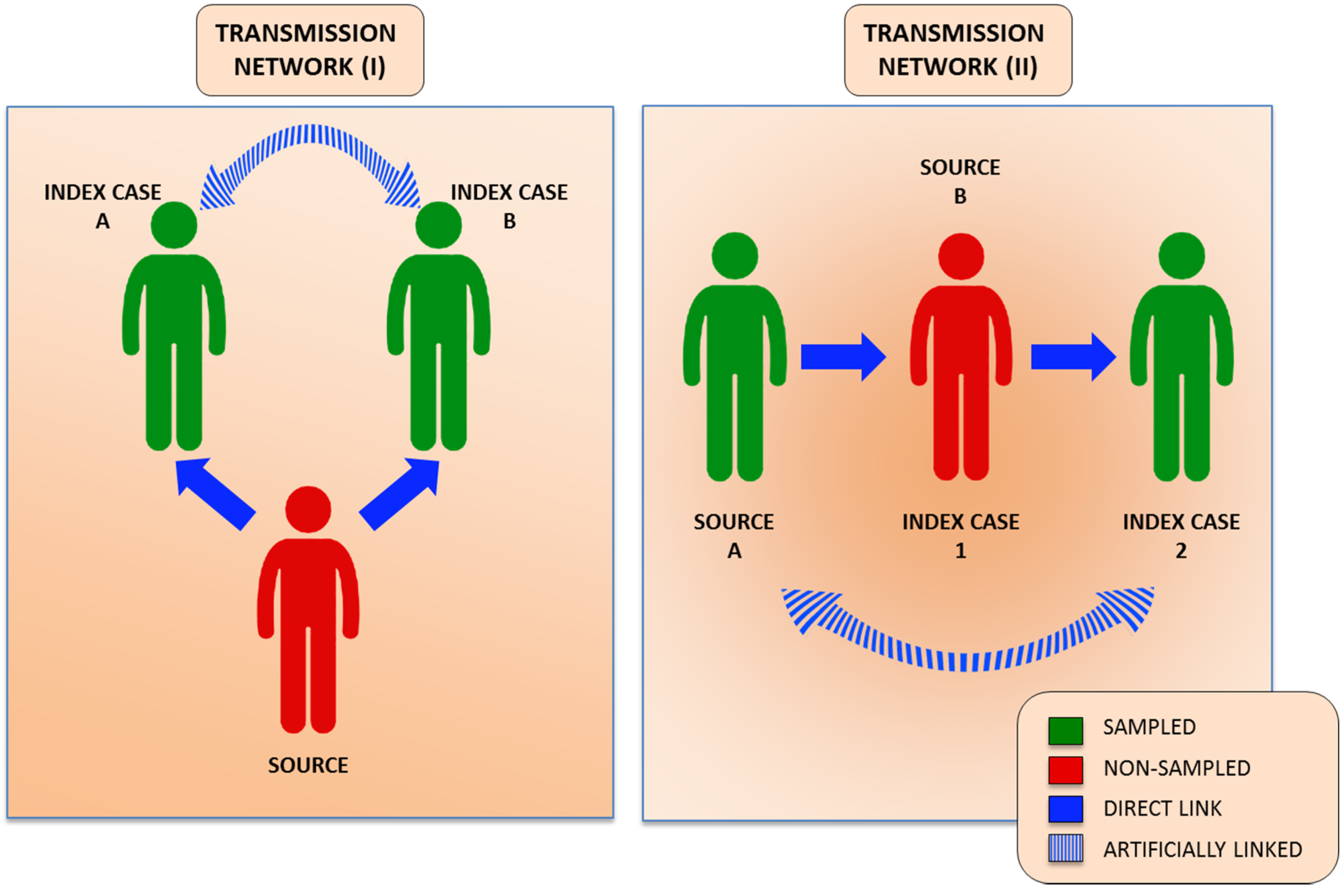{kind=link}
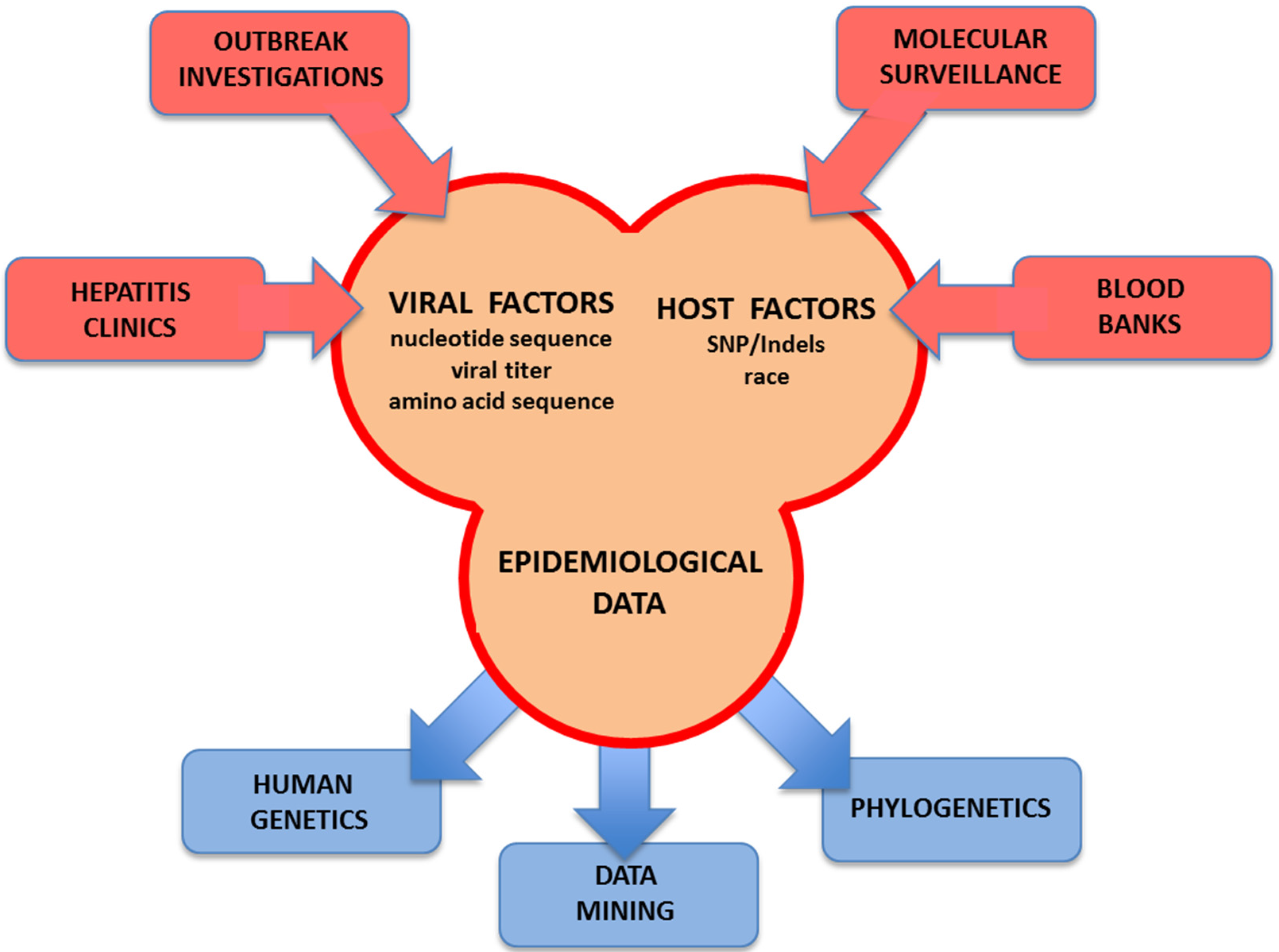{kind=link}
Abstract
:1. Introduction
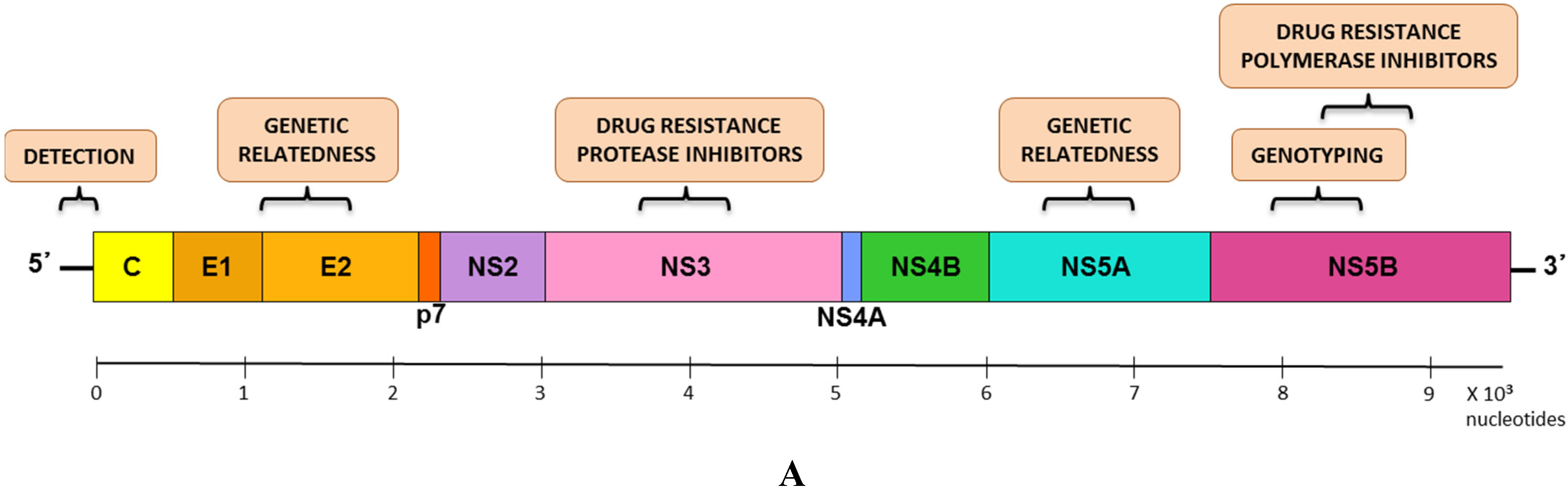

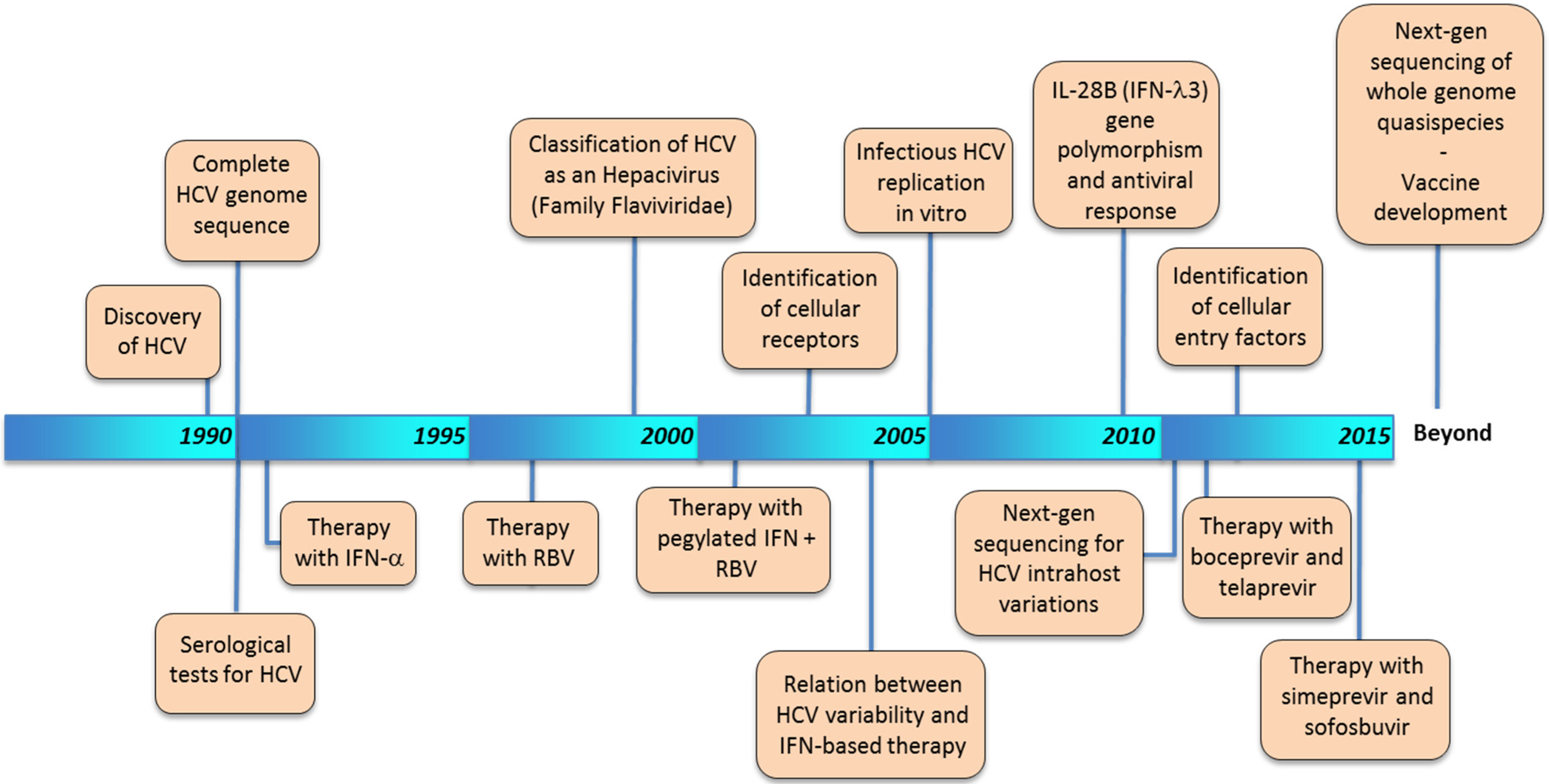
2. Historical Timeline of HCV Characterization

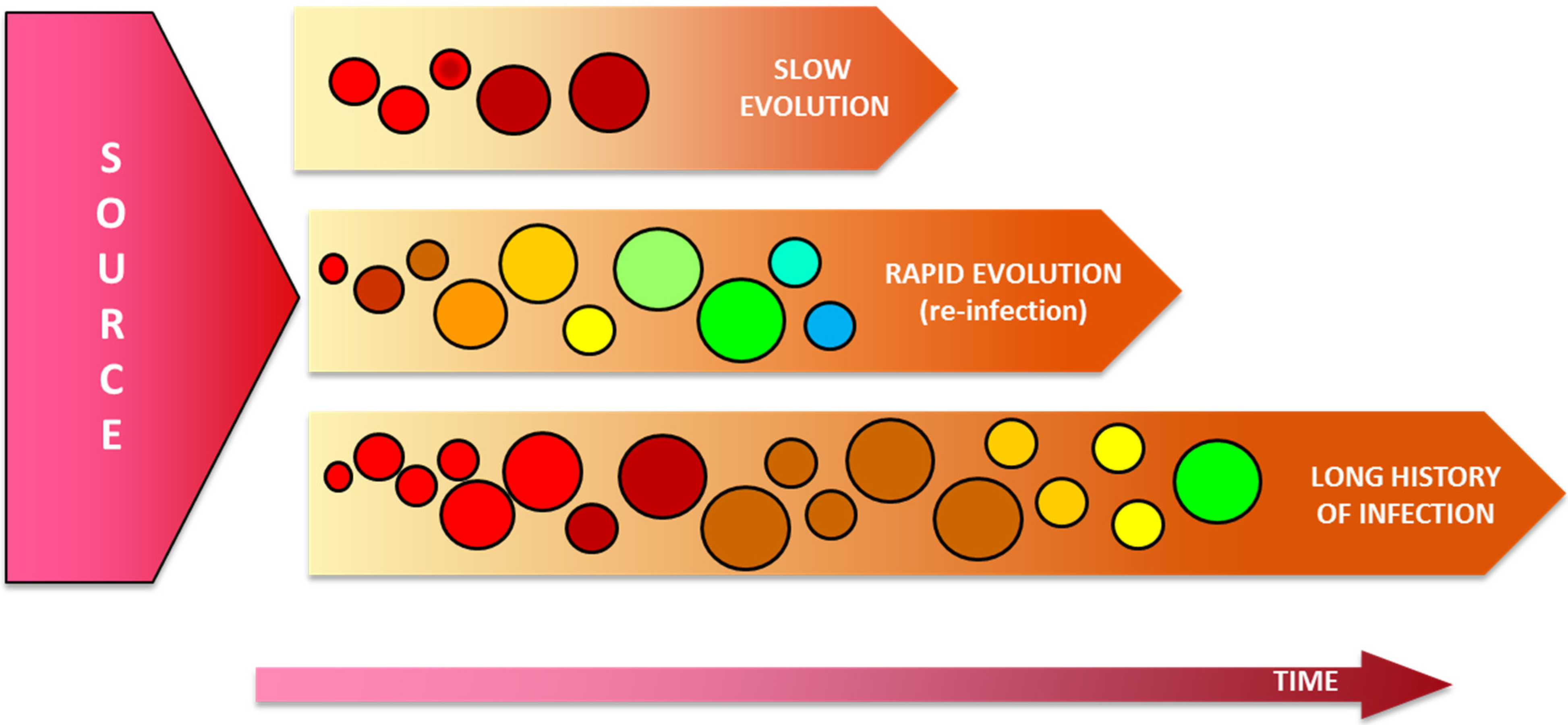
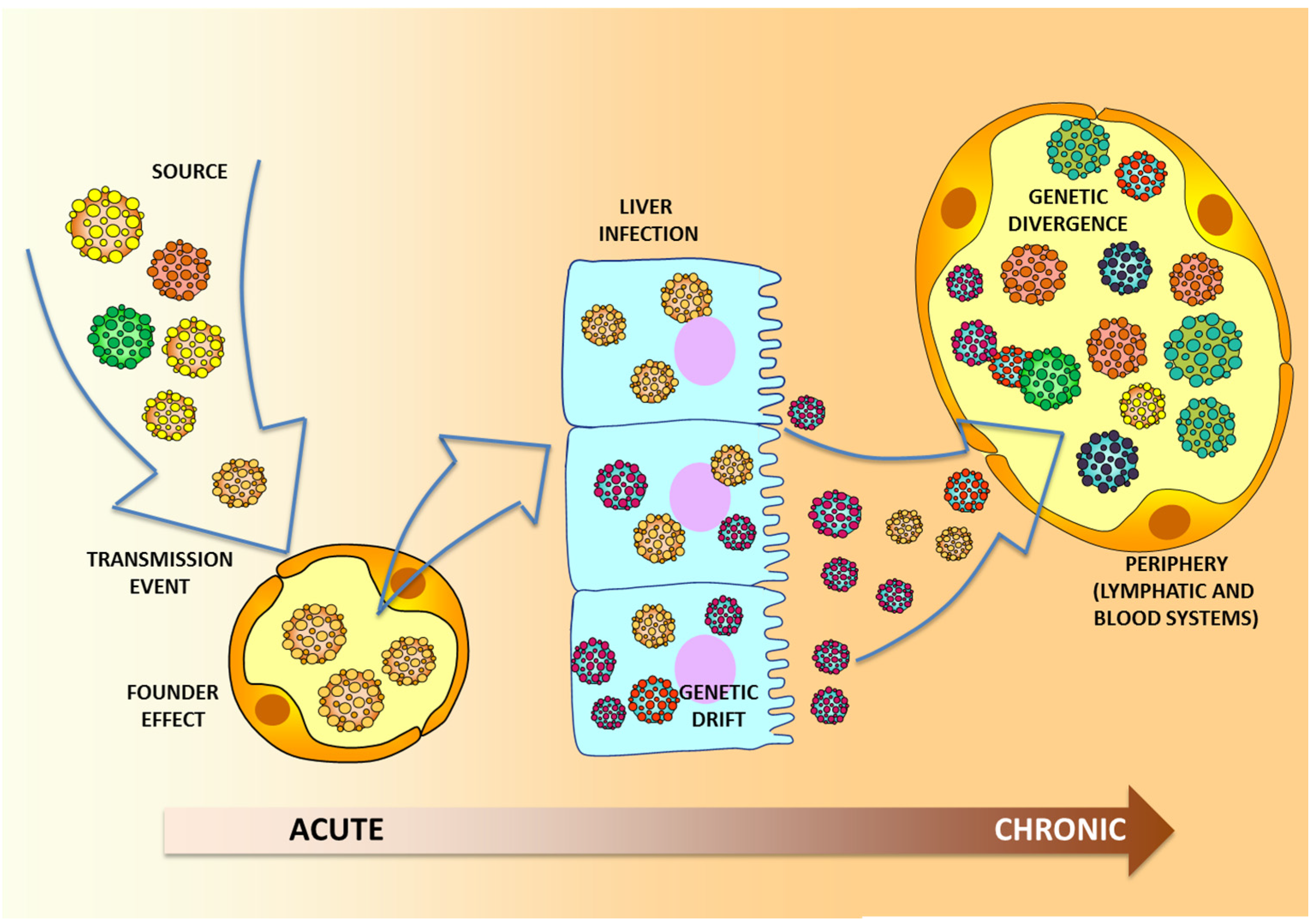
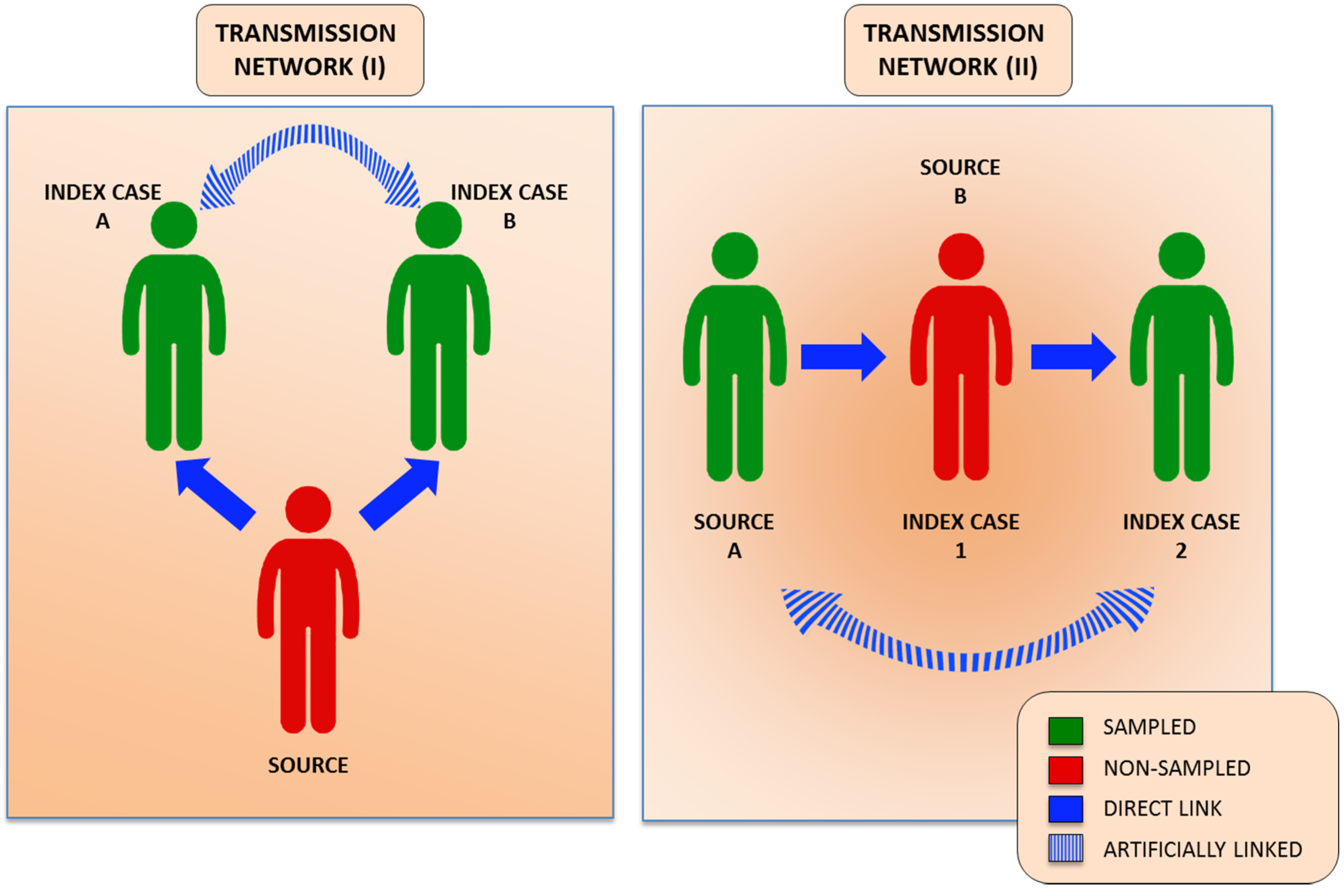
3. Molecular Aspects of HCV Transmission
4. Human Genomics in HCV Advanced Molecular Surveillance
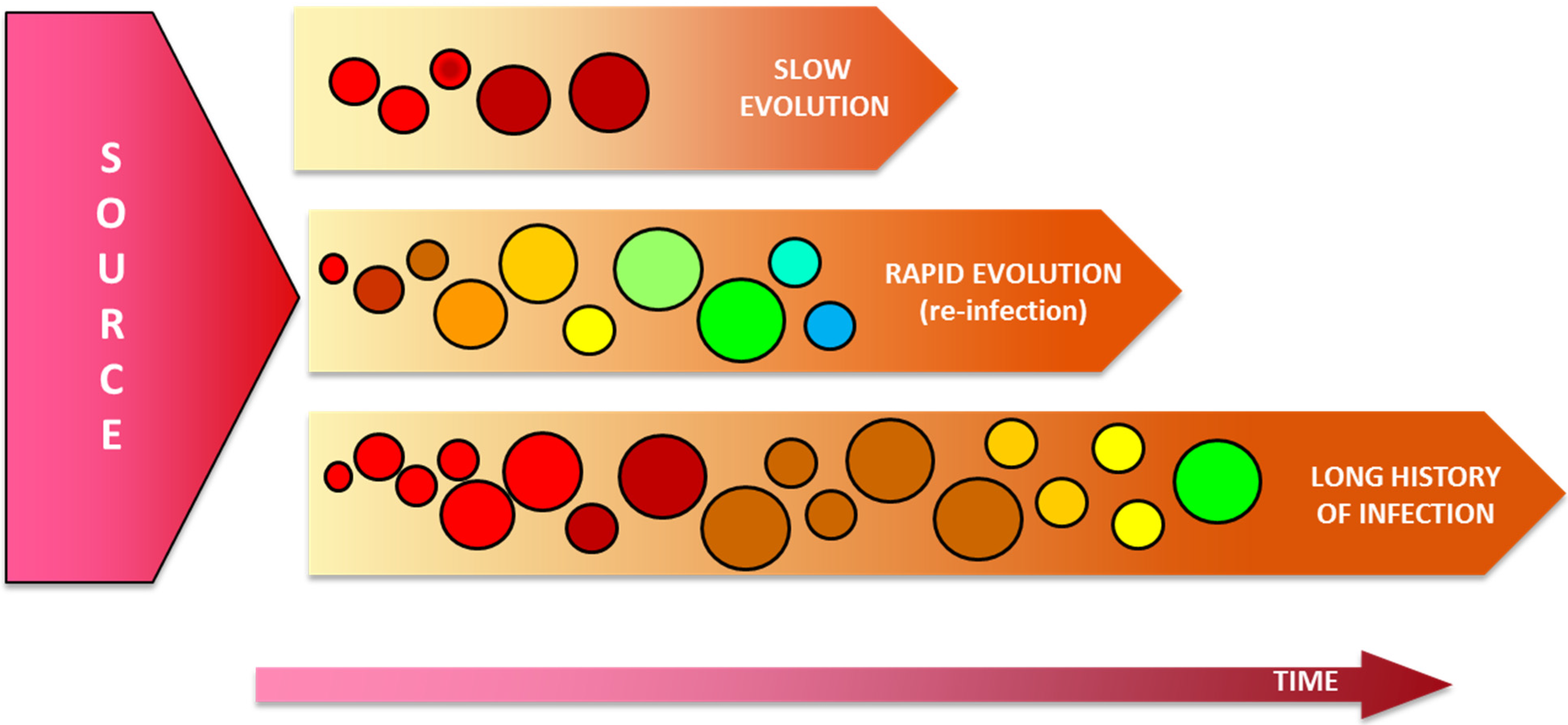
5. Assessment of the HCV Intrahost Genetic Variability
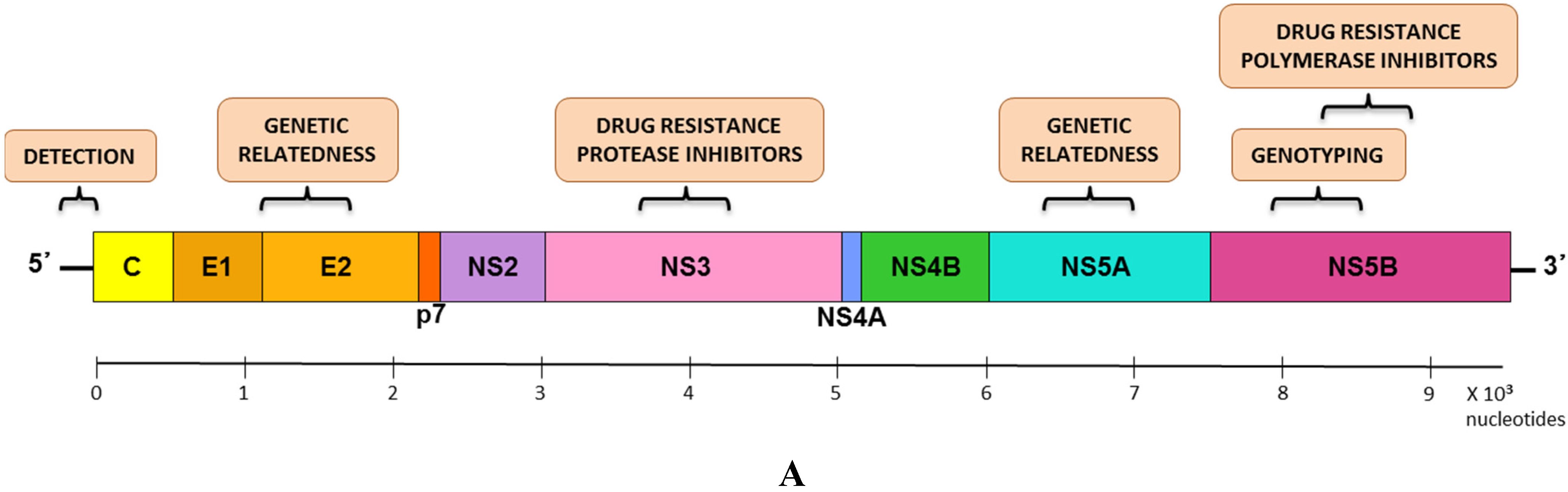
5.1. Genomic Regions
5.2. Single-Stranded Conformation Polymorphism (SSCP)
5.3. Heteroduplex Gel Shift Assays
5.4. Population Sequencing
Bacterial Cloning
5.5. End-Point Limiting-Dilution PCR (EPLD-PCR)
5.6. Mass Spectrometry (MS)
5.7. Next Generation Sequencing (NGS)
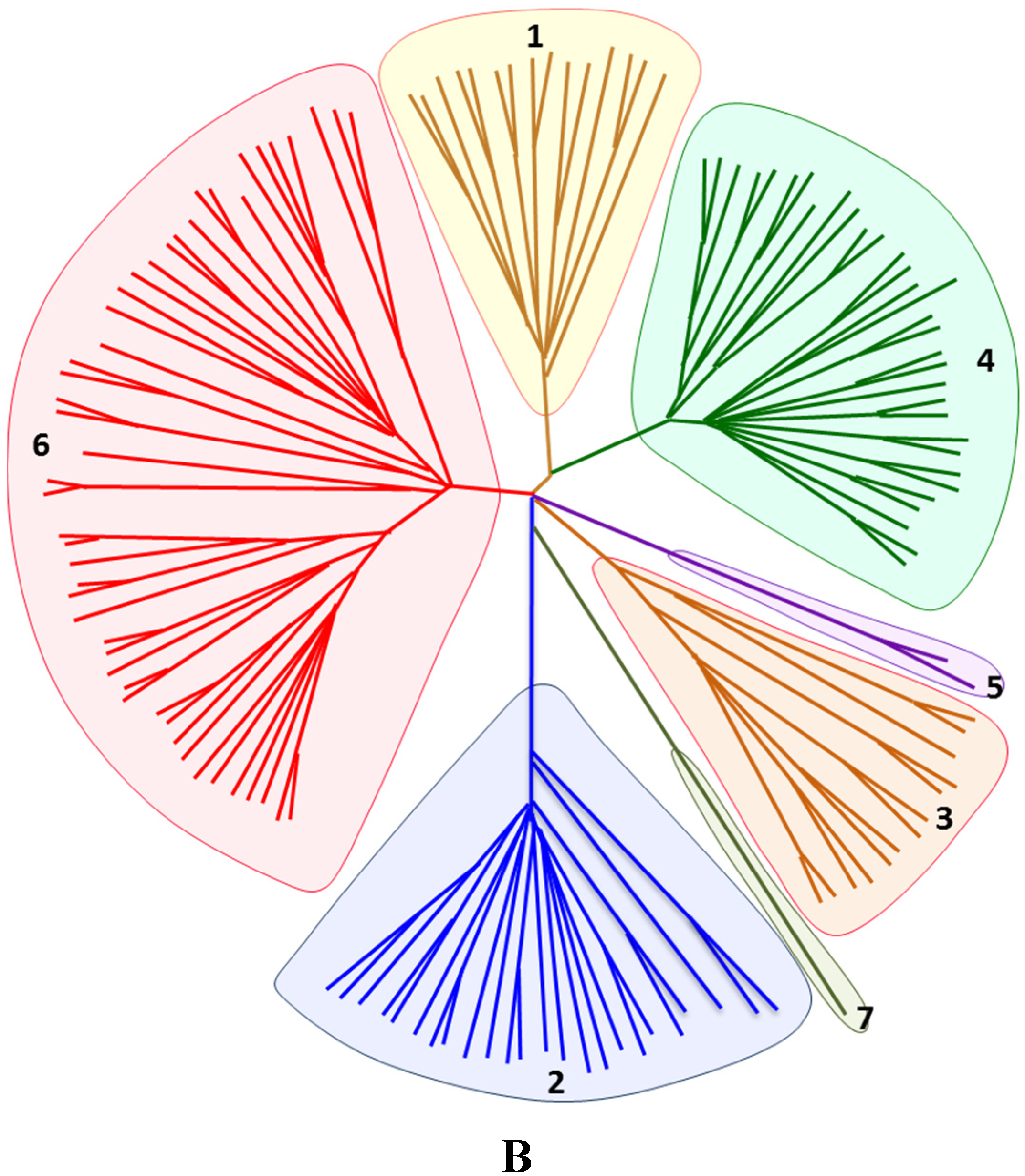
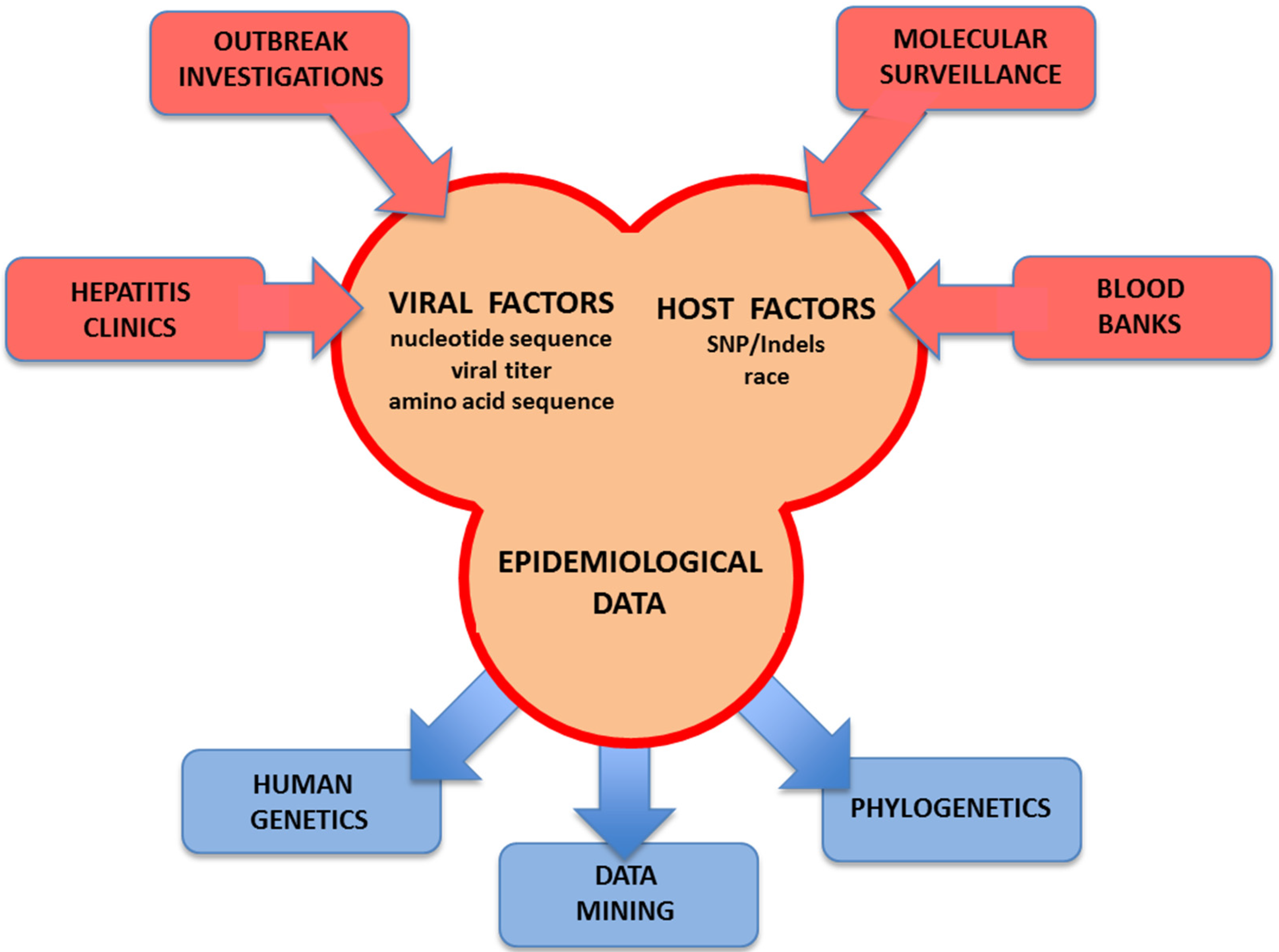
6. Bioinformatics, Phylogenetics and Data Mining
6.1. Bioinformatics
6.2. Phylogenetics
6.3. Databases and Data Mining
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohd Hanafiah, K.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29 (Suppl. 1), 74–81. [Google Scholar] [CrossRef] [PubMed]
- Lauer, G.M.; Walker, B.D. Hepatitis C virus infection. N. Engl. J. Med. 2001, 345, 41–52. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Bacon, B.R. Chronic hepatitis C: An age wave of disease burden. Am. J. Manag. Care 2005, 11 (Suppl. 10), S286–S295. [Google Scholar] [PubMed]
- Stanley, M.L.; Walker, C.; Alter, M.J.; Yi, M. Hepatitis C Virus. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 1, p. 1280. [Google Scholar]
- Chevaliez, S.; Pawlotsky, J.M. HCV Genome and Life Cycle. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Agha, S.; Tanaka, Y.; Saudy, N.; Kurbanov, F.; Abo-Zeid, M.; El-Malky, M.; Khalaf, M.; Ohta, N.; Yoshizawa, H.; Mizokami, M. Reliability of hepatitis C virus core antigen assay for detection of viremia in HCV genotypes 1, 2, 3, and 4 infected blood donors: A collaborative study between Japan, Egypt, and Uzbekistan. J. Med. Virol. 2004, 73, 216–222. [Google Scholar] [CrossRef] [PubMed]
- McOmish, F.; Yap, P.L.; Dow, B.C.; Follett, E.A.; Seed, C.; Keller, A.J.; Cobain, T.J.; Krusius, T.; Kolho, E.; Naukkarinen, R.; et al. Geographical distribution of hepatitis C virus genotypes in blood donors: An international collaborative survey. J. Clin. Microbiol. 1994, 32, 884–892. [Google Scholar]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Yang, P.M.; Sheu, J.C.; Wang, T.H.; Chen, D.S. Genotypes of hepatitis C virus in Taiwan and the progression of liver disease. J. Clin. Gastroenterol. 1995, 21, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 1–15. [Google Scholar] [PubMed]
- Iles, J.C.; Raghwani, J.; Harrison, G.L.; Pepin, J.; Djoko, C.F.; Tamoufe, U.; LeBreton, M.; Schneider, B.S.; Fair, J.N.; Tshala, F.M.; et al. Phylogeography and epidemic history of hepatitis C virus genotype 4 in Africa. Virology 2014, 464–465, 233–243. [Google Scholar] [CrossRef]
- Pybus, O.G.; Barnes, E.; Taggart, R.; Lemey, P.; Markov, P.V.; Rasachak, B.; Syhavong, B.; Phetsouvanah, R.; Sheridan, I.; Humphreys, I.S.; et al. Genetic history of hepatitis C virus in East Asia. J. Virol. 2009, 83, 1071–1082. [Google Scholar] [CrossRef]
- Cruz-Rivera, M.; Carpio-Pedroza, J.C.; Escobar-Gutierrez, A.; Lozano, D.; Vergara-Castaneda, A.; Rivera-Osorio, P.; Martinez-Guarneros, A.; Chacon, C.A.; Fonseca-Coronado, S.; Vaughan, G. Rapid hepatitis C virus divergence among chronically infected individuals. J. Clin. Microbiol. 2013, 51, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Honegger, J.R.; Kim, S.; Price, A.A.; Kohout, J.A.; McKnight, K.L.; Prasad, M.R.; Lemon, S.M.; Grakoui, A.; Walker, C.M. Loss of immune escape mutations during persistent HCV infection in pregnancy enhances replication of vertically transmitted viruses. Nat. Med. 2013, 19, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Martell, M.; Esteban, J.I.; Quer, J.; Genesca, J.; Weiner, A.; Esteban, R.; Guardia, J.; Gomez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229. [Google Scholar] [PubMed]
- Von Hahn, T.; Yoon, J.C.; Alter, H.; Rice, C.M.; Rehermann, B.; Balfe, P.; McKeating, J.A. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 2007, 132, 667–678. [Google Scholar]
- Ralston, R.; Jacobson, I.; Scull, M. The conundrum of relapse in STAT-C therapy: Does HCV play the Red Queen or Rip Van Winkle? Semin. Liver Dis. 2011, 31, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Khudyakov, Y. Molecular surveillance of hepatitis C. Antivir. Ther. 2012, 17, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Gray, E.R.; Kundu, S.; Cooray, S.; Poulsen, A.; Aaby, P.; Breuer, J. Evolution of cocirculating varicella-zoster virus genotypes during a chickenpox outbreak in Guinea-Bissau. J. Virol. 2014, 88, 13936–13946. [Google Scholar] [CrossRef] [PubMed]
- Krugman, S.; Ward, R.; Giles, J.P. The natural history of infectious hepatitis. Am. J. Med. 1962, 32, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purceli, R.H. Hepatitis A: Detection by immune electron microscopy of a viruslike antigen associated with acute illness. Science 1973, 182, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Bayer, M.E.; Blumberg, B.S.; Werner, B. Particles associated with Australia antigen in the sera of patients with leukaemia, Down’s Syndrome and hepatitis. Nature 1968, 218, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, F.B.; Gitnick, G.L.; Aach, R.D.; Szmuness, W.; Mosley, J.W.; Stevens, C.E.; Peters, R.L.; Weiner, J.M.; Werch, J.B.; Lander, J.J. Non-A, non-B hepatitis transmission in chimpanzees: A project of the transfusion-transmitted viruses study group. Intervirology 1978, 10, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, T.; Saito, I.; Katayama, T.; Kikuchi, S.; Tateda, A.; Houghton, M.; Choo, Q.L.; Kuo, G. Detection of antibody against antigen expressed by molecularly cloned hepatitis C virus cDNA: Application to diagnosis and blood screening for posttransfusion hepatitis. Proc. Natl. Acad. Sci. USA 1990, 87, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Takeuchi, K.; Boonmar, S.; Katayama, T.; Choo, Q.L.; Kuo, G.; Weiner, A.J.; Bradley, D.W.; Houghton, M.; Saito, I.; et al. A cDNA fragment of hepatitis C virus isolated from an implicated donor of post-transfusion non-A, non-B hepatitis in Japan. Nucleic Acids Res. 1989, 17, 10367–10372. [Google Scholar] [CrossRef]
- Choo, Q.L.; Richman, K.H.; Han, J.H.; Berger, K.; Lee, C.; Dong, C.; Gallegos, C.; Coit, D.; Medina-Selby, R.; Barr, P.J.; et al. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 2451–2455. [Google Scholar] [CrossRef]
- Kato, N.; Hijikata, M.; Ootsuyama, Y.; Nakagawa, M.; Ohkoshi, S.; Sugimura, T.; Shimotohno, K. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc. Natl. Acad. Sci. USA 1990, 87, 9524–9528. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Hijikata, M.; Ootsuyama, Y.; Nakagawa, M.; Ohkoshi, S.; Shimotohno, K. Sequence diversity of hepatitis C viral genomes. Mol. Biol. Med. 1990, 7, 495–501. [Google Scholar] [PubMed]
- Grakoui, A.; McCourt, D.W.; Wychowski, C.; Feinstone, S.M.; Rice, C.M. Characterization of the hepatitis C virus-encoded serine proteinase: Determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 1993, 67, 2832–2843. [Google Scholar] [PubMed]
- Hijikata, M.; Mizushima, H.; Akagi, T.; Mori, S.; Kakiuchi, N.; Kato, N.; Tanaka, T.; Kimura, K.; Shimotohno, K. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J. Virol. 1993, 67, 4665–4675. [Google Scholar] [PubMed]
- Eckart, M.R.; Selby, M.; Masiarz, F.; Lee, C.; Berger, K.; Crawford, K.; Kuo, C.; Kuo, G.; Houghton, M.; Choo, Q.L. The hepatitis C virus encodes a serine protease involved in processing of the putative nonstructural proteins from the viral polyprotein precursor. Biochem. Biophys. Res. Commun. 1993, 192, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Suzich, J.A.; Tamura, J.K.; Palmer-Hill, F.; Warrener, P.; Grakoui, A.; Rice, C.M.; Feinstone, S.M.; Collett, M.S. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol. 1993, 67, 6152–6158. [Google Scholar] [PubMed]
- Lohmann, V.; Korner, F.; Herian, U.; Bartenschlager, R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 1997, 71, 8416–8428. [Google Scholar] [PubMed]
- Simmonds, P. Variability of hepatitis C virus. Hepatology 1995, 21, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Purcell, R.H.; Miller, R.H. Sequence analysis of the core gene of 14 hepatitis C virus genotypes. Proc. Natl. Acad. Sci. USA 1994, 91, 8239–8243. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.; Myers, G.; Howard, C.; Brettin, T.; Bukh, J.; Gaschen, B.; Gojobori, T.; Maertens, G.; Mizokami, M.; Nainan, O.; et al. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: Proposals for standardization. International Committee on Virus Taxonomy. Arch. Virol. 1998, 143, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; McHutchison, J.G. Initial treatment for chronic hepatitis C: Current therapies and their optimal dosing and duration. Clevel. Clin. J. Med. 2004, 71 (Suppl. 3), S8–S12. [Google Scholar] [CrossRef]
- Perales, C.; Beach, N.M.; Sheldon, J.; Domingo, E. Molecular basis of interferon resistance in hepatitis C virus. Curr. Opin. Virol. 2014, 8, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Lohmann, V.; Rutter, G.; Kurpanek, K.; Bartenschlager, R. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 2001, 75, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient initiation of HCV RNA replication in cell culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Luik, P.; Chew, C.; Aittoniemi, J.; Chang, J.; Wentworth, P., Jr.; Dwek, R.A.; Biggin, P.C.; Venien-Bryan, C.; Zitzmann, N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 12712–12716. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLOS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Luciani, F.; McElroy, K.; Gaudieri, S.; Pham, S.T.; Chopra, A.; Cameron, B.; Maher, L.; Dore, G.J.; White, P.A.; Lloyd, A.R. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLOS Pathog. 2011, 7, e1002243. [Google Scholar] [CrossRef] [PubMed]
- Nasu, A.; Marusawa, H.; Ueda, Y.; Nishijima, N.; Takahashi, K.; Osaki, Y.; Yamashita, Y.; Inokuma, T.; Tamada, T.; Fujiwara, T.; et al. Genetic heterogeneity of hepatitis C virus in association with antiviral therapy determined by ultra-deep sequencing. PLOS ONE 2011, 6, e24907. [Google Scholar] [CrossRef]
- Ninomiya, M.; Ueno, Y.; Funayama, R.; Nagashima, T.; Nishida, Y.; Kondo, Y.; Inoue, J.; Kakazu, E.; Kimura, O.; Nakayama, K.; et al. Use of illumina deep sequencing technology to differentiate hepatitis C virus variants. J. Clin. Microbiol. 2012, 50, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Forbi, J.C.; Purdy, M.A.; Campo, D.S.; Vaughan, G.; Dimitrova, Z.E.; Ganova-Raeva, L.M.; Xia, G.L.; Khudyakov, Y.E. Epidemic history of hepatitis C virus infection in two remote communities in Nigeria, West Africa. J. Gen. Virol. 2012, 93, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Svarovskaia, E.S.; Martin, R.; McHutchison, J.G.; Miller, M.D.; Mo, H. Abundant drug-resistant NS3 mutants detected by deep sequencing in hepatitis C virus-infected patients undergoing NS3 protease inhibitor monotherapy. J. Clin. Microbiol. 2012, 50, 3267–3274. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.; Schelhorn, S.E.; Fitting, D.; Mihm, U.; Susser, S.; Welker, M.W.; Fuller, C.; Daumer, M.; Teuber, G.; Wedemeyer, H.; et al. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J. Virol. 2013, 87, 6172–6181. [Google Scholar] [CrossRef] [PubMed]
- Akuta, N.; Suzuki, F.; Seko, Y.; Kawamura, Y.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Hara, T.; Saitoh, S.; et al. Emergence of telaprevir-resistant variants detected by ultra-deep sequencing after triple therapy in patients infected with HCV genotype 1. J. Med. Virol. 2013, 85, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Trimoulet, P.; Pinson, P.; Papuchon, J.; Foucher, J.; Vergniol, J.; Chermak, F.; Wittkop, L.; Castaing, N.; Merrouche, W.; Reigadas, S.; et al. Dynamic and rapid changes in viral quasispecies by UDPS in chronic hepatitis C patients receiving telaprevir-based therapy. Antivir. Ther. 2013, 18, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Caraballo Cortes, K.; Zagordi, O.; Laskus, T.; Ploski, R.; Bukowska-Osko, I.; Pawelczyk, A.; Berak, H.; Radkowski, M. Ultradeep pyrosequencing of hepatitis C virus hypervariable region 1 in quasispecies analysis. BioMed Res. Int. 2013, 2013, e626083. [Google Scholar]
- Akuta, N.; Suzuki, F.; Fukushima, T.; Kawamura, Y.; Sezaki, H.; Suzuki, Y.; Hosaka, T.; Kobayashi, M.; Hara, T.; Saitoh, S.; et al. Prediction of treatment efficacy and telaprevir-resistant variants after triple therapy in patients infected with hepatitis C virus genotype 1. J. Clin. Microbiol. 2013, 51, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Kirst, M.E.; Li, E.C.; Wang, C.X.; Dong, H.J.; Liu, C.; Fried, M.W.; Nelson, D.R.; Wang, G.P. Deep sequencing analysis of HCV NS3 resistance-associated variants and mutation linkage in liver transplant recipients. PLOS ONE 2013, 8, e69698. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Maekawa, S.; Takano, S.; Komatsu, N.; Tatsumi, A.; Asakawa, Y.; Shindo, K.; Amemiya, F.; Nakayama, Y.; Inoue, T.; et al. Deep-sequencing analysis of the association between the quasispecies nature of the hepatitis C virus core region and disease progression. J. Virol. 2013, 87, 12541–12551. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, B.; Giombini, E.; Zaccaro, P.; Selleri, M.; Rozera, G.; Abbate, I.; Comandini, U.V.; Ippolito, G.; Solmone, M.; Capobianchi, M.R. Extent of HCV NS3 protease variability and resistance-associated mutations assessed by next generation sequencing in HCV monoinfected and HIV/HCV coinfected patients. Virus Res. 2013, 177, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Kanda, T.; Nakamoto, S.; Jiang, X.; Miyamura, T.; Nakatani, S.M.; Ono, S.K.; Takahashi-Nakaguchi, A.; Gonoi, T.; Yokosuka, O. Prevalence of hepatitis C virus subgenotypes 1a and 1b in Japanese patients: Ultra-deep sequencing analysis of HCV NS5B genotype-specific region. PLOS ONE 2013, 8, e73615. [Google Scholar] [CrossRef] [PubMed]
- Campo, D.S.; Skums, P.; Dimitrova, Z.; Vaughan, G.; Forbi, J.C.; Teo, C.G.; Khudyakov, Y.; Lau, D.T. Drug resistance of a viral population and its individual intrahost variants during the first 48 h of therapy. Clin. Pharmacol. Ther. 2014, 95, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, T.; Hughes, J.; Main, J.; McLauchlan, J.; Thursz, M.; Thomson, E. Next generation sequencing sheds light on the natural history of hepatitis C infection in patients that fail treatment. Hepatology 2014, 61, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Zagordi, O.; Bhattacharya, A.; Eriksson, N.; Beerenwinkel, N. ShoRAH: Estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinform. 2011, 12, e119. [Google Scholar] [CrossRef]
- Mancuso, N.; Tork, B.; Skums, P.; Ganova-Raeva, L.; Mandoiu, I.; Zelikovsky, A. Reconstructing viral quasispecies from NGS amplicon reads. Silico Biol. 2011, 11, 237–249. [Google Scholar]
- Skums, P.; Mancuso, N.; Artyomenko, A.; Tork, B.; Mandoiu, I.; Khudyakov, Y.; Zelikovsky, A. Reconstruction of viral population structure from next-generation sequencing data using multicommodity flows. BMC Bioinform. 2013, 14 (Suppl. 9), eS2. [Google Scholar]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nishida, N.; Sugiyama, M.; Kurosaki, M.; Matsuura, K.; Sakamoto, N.; Nakagawa, M.; Korenaga, M.; Hino, K.; Hige, S.; et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009, 41, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Tungol, A.; Rademacher, K.; Schafer, J.A. Formulary management of the protease inhibitors boceprevir and telaprevir for chronic hepatitis C virus. J. Manag. Care Pharm. 2011, 17, 685–694. [Google Scholar] [PubMed]
- Chao, D.; Botwin, G.J.; Morgan, T.R. Update on recently approved treatments for hepatitis C. Curr. Treat. Options Gastroenterol. 2014, 12, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Mangia, A.; Wyles, D.; Rodriguez-Torres, M.; Hassanein, T.; Gordon, S.C.; Schultz, M.; Davis, M.N.; Kayali, Z.; Reddy, K.R.; et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 2013, 368, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H.; Ding, X.; Svarovskaia, E.; Symonds, W.T.; Hindes, R.G.; Berrey, M.M. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Izumi, N.; Kumada, H.; Okanoue, T.; Tsubouchi, H.; Yatsuhashi, H.; Kato, M.; Ki, R.; Komada, Y.; Seto, C.; et al. Simeprevir with peginterferon/ribavirin for treatment-naive hepatitis C genotype 1 patients in Japan: CONCERTO-1, a phase III trial. J. Hepatol. 2014, 61, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. New hepatitis C therapies: The toolbox, strategies, and challenges. Gastroenterology 2014, 146, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.; Folgori, A.; Capone, S.; Swadling, L.; Aston, S.; Kurioka, A.; Meyer, J.; Huddart, R.; Smith, K.; Townsend, R.; et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Trans. Med. 2012, 4, e115ra1. [Google Scholar] [CrossRef]
- Honegger, J.R.; Zhou, Y.; Walker, C.M. Will there be a vaccine to prevent HCV infection? Semin. Liver Dis. 2014, 34, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.L. Commentary: Modelling the epidemiology of hepatitis C and its complications. Int. J. Epidemiol. 2003, 32, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.O. Quasispecies theory in the context of population genetics. BMC Evol. Biol. 2005, 5, e44. [Google Scholar] [CrossRef]
- Gismondi, M.I.; Becker, P.D.; Diaz Carrasco, J.M.; Guzman, C.A.; Campos, R.H.; Preciado, M.V. Evolution of hepatitis C virus hypervariable region 1 in immunocompetent children born to HCV-infected mothers. J. Viral Hepat. 2009, 16, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Gismondi, M.I.; Diaz Carrasco, J.M.; Valva, P.; Becker, P.D.; Guzman, C.A.; Campos, R.H.; Preciado, M.V. Dynamic changes in viral population structure and compartmentalization during chronic hepatitis C virus infection in children. Virology 2013, 447, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.E.; Schaefer, M.K.; Labus, B.J.; Sands, L.; Rowley, P.; Azzam, I.A.; Armour, P.; Khudyakov, Y.E.; Lin, Y.; Xia, G.; et al. Hepatitis C virus infections from unsafe injection practices at an endoscopy clinic in Las Vegas, Nevada, 2007–2008. Clin. Infect. Dis. 2010, 51, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Warner, A.E.; Schaefer, M.K.; Patel, P.R.; Drobeniuc, J.; Xia, G.; Lin, Y.; Khudyakov, Y.; Vonderwahl, C.W.; Miller, L.; Thompson, N.D. Outbreak of hepatitis C virus infection associated with narcotics diversion by an hepatitis C virus-infected surgical technician. Am. J. Infect. Control 2015, 43, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Candelas, F.; Bracho, M.A.; Wrobel, B.; Moya, A. Molecular evolution in court: Analysis of a large hepatitis C virus outbreak from an evolving source. BMC Biol. 2013, 11, e76. [Google Scholar] [CrossRef]
- Escobar-Gutierrez, A.; Vazquez-Pichardo, M.; Cruz-Rivera, M.; Rivera-Osorio, P.; Carpio-Pedroza, J.C.; Ruiz-Pacheco, J.A.; Ruiz-Tovar, K.; Vaughan, G. Identification of hepatitis C virus transmission using a next-generation sequencing approach. J. Clin. Microbiol. 2012, 50, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.C.; Fanning, L.; Wang, X.H.; Netski, D.M.; Kenny-Walsh, E.; Thomas, D.L. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J. Exp. Med. 2005, 201, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Gutierrez, A.; Soudeyns, H.; Larouche, A.; Carpio-Pedroza, J.C.; Martinez-Guarneros, A.; Vazquez-Chacon, C.A.; Fonseca-Coronado, S.; Yamasaki, L.H.; Ruiz-Tovar, K.; Cruz-Rivera, M.Y. Vertical transmission of hepatitis C virus: A tale of multiple outcomes. Infect. Genet. Evol. 2013, 20, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Larouche, A.; Gaetan, G.; El-Bilali, N.; Quesnel-Vallieres, M.; Martin, S.R.; Alvarez, F.; Shoukry, N.H.; Soudeyns, H. Seronegative hepatitis C virus infection in a child infected via mother-to-child transmission. J. Clin. Microbiol. 2012, 50, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Sherrill-Mix, S.A.; Chang, K.M.; Quince, C.; Bushman, F.D. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J. Virol. 2010, 84, 6218–6228. [Google Scholar] [CrossRef] [PubMed]
- Cubero, M.; Gregori, J.; Esteban, J.I.; Garcia-Cehic, D.; Bes, M.; Perales, C.; Domingo, E.; Rodriguez-Frias, F.; Sauleda, S.; Casillas, R.; et al. Identification of host and viral factors involved in a dissimilar resolution of a hepatitis C virus infection. Liver Int. 2014, 34, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Manrubia, S.C.; Escarmis, C.; Domingo, E.; Lazaro, E. High mutation rates, bottlenecks, and robustness of RNA viral quasispecies. Gene 2005, 347, 273–282. [Google Scholar] [CrossRef] [PubMed]
- D’Arienzo, V.; Moreau, A.; D’Alteroche, L.; Gissot, V.; Blanchard, E.; Gaudy-Graffin, C.; Roch, E.; Dubois, F.; Giraudeau, B.; Plantier, J.C.; et al. Sequence and functional analysis of the envelope glycoproteins of hepatitis C virus variants selectively transmitted to a new host. J. Virol. 2013, 87, 13609–13618. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Stoddard, M.B.; Wang, S.; Blair, L.M.; Giorgi, E.E.; Parrish, E.H.; Learn, G.H.; Hraber, P.; Goepfert, P.A.; Saag, M.S.; et al. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLOS Pathog. 2012, 8, e1002880. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Ootsuyama, Y.; Sekiya, H.; Ohkoshi, S.; Nakazawa, T.; Hijikata, M.; Shimotohno, K. Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J. Virol. 1994, 68, 4776–4784. [Google Scholar] [PubMed]
- Van Doorn, L.J.; Quint, W.; Tsiquaye, K.; Voermans, J.; Paelinck, D.; Kos, T.; Maertens, G.; Schellekens, H.; Murray, K. Longitudinal analysis of hepatitis C virus infection and genetic drift of the hypervariable region. J. Infect. Dis. 1994, 169, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Kojima, M.; Okada, S.; Yoshizawa, H.; Iizuka, H.; Tanaka, T.; Muchmore, E.E.; Peterson, D.A.; Ito, Y.; Mishiro, S. Genetic drift of hepatitis C virus during an 8.2-year infection in a chimpanzee: Variability and stability. Virology 1992, 190, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Allain, J.P.; Dong, Y.; Vandamme, A.M.; Moulton, V.; Salemi, M. Evolutionary rate and genetic drift of hepatitis C virus are not correlated with the host immune response: Studies of infected donor-recipient clusters. J. Virol. 2000, 74, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Freitas, I.T.; Zhu, C.; Zheng, W.; Hall, W.W.; Higgins, D.G. Recombination in hepatitis C virus: Identification of four novel naturally occurring inter-subtype recombinants. PLOS ONE 2012, 7, e41997. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Evans, M.J.; von Hahn, T.; Jones, C.T.; Stamataki, Z.; McKeating, J.A.; Lindenbach, B.D.; Rice, C.M. Superinfection exclusion in cells infected with hepatitis C virus. J. Virol. 2007, 81, 3693–3703. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Ott, M.; Greene, W.C. Evasion of superinfection exclusion and elimination of primary viral RNA by an adapted strain of hepatitis C virus. J. Virol. 2013, 87, 13354–13369. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Wissing, S.; Herker, E.; Ott, M.; Greene, W.C. Rapid intracellular competition between hepatitis C viral genomes as a result of mitosis. J. Virol. 2013, 87, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, O.; Norder, H.; Mukomolov, S.; Magnius, L.O. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J. Virol. 2002, 76, 4034–4043. [Google Scholar] [CrossRef] [PubMed]
- Sentandreu, V.; Jimenez-Hernandez, N.; Torres-Puente, M.; Bracho, M.A.; Valero, A.; Gosalbes, M.J.; Ortega, E.; Moya, A.; Gonzalez-Candelas, F. Evidence of recombination in intrapatient populations of hepatitis C virus. PLOS ONE 2008, 3, e3239. [Google Scholar] [CrossRef] [PubMed]
- Colina, R.; Casane, D.; Vasquez, S.; Garcia-Aguirre, L.; Chunga, A.; Romero, H.; Khan, B.; Cristina, J. Evidence of intratypic recombination in natural populations of hepatitis C virus. J. Gen. Virol. 2004, 85, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, O.; Norder, H.; Magnius, L.O. Full-length open reading frame of a recombinant hepatitis C virus strain from St Petersburg: Proposed mechanism for its formation. J. Gen. Virol. 2004, 85, 1853–1857. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Candelas, F.; Lopez-Labrador, F.X.; Bracho, M.A. Recombination in hepatitis C virus. Viruses 2011, 3, 2006–2024. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Campo, D.S.; Dimitrova, Z.E.; Xia, G.L.; Purdy, M.A.; Khudyakov, Y.E. Temporal variations in the hepatitis C virus intrahost population during chronic infection. J. Virol. 2011, 85, 6369–6380. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.R.; Salemi, M.; Klenerman, P.; Pybus, O.G. A new evolutionary model for hepatitis C virus chronic infection. PLOS Pathog. 2012, 8, e1002656. [Google Scholar] [CrossRef] [PubMed]
- Goncalves Rossi, L.M.; Rahal, P. Challenges in molecular epidemiology of hepatitis C virus. J. Clin. Virol. 2014, 60, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.C. Antibody- and genome-based identification of recent HCV infection. Antivir. Ther. 2012, 17, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, A.M.; Pybus, O.G. Viral phylogeny in court: The unusual case of the Valencian anesthetist. BMC Biol. 2013, 11, e83. [Google Scholar] [CrossRef]
- Abecasis, A.B.; Geretti, A.M.; Albert, J.; Power, L.; Weait, M.; Vandamme, A.M. Science in court: The myth of HIV fingerprinting. Lancet Infect. Dis. 2011, 11, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Roque-Afonso, A.M.; Ducoulombier, D.; Di Liberto, G.; Kara, R.; Gigou, M.; Dussaix, E.; Samuel, D.; Feray, C. Compartmentalization of hepatitis C virus genotypes between plasma and peripheral blood mononuclear cells. J. Virol. 2005, 79, 6349–6357. [Google Scholar] [CrossRef] [PubMed]
- Indolfi, G.; Azzari, C.; Resti, M. Hepatitis: Immunoregulation in pregnancy and perinatal transmission of HCV. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Manzoor, S.; Ashraf, J.; Khalid, M.; Tariq, M.; Khaliq, H.M.; Azam, S. Role of viral and host factors in interferon based therapy of hepatitis C virus infection. Virol. J. 2013, 10, 299. [Google Scholar] [CrossRef] [PubMed]
- Villano, S.A.; Vlahov, D.; Nelson, K.E.; Cohn, S.; Thomas, D.L. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology 1999, 29, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Micallef, J.M.; Kaldor, J.M.; Dore, G.J. Spontaneous viral clearance following acute hepatitis C infection: A systematic review of longitudinal studies. J. Viral Hepat. 2006, 13, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ueno, Y. Transmission of hepatitis C virus: Self-limiting hepatitis or chronic hepatitis? World J. Gastroenterol. 2013, 19, 6957–6961. [Google Scholar] [CrossRef] [PubMed]
- Diepolder, H.M.; Zachoval, R.; Hoffmann, R.M.; Wierenga, E.A.; Santantonio, T.; Jung, M.C.; Eichenlaub, D.; Pape, G.R. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 1995, 346, 1006–1007. [Google Scholar] [CrossRef] [PubMed]
- Ulsenheimer, A.; Gerlach, J.T.; Gruener, N.H.; Jung, M.C.; Schirren, C.A.; Schraut, W.; Zachoval, R.; Pape, G.R.; Diepolder, H.M. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology 2003, 37, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Fisicaro, P.; Tola, D.; Orlandini, A.; Sacchelli, L.; Mori, C.; Missale, G.; Ferrari, C. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 2006, 44, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.; Mihalik, K.; Tilton, J.C.; Williams, O.; Merchlinsky, M.; Connors, M.; Feinstone, S.M.; Major, M.E. CD4+ immune escape and subsequent T-cell failure following chimpanzee immunization against hepatitis C virus. Hepatology 2006, 44, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; McGavern, D.B.; Oldstone, M.B. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J. Clin. Investig. 2006, 116, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Bes, M.; Sauleda, S.; Casamitjana, N.; Piron, M.; Campos-Varela, I.; Quer, J.; Cubero, M.; Puig, L.; Guardia, J.; Esteban, J.I. Reversal of nonstructural protein 3-specific CD4(+) T cell dysfunction in patients with persistent hepatitis C virus infection. J. Viral Hepat. 2012, 19, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Thursz, M.; Yallop, R.; Goldin, R.; Trepo, C.; Thomas, H.C. Influence of MHC class II genotype on outcome of infection with hepatitis C virus. Lancet 1999, 354, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Astemborski, J.; Rai, R.M.; Anania, F.A.; Schaeffer, M.; Galai, N.; Nolt, K.; Nelson, K.E.; Strathdee, S.A.; Johnson, L.; et al. The natural history of hepatitis C virus infection: Host, viral, and environmental factors. JAMA 2000, 284, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.; Thomas, D.L.; Goedert, J.J.; Vlahov, D.; Nelson, K.E.; Hilgartner, M.W.; O’Brien, S.J.; Karacki, P.; Marti, D.; Astemborski, J.; et al. Racial differences in HLA class II associations with hepatitis C virus outcomes. J. Infect. Dis. 2001, 184, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, S.I.; Thio, C.L.; Martin, M.P.; Brooks, C.R.; Gao, X.; Astemborski, J.; Cheng, J.; Goedert, J.J.; Vlahov, D.; Hilgartner, M.; et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 2004, 305, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Grody, W.W. Molecular genetic risk screening. Ann. Rev. Med. 2003, 54, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.; van Booven, D.J.; McLeod, H.L. Global pharmacogenetics: Giving the genome to the masses. Pharmacogenomics 2006, 7, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Mosbruger, T.L.; Duggal, P.; Goedert, J.J.; Kirk, G.D.; Hoots, W.K.; Tobler, L.H.; Busch, M.; Peters, M.G.; Rosen, H.R.; Thomas, D.L.; et al. Large-scale candidate gene analysis of spontaneous clearance of hepatitis C virus. J. Infect. Dis. 2010, 201, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.; Goedert, J.J.; Mosbruger, T.; Vlahov, D.; Strathdee, S.A.; O’Brien, S.J.; Astemborski, J.; Thomas, D.L. An analysis of tumor necrosis factor alpha gene polymorphisms and haplotypes with natural clearance of hepatitis C virus infection. Genes Immun. 2004, 5, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Lio, D.; Caruso, C.; di Stefano, R.; Colonna Romano, G.; Ferraro, D.; Scola, L.; Crivello, A.; Licata, A.; Valenza, L.M.; Candore, G.; et al. IL-10 and TNF-alpha polymorphisms and the recovery from HCV infection. Hum. Immunol. 2003, 64, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Vassalli, P. The pathophysiology of tumor necrosis factors. Ann. Rev. Immunol. 1992, 10, 411–452. [Google Scholar] [CrossRef]
- Koziel, M.J.; Dudley, D.; Afdhal, N.; Grakoui, A.; Rice, C.M.; Choo, Q.L.; Houghton, M.; Walker, B.D. HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J. Clin. Investig. 1995, 96, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Saito, T.; Yoshimura, M.; Yixuan, S.; Baba, M.; Ji, G.; Muramatsu, M.; Kawata, S. Association of transforming growth factor-beta 1 functional polymorphisms with natural clearance of hepatitis C virus. J. Infect. Dis. 2006, 193, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, H.; Borg, B.B.; Su, X.; Rhodes, S.L.; Yang, K.; Tong, X.; Tang, G.; Howell, C.D.; Rosen, H.R.; et al. A functional SNP of interferon-gamma gene is important for interferon-alpha-induced and spontaneous recovery from hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2007, 104, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Rook, A.H.; Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Sporn, M.B.; Burlington, D.B.; Lane, H.C.; Fauci, A.S. Effects of transforming growth factor beta on the functions of natural killer cells: Depressed cytolytic activity and blunting of interferon responsiveness. J. Immunol. 1986, 136, 3916–3920. [Google Scholar] [PubMed]
- Okumoto, K.; Hattori, E.; Tamura, K.; Kiso, S.; Watanabe, H.; Saito, K.; Saito, T.; Togashi, H.; Kawata, S. Possible contribution of circulating transforming growth factor-beta1 to immunity and prognosis in unresectable hepatocellular carcinoma. Liver Int. 2004, 24, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Frese, M.; Schwarzle, V.; Barth, K.; Krieger, N.; Lohmann, V.; Mihm, S.; Haller, O.; Bartenschlager, R. Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology 2002, 35, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Woollard, D.J.; Grakoui, A.; Shoukry, N.H.; Murthy, K.K.; Campbell, K.J.; Walker, C.M. Characterization of HCV-specific Patr class II restricted CD4+ T cell responses in an acutely infected chimpanzee. Hepatology 2003, 38, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Piazzolla, G.; Tortorella, C.; Schiraldi, O.; Antonaci, S. Relationship between interferon-gamma, interleukin-10, and interleukin-12 production in chronic hepatitis C and in vitro effects of interferon-alpha. J. Clin. Immunol. 2000, 20, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Edwards-Smith, C.J.; Jonsson, J.R.; Purdie, D.M.; Bansal, A.; Shorthouse, C.; Powell, E.E. Interleukin-10 promoter polymorphism predicts initial response of chronic hepatitis C to interferon alfa. Hepatology 1999, 30, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Lauwers, G.Y.; Lau, J.Y.; Davis, G.L. Interleukin 10 treatment reduces fibrosis in patients with chronic hepatitis C: A pilot trial of interferon nonresponders. Gastroenterology 2000, 118, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Matsui, A.; Inao, M.; Nagoshi, S.; Nagano, M.; Ito, N.; Egashira, T.; Hashimoto, M.; Mishiro, S.; Mochida, S.; et al. SNPs in the promoter region of the osteopontin gene as a marker predicting the efficacy of interferon-based therapies in patients with chronic hepatitis C. J. Gastroenterol. 2005, 40, 381–388. [Google Scholar] [CrossRef] [PubMed]
- McKee, M.D.; Nanci, A. Osteopontin: An interfacial extracellular matrix protein in mineralized tissues. Connect. Tissue Res. 1996, 35, 197–205. [Google Scholar] [CrossRef] [PubMed]
- El Awady, M.K.; Anany, M.A.; Esmat, G.; Zayed, N.; Tabll, A.A.; Helmy, A.; el Zayady, A.R.; Abdalla, M.S.; Sharada, H.M.; el Raziky, M.; et al. Single nucleotide polymorphism at exon 7 splice acceptor site of OAS1 gene determines response of hepatitis C virus patients to interferon therapy. J. Gastroenterol. Hepatol. 2011, 26, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Hamano, E.; Hijikata, M.; Itoyama, S.; Quy, T.; Phi, N.C.; Long, H.T.; Ha, L.D.; Ban, V.V.; Matsushita, I.; Yanai, H.; et al. Polymorphisms of interferon-inducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem. Biophys. Res. Commun. 2005, 329, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Alves Pedroso, M.L.; Boldt, A.B.; Pereira-Ferrari, L.; Steffensen, R.; Strauss, E.; Jensenius, J.C.; Ioshii, S.O.; Messias-Reason, I. Mannan-binding lectin MBL2 gene polymorphism in chronic hepatitis C: Association with the severity of liver fibrosis and response to interferon therapy. Clin. Exp. Immunol. 2008, 152, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; McKenzie, I.F. Role of the mannose receptor in the immune response. Curr. Mol. Med. 2001, 1, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.H.; Dellgren, C.; Hamming, O.J.; Vends, S.; Paludan, S.R.; Hartmann, R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J. Biol. Chem. 2009, 284, 20869–20875. [Google Scholar] [CrossRef] [PubMed]
- Iversen, M.B.; Ank, N.; Melchjorsen, J.; Paludan, S.R. Expression of type III interferon (IFN) in the vaginal mucosa is mediated primarily by dendritic cells and displays stronger dependence on NF-kappaB than type I IFNs. J. Virol. 2010, 84, 4579–4586. [Google Scholar] [CrossRef] [PubMed]
- Marcello, T.; Grakoui, A.; Barba-Spaeth, G.; Machlin, E.S.; Kotenko, S.V.; MacDonald, M.R.; Rice, C.M. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006, 131, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Megjugorac, N.J.; Gallagher, G.E.; Gallagher, G. Modulation of human plasmacytoid DC function by IFN-lambda1 (IL-29). J. Leukoc. Biol. 2009, 86, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Mennechet, F.J.; Uze, G. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood 2006, 107, 4417–4423. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, K.; Witte, E.; Proesch, S.; Schulze-Tanzil, G.; Nasilowska, K.; Thilo, J.; Asadullah, K.; Sterry, W.; Volk, H.D.; et al. Maturing dendritic cells are an important source of IL-29 and IL-20 that may cooperatively increase the innate immunity of keratinocytes. J. Leukoc. Biol. 2008, 83, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Preciado, M.V.; Valva, P.; Escobar-Gutierrez, A.; Rahal, P.; Ruiz-Tovar, K.; Yamasaki, L.; Vazquez-Chacon, C.; Martinez-Guarneros, A.; Carpio-Pedroza, J.C.; Fonseca-Coronado, S.; et al. Hepatitis C virus molecular evolution: Transmission, disease progression and antiviral therapy. World J. Gastroenterol. 2014, 20, 15992–16013. [Google Scholar] [PubMed]
- Cruz-Rivera, M.; Forbi, J.C.; Yamasaki, L.H.; Vazquez-Chacon, C.A.; Martinez-Guarneros, A.; Carpio-Pedroza, J.C.; Escobar-Gutierrez, A.; Ruiz-Tovar, K.; Fonseca-Coronado, S.; Vaughan, G. Molecular epidemiology of viral diseases in the era of next generation sequencing. J. Clin. Virol. 2013, 57, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Lauck, M.; Alvarado-Mora, M.V.; Becker, E.A.; Bhattacharya, D.; Striker, R.; Hughes, A.L.; Carrilho, F.J.; O’Connor, D.H.; Pinho, J.R. Analysis of hepatitis C virus intrahost diversity across the coding region by ultradeep pyrosequencing. J. Virol. 2012, 86, 3952–3960. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Gunthard, H.F.; Roth, V.; Metzner, K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 2012, 3, e329. [Google Scholar] [CrossRef]
- Ganova-Raeva, L.M.; Dimitrova, Z.E.; Campo, D.S.; Khudyakov, Y. Application of mass spectrometry to molecular surveillance of hepatitis B and C viral infections. Antivir. Ther. 2012, 17, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Ganova-Raeva, L.M.; Dimitrova, Z.E.; Campo, D.S.; Lin, Y.; Ramachandran, S.; Xia, G.L.; Honisch, C.; Cantor, C.R.; Khudyakov, Y.E. Detection of hepatitis C virus transmission by use of DNA mass spectrometry. J. Infect. Dis. 2013, 207, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Coronado, S.; Escobar-Gutierrez, A.; Ruiz-Tovar, K.; Cruz-Rivera, M.Y.; Rivera-Osorio, P.; Vazquez-Pichardo, M.; Carpio-Pedroza, J.C.; Ruiz-Pacheco, J.A.; Cazares, F.; Vaughan, G. Specific detection of naturally occurring hepatitis C virus mutants with resistance to telaprevir and boceprevir (protease inhibitors) among treatment-naive infected individuals. J. Clin. Microbiol. 2012, 50, 281–287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fonseca-Coronado, S.; Vaughan, G.; Cruz-Rivera, M.Y.; Carpio-Pedroza, J.C.; Ruiz-Tovar, K.; Ruiz-Pacheco, J.A.; Escobar-Gutierrez, A. Interleukin-28B genotyping by melt-mismatch amplification mutation assay PCR analysis using single nucleotide polymorphisms rs12979860 and rs8099917, a useful tool for prediction of therapy response in hepatitis C patients. J. Clin. Microbiol. 2011, 49, 2706–2710. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Kurosaki, M.; Tanaka, Y.; Marumo, F.; Sato, C. Fluctuation of hepatitis C virus quasispecies in persistent infection and interferon treatment revealed by single-strand conformation polymorphism analysis. J. Gen. Virol. 1994, 75, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Enomoto, N.; Marumo, F.; Sato, C. Evolution and selection of hepatitis C virus variants in patients with chronic hepatitis C. Virology 1994, 205, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Moribe, T.; Hayashi, N.; Kanazawa, Y.; Mita, E.; Fusamoto, H.; Negi, M.; Kaneshige, T.; Igimi, H.; Kamada, T.; Uchida, K. Hepatitis C viral complexity detected by single-strand conformation polymorphism and response to interferon therapy. Gastroenterology 1995, 108, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K. PCR-SSCP: A simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991, 1, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Vera-Otarola, J.; Barria, M.I.; Leon, U.; Carvallo, P.; Soza, A.; Lopez-Lastra, M. Is single-strand conformation polymorphism analysis of the full 5' untranslated region an adequate approach to study hepatitis C virus quasispecies distribution? J. Virol. 2009, 83, 9018–9021. [Google Scholar] [CrossRef] [PubMed]
- Laskus, T.; Wang, L.F.; Rakela, J.; Vargas, H.; Pinna, A.D.; Tsamandas, A.C.; Demetris, A.J.; Fung, J. Dynamic behavior of hepatitis C virus in chronically infected patients receiving liver graft from infected donors. Virology 1996, 220, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kukita, Y.; Tahira, T.; Sommer, S.S.; Hayashi, K. SSCP analysis of long DNA fragments in low pH gel. Hum. Mutat. 1997, 10, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.L.; Gordon, C.J. Tracking changes in HIV-1 envelope quasispecies using DNA heteroduplex analysis. Methods 1997, 12, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Polyak, S.J.; Day, T.D.; Gretch, D.R. Characterization of simple and complex hepatitis C virus quasispecies by heteroduplex gel shift analysis: Correlation with nucleotide sequencing. J. Gen. Virol. 1995, 76, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.A.; Spencer, M.; Matsumura, S.; Nisbet, R.E.; Howe, C.J. How many clones need to be sequenced from a single forensic or ancient DNA sample in order to determine a reliable consensus sequence? Nucleic Acids Res. 2005, 33, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, G.W.; Laskus, T.; Vargas, H.E. Hepatitis C virus quasispecies: Misunderstood and mistreated? Liver Transplant. 2003, 9, 1048–1052. [Google Scholar] [CrossRef]
- Stein, C.A.; Levantis, P.; Oxford, J.S. Sequence analysis of proviral HIV RT amplified directly by a semi-quantitative technique from AZT treated patients. J. Med. Virol. 1994, 44, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Ison, M.G.; Llata, E.; Conover, C.S.; Friedewald, J.J.; Gerber, S.I.; Grigoryan, A.; Heneine, W.; Millis, J.M.; Simon, D.M.; Teo, C.G.; et al. Transmission of human immunodeficiency virus and hepatitis C virus from an organ donor to four transplant recipients. Am. J. Transplant. 2011, 11, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Zhai, X.; Thai, H.; Campo, D.S.; Xia, G.; Ganova-Raeva, L.M.; Drobeniuc, J.; Khudyakov, Y.E. Evaluation of intra-host variants of the entire hepatitis B virus genome. PLOS ONE 2011, 6, e25232. [Google Scholar] [CrossRef] [PubMed]
- Leushner, J.; Chiu, N.H. Automated mass spectrometry: A revolutionary technology for clinical diagnostics. Mol. Diagn. 2000, 5, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, Z.; Campo, D.S.; Ramachandran, S.; Vaughan, G.; Ganova-Raeva, L.; Lin, Y.; Forbi, J.C.; Xia, G.; Skums, P.; Pearlman, B.; et al. Evaluation of viral heterogeneity using next-generation sequencing, end-point limiting-dilution and mass spectrometry. Silico biol. 2011, 11, 183–192. [Google Scholar]
- Bocker, S. SNP and mutation discovery using base-specific cleavage and MALDI-TOF mass spectrometry. Bioinformatics 2003, 19 (Suppl. 1), i44–i53. [Google Scholar] [CrossRef] [PubMed]
- Yea, C.; Bukh, J.; Ayers, M.; Roberts, E.; Krajden, M.; Tellier, R. Monitoring of hepatitis C virus quasispecies in chronic infection by matrix-assisted laser desorption ionization-time of flight mass spectrometry mutation detection. J. Clin. Microbiol. 2007, 45, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, R.S.; Li, F.; Sampath, R.; Yasuda, I.J.; Carolan, H.E.; Wolfe, J.M.; Brown, M.K.; Alexander, R.C.; Williams-Hill, D.M.; Martin, W.B. Rapid detection and differentiation of human noroviruses using RT-PCR coupled to electrospray ionization mass spectrometry. Food Microbiol. 2014, 44, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Dunne, E.M.; Ong, E.K.; Moser, R.J.; Siba, P.M.; Phuanukoonnon, S.; Greenhill, A.R.; Robins-Browne, R.M.; Mulholland, E.K.; Satzke, C. Multilocus sequence typing of Streptococcus pneumoniae by use of mass spectrometry. J. Clin. Microbiol. 2011, 49, 3756–3760. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Eloit, M. The diagnosis of infectious diseases by whole genome next generation sequencing: A new era is opening. Front. Cell. Infect. Microbiol. 2014, 4, e25. [Google Scholar] [CrossRef]
- Tombacz, D.; Sharon, D.; Olah, P.; Csabai, Z.; Snyder, M.; Boldogkoi, Z. Strain Kaplan of Pseudorabies Virus Genome Sequenced by PacBio Single-Molecule Real-Time Sequencing Technology. Genome Announc. 2014, 2, 8–14. [Google Scholar]
- Giallonardo, F.D.; Topfer, A.; Rey, M.; Prabhakaran, S.; Duport, Y.; Leemann, C.; Schmutz, S.; Campbell, N.K.; Joos, B.; Lecca, M.R.; et al. Full-length haplotype reconstruction to infer the structure of heterogeneous virus populations. Nucleic Acids Res. 2014, 42, e115. [Google Scholar] [CrossRef] [PubMed]
- Brinzevich, D.; Young, G.R.; Sebra, R.; Ayllon, J.; Maio, S.M.; Deikus, G.; Chen, B.K.; Fernandez-Sesma, A.; Simon, V.; Mulder, L.C. HIV-1 interacts with human endogenous retrovirus K (HML-2) envelopes derived from human primary lymphocytes. J. Virol. 2014, 88, 6213–6223. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Frias, F.; Tabernero, D.; Quer, J.; Esteban, J.I.; Ortega, I.; Domingo, E.; Cubero, M.; Camos, S.; Ferrer-Costa, C.; Sanchez, A.; et al. Ultra-deep pyrosequencing detects conserved genomic sites and quantifies linkage of drug-resistant amino acid changes in the hepatitis B virus genome. PLOS ONE 2012, 7, e37874. [Google Scholar] [CrossRef] [PubMed]
- Aw, T.G.; Howe, A.; Rose, J.B. Metagenomic approaches for direct and cell culture evaluation of the virological quality of wastewater. J. Virol. Methods 2014, 210C, 15–21. [Google Scholar] [CrossRef]
- Idris, A.; Al-Saleh, M.; Piatek, M.J.; Al-Shahwan, I.; Ali, S.; Brown, J.K. Viral metagenomics: Analysis of begomoviruses by illumina high-throughput sequencing. Viruses 2014, 6, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Zhuang, Q.Y.; Wang, K.C.; Liu, S.; Shao, J.Z.; Jiang, W.M.; Hou, G.Y.; Li, J.P.; Yu, J.M.; Li, Y.P.; et al. Identification and survey of a novel avian coronavirus in ducks. PLOS ONE 2013, 8, e72918. [Google Scholar] [CrossRef] [PubMed]
- Callegaro, A.; Di Filippo, E.; Astuti, N.; Ortega, P.A.; Rizzi, M.; Farina, C.; Valenti, D.; Maggiolo, F. Early clinical response and presence of viral resistant minority variants: A proof of concept study. J. Int. AIDS Soc. 2014, 17, e19759. [Google Scholar] [CrossRef]
- Gregori, J.; Esteban, J.I.; Cubero, M.; Garcia-Cehic, D.; Perales, C.; Casillas, R.; Alvarez-Tejado, M.; Rodriguez-Frias, F.; Guardia, J.; Domingo, E.; et al. Ultra-deep pyrosequencing (UDPS) data treatment to study amplicon HCV minor variants. PLOS ONE 2013, 8, e83361. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Salicru, M.; Domingo, E.; Sanchez, A.; Esteban, J.I.; Rodriguez-Frias, F.; Quer, J. Inference with viral quasispecies diversity indices: Clonal and NGS approaches. Bioinformatics 2014, 30, 1104–1111. [Google Scholar] [CrossRef]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Sodergren, E.; Weinstock, G.M.; Storch, G.A. Sequence analysis of the human virome in febrile and afebrile children. PLOS ONE 2012, 7, e27735. [Google Scholar] [CrossRef] [PubMed]
- Quer, J.; Gregori, J.; Rodriguez-Frias, F.; Buti, M.; Madejon, A.; Perez-del-Pulgar, S.; Garcia-Cehic, D.; Casillas, R.; Blasi, M.; Homs, M.; et al. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J. Clin. Microbiol. 2015, 53, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Marz, M.; Beerenwinkel, N.; Drosten, C.; Fricke, M.; Frishman, D.; Hofacker, I.L.; Hoffmann, D.; Middendorf, M.; Rattei, T.; Stadler, P.F.; et al. Challenges in RNA virus bioinformatics. Bioinformatics 2014, 30, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Hraber, P.; Thurmond, J.; Yusim, K. The hepatitis C sequence database in Los Alamos. Nucleic Acids Res. 2008, 36, D512–D516. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Yusim, K.; Boykin, L.; Richardson, R. The Los Alamos hepatitis C sequence database. Bioinformatics 2005, 21, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef]
- Salvatierra, K.; Fareleski, S.; Forcada, A.; Lopez-Labrador, F.X. Hepatitis C virus resistance to new specifically-targeted antiviral therapy: A public health perspective. World J. Virol. 2013, 2, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Scornavacca, C. A survey of combinatorial methods for phylogenetic networks. Genome Biol. Evol. 2011, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Scornavacca, C.; Zickmann, F.; Huson, D.H. Tanglegrams for rooted phylogenetic trees and networks. Bioinformatics 2011, 27, i248–i256. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, B.; Torres-Puente, M.; Jimenez, N.; Bracho, M.A.; Garcia-Robles, I.; Moya, A.; Gonzalez-Candelas, F. Analysis of the overdispersed clock in the short-term evolution of hepatitis C virus: Using the E1/E2 gene sequences to infer infection dates in a single source outbreak. Mol. Biol. Evol. 2006, 23, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, T.; Pybus, O.G.; Rambaut, A.; Salemi, M.; Cassol, S.; Ciccozzi, M.; Rezza, G.; Gattinara, G.C.; D’Arrigo, R.; Amicosante, M.; et al. Molecular epidemiology: HIV-1 and HCV sequences from Libyan outbreak. Nature 2006, 444, 836–837. [Google Scholar] [CrossRef] [PubMed]
- Niesters, H.G.; Rossen, J.W.; van der Avoort, H.; Baas, D.; Benschop, K.; Claas, E.C.; Kroneman, A.; van Maarseveen, N.; Pas, S.; van Pelt, W.; et al. Laboratory-based surveillance in the molecular era: The TYPENED model, a joint data-sharing platform for clinical and public health laboratories. Euro Surveill. 2013, 18, 20387. [Google Scholar] [PubMed]
- Squires, R.B.; Noronha, J.; Hunt, V.; Garcia-Sastre, A.; Macken, C.; Baumgarth, N.; Suarez, D.; Pickett, B.E.; Zhang, Y.; Larsen, C.N.; et al. Influenza research database: An integrated bioinformatics resource for influenza research and surveillance. Influenza Respir. Viruses 2012, 6, 404–416. [Google Scholar] [CrossRef]
- Gaschen, B.; Kuiken, C.; Korber, B.; Foley, B. Retrieval and on-the-fly alignment of sequence fragments from the HIV database. Bioinformatics 2001, 17, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus pathogen database and analysis resource (ViPR): A comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef] [PubMed]
- Janies, D.A.; Voronkin, I.O.; Das, M.; Hardman, J.; Treseder, T.W.; Studer, J. Genome informatics of influenza A: From data sharing to shared analytical capabilities. Anim. Health Res. Rev. 2010, 11, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Heisey-Grove, D.M.; Church, D.R.; Haney, G.A.; Demaria, A., Jr. Enhancing surveillance for hepatitis C through public health informatics. Public Health Rep. 2011, 126, 13–18. [Google Scholar] [PubMed]
- Glasgow, J.; Jurisica, I.I.; Ng, R. Data mining and knowledge discovery in molecular databases. Pac. Symp. Biocomput. 2000, 5, 365–366. [Google Scholar]
- Shang, G.; Richardson, A.; Gahan, M.E.; Easteal, S.; Ohms, S.; Lidbury, B.A. Predicting the presence of hepatitis B virus surface antigen in Chinese patients by pathology data mining. J. Med. Virol. 2013, 85, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Zayed, N.; Awad, A.B.; El-Akel, W.; Doss, W.; Awad, T.; Radwan, A.; Mabrouk, M. The assessment of data mining for the prediction of therapeutic outcome in 3719 Egyptian patients with chronic hepatitis C. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Hiramatsu, N.; Sakamoto, M.; Suzuki, Y.; Iwasaki, M.; Tamori, A.; Matsuura, K.; Kakinuma, S.; Sugauchi, F.; Sakamoto, N.; et al. Data mining model using simple and readily available factors could identify patients at high risk for hepatocellular carcinoma in chronic hepatitis C. J. Hepatol. 2012, 56, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Lara, J.; Lopez-Labrador, F.; Gonzalez-Candelas, F.; Berenguer, M.; Khudyakov, Y.E. Computational models of liver fibrosis progression for hepatitis C virus chronic infection. BMC Bioinform. 2014, 15 (Suppl. 8), eS5. [Google Scholar]
- Lara, J.; Tavis, J.E.; Donlin, M.J.; Lee, W.M.; Yuan, H.J.; Pearlman, B.L.; Vaughan, G.; Forbi, J.C.; Xia, G.L.; Khudyakov, Y.E. Coordinated evolution among hepatitis C virus genomic sites is coupled to host factors and resistance to interferon. Silico Biol. 2011, 11, 213–224. [Google Scholar]
- Lara, J.; Xia, G.; Purdy, M.; Khudyakov, Y. Coevolution of the hepatitis C virus polyprotein sites in patients on combined pegylated interferon and ribavirin therapy. J. Virol. 2011, 85, 3649–3663. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Sakamoto, N.; Iwasaki, M.; Sakamoto, M.; Suzuki, Y.; Hiramatsu, N.; Sugauchi, F.; Yatsuhashi, H.; Izumi, N. Pretreatment prediction of response to peginterferon plus ribavirin therapy in genotype 1 chronic hepatitis C using data mining analysis. J. Gastroenterol. 2011, 46, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Kurosaki, M.; Sakamoto, N.; Iwasaki, M.; Sakamoto, M.; Suzuki, Y.; Sugauchi, F.; Tamori, A.; Kakinnuma, S.; Matsuura, K.; et al. Pretreatment prediction of anemia progression by pegylated interferon alpha-2b plus ribavirin combination therapy in chronic hepatitis C infection: Decision-tree analysis. J. Gastroenterol. 2011, 46, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Sakamoto, N.; Iwasaki, M.; Sakamoto, M.; Suzuki, Y.; Hiramatsu, N.; Sugauchi, F.; Tamori, A.; Nakagawa, M.; Izumi, N. Sequences in the interferon sensitivity-determining region and core region of hepatitis C virus impact pretreatment prediction of response to PEG-interferon plus ribavirin: Data mining analysis. J. Med. Virol. 2011, 83, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.C.; Astrakhantseva, I.V.; Fields, H.A.; Kamili, S. Distinguishing acute from chronic hepatitis C virus (HCV) infection based on antibody reactivities to specific HCV structural and nonstructural proteins. J. Clin. Microbiol. 2011, 49, 54–57. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, L.M.G.; Escobar-Gutierrez, A.; Rahal, P. Advanced Molecular Surveillance of Hepatitis C Virus. Viruses 2015, 7, 1153-1188. https://doi.org/10.3390/v7031153
Rossi LMG, Escobar-Gutierrez A, Rahal P. Advanced Molecular Surveillance of Hepatitis C Virus. Viruses. 2015; 7(3):1153-1188. https://doi.org/10.3390/v7031153
Chicago/Turabian StyleRossi, Livia Maria Gonçalves, Alejandro Escobar-Gutierrez, and Paula Rahal. 2015. "Advanced Molecular Surveillance of Hepatitis C Virus" Viruses 7, no. 3: 1153-1188. https://doi.org/10.3390/v7031153
APA StyleRossi, L. M. G., Escobar-Gutierrez, A., & Rahal, P. (2015). Advanced Molecular Surveillance of Hepatitis C Virus. Viruses, 7(3), 1153-1188. https://doi.org/10.3390/v7031153