
Evaluation of Virus Inactivation by Formaldehyde to Enhance Biosafety of Diagnostic Electron Microscopy

Abstract
:1. Introduction
2. Experimental Section
2.1. Virus Stocks and Cell Culture Systems
2.2. Virus Propagation and Harvesting
2.2.1. Vaccinia Virus
2.2.2. Human Adenovirus
2.2.3. Murine Norovirus
2.3. Inactivation of Virus Suspensions

{kind=link}
| Temperature (°C) | Incubation Time (h) | Virus Titer (PFU/mL) | Virus Titer after Inactivation (PFU/mL) | Reduction at log10 Scale |
|---|---|---|---|---|
| 4 | 2 | 1.2 × 108 | 1.3 × 103 | 4.9 |
| 4 | 2 | 1.2 × 108 | 1.8 × 103 | 4.7 |
| 4 | 48 | 1.2 × 108 | 2.6 × 103 | 4.5 |
| 4 | 48 | 1.2 × 108 | 2.1 × 103 | 4.6 |
| 4 | 168 | 1.2 × 108 | 1.4 × 103 | 4.9 |
| 4 | 168 | 1.2 × 108 | 9.8 × 102 | 5.2 |
| 25 | 2 | 1.6 × 108 | no plaques | complete |
| 25 | 2 | 1.6 × 108 | no plaques | complete |
| 25 (*) | 3 | 1.3 × 109 | 1.1 × 101 | 8.2 |
| 25 (*) | 3 | 1.3 × 109 | no plaques | complete |
| 25 | 4 | 1.4 × 108 | 2.5 × 102 (***) | 5.6 |
| 25 | 4 | 1.4 × 108 | no plaques | complete |
| 25 | 4 | 1.4 × 108 | no plaques | complete |
| 25 | 4 | 1.4 × 108 | no plaques | complete |
| 25 | 4 | 1.6 × 108 | no plaques | complete |
| 25 | 4 | 1.6 × 108 | no plaques | complete |
| 25 | 6 | 1.4 × 108 | 2.5 × 102 (***) | 5.6 |
| 25 | 6 | 1.4 × 108 | no plaques | complete |
| 25 | 6 | 1.4 × 108 | no plaques | complete |
| 25 | 6 | 1.4 × 108 | no plaques | complete |
| 25 | 6 | 1.3 × 109 | no plaques | complete |
| 25 | 6 | 1.3 × 109 | no plaques | complete |
| 25 | 24 | 1.3 × 109 | no plaques | complete |
| 25 | 24 | 1.3 × 109 | no plaques | complete |
| 25/37 | 0.5/0.5 | 1.6 × 108 | no plaques | complete |
| 25/37 | 0.5/0.5 | 1.6 × 108 | no plaques | complete |
| 25/60 | 1/2 | 1.3 × 109 | no plaques | complete |
| 25/60 | 1/2 | 1.3 × 109 | no plaques | complete |
| 37 | 0.5 | 1.4 × 108 | no plaques | complete |
| 37 | 0.5 | 1.4 × 108 | no plaques | complete |
| 37 | 1 | 1.6 × 108 | no plaques | complete |
| 37 | 1 | 1.6 × 108 | no plaques | complete |
| 37 (**) | 0.5 | 1.4 × 108 | 1.5 × 103 (***) | 4.9 |
| 37 (**) | 0.5 | 1.4 × 108 | no plaques | complete |
| 37 (**) | 0.5 | 1.4 × 108 | no plaques | complete |
| 37 (**) | 0.5 | 1.4 × 108 | no plaques | complete |
| Treatment | Temperature (°C) | Incubation Time (h) | Virus Titer (log10 TCID50/mL) |
|---|---|---|---|
| - | 4 | 1 | 8.9 |
| 2% FA | 4 | 1 | no CPE |
| 2% FA | 4 | 1 | no CPE |
| - | 25 | 1 | 8.5 |
| 2% FA | 25 | 1 | no CPE |
| 2% FA | 25 | 1 | no CPE |
| - | 25/37 | 0.5/0.5 | 8.9 |
| 2% FA | 25/37 | 0.5/0.5 | no CPE |
| 2% FA | 25/37 | 0.5/0.5 | no CPE |
| Treatment | Temperature (°C) | Incubation Time (h) | Virus Titer (log10 TCID50/mL) |
|---|---|---|---|
| - | 4 | 1 | 7.2 |
| 2% FA | 4 | 1 | 4.4 |
| 2% FA | 4 | 1 | 4 |
| - | 25 | 1 | 7.6 |
| 2% FA | 25 | 1 | no CPE |
| 2% FA | 25 | 1 | no CPE |
| - | 25/37 | 0.5/0.5 | 7.4 |
| 2% FA | 25/37 | 0.5/0.5 | no CPE |
| 2% FA | 25/37 | 0.5/0.5 | no CPE |
| Temperature (°C) | Incubation Time (h) | Number of Tests Showing Plaques after Treatment (n = 10) |
|---|---|---|
| 4 | 1 | 1 |
| 25° | 24 | 0 |
| 25/37° | 0.5/0.5 | 0 |
2.4. Determination of Virus Infectivity
2.4.1. Plaque Assays of Vaccinia Virus Infected Vero Cells
2.4.2. Determination of Tissue Culture Infective Dose 50 (TCID50) for Murine Norovirus and Human Adenovirus
2.5. Negative Staining Transmission Electron Microscopy
3. Results
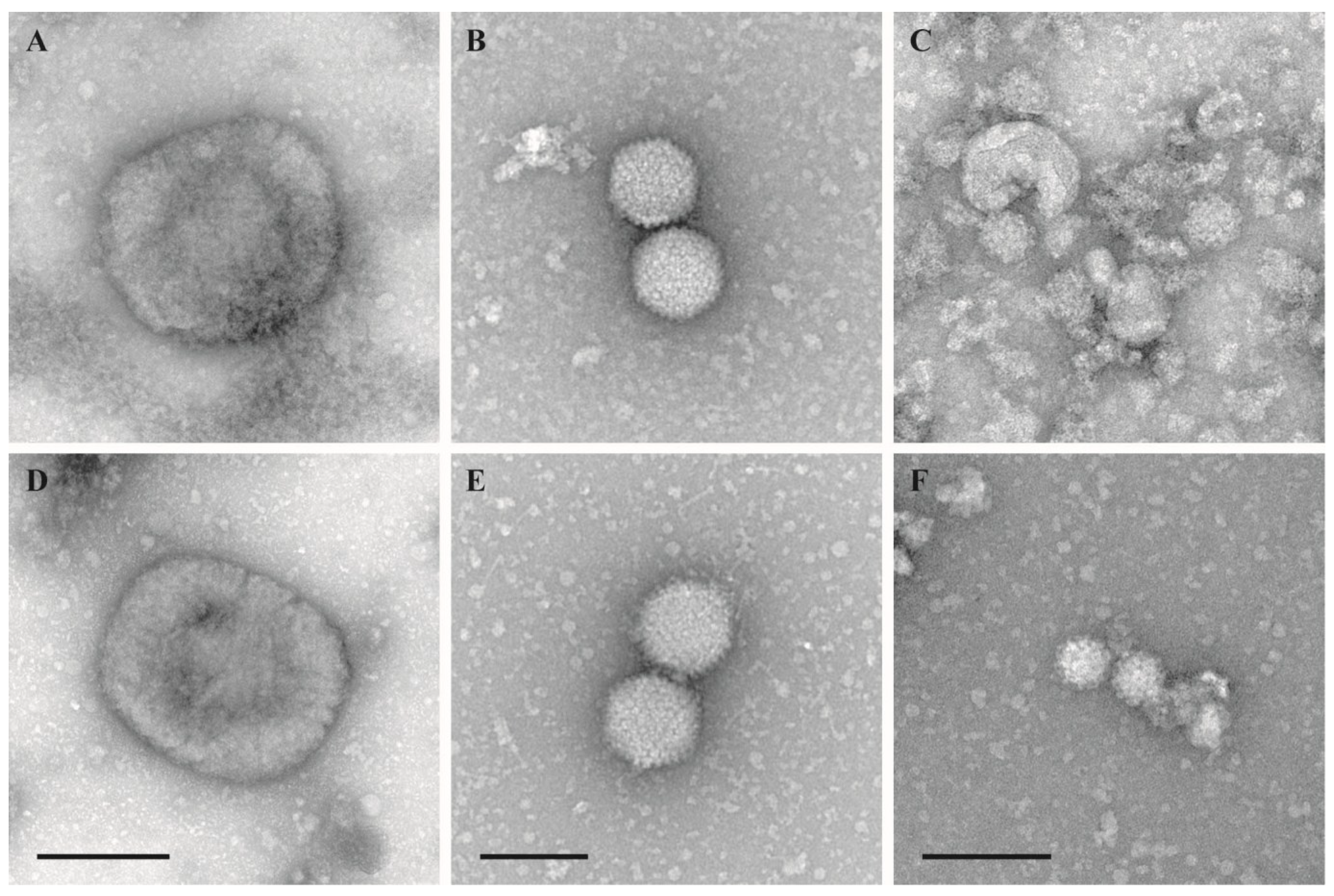
4. Discussion
- (1)
- The inactivation of viruses by FA has been evaluated by using three different test viruses, and results showed that the inactivation efficiency is highly dependent on the virus type, as expected (e.g., [36]). While our results allow already generalized conclusions, tests using other viruses, even more resistant viruses than poxviruses (e.g., small non-enveloped viruses, like parvo- and circoviruses [34,37]), must be performed to prove the inactivation efficiency of the so far successful protocols.
- (2)
- The inactivation tests have been conducted using non–purified cell culture supernatants. Although these samples contain biological macromolecules besides virus particles released by the cells in culture, concentration of such material is rather low in comparison to some diagnostic samples, e.g., serum or stool. Therefore, additional experiments, using defined loads of protein or other biological material (e.g., urine, cell or tissue homogenates), must be performed to determine the possible interference of the inactivation by high concentrations of other biological material.
- (3)
- Concentration of viruses may also be a critical factor for inactivation efficiency. To produce high concentrations and sufficient amount of active test viruses is difficult and laborious. In our tests, we used concentrations up to 109 PFU/mL of the highly stable poxviruses. Although the situation of diagnosing a sample with higher virus load seems rather unlikely, it should be investigated whether the proposed inactivation protocols are efficient at even higher concentrations or not.
- (4)
- Our results showed a slight variability in the inactivation efficiency, even under ideal experimental conditions. The poxvirus suspension used for the reproduction experiments (Table 4) was more sensitive for FA inactivation at low temperature than the poxvirus suspension used in the preliminary experiments (Table 1). Variability may be due to differences in the sensitivity of varying virus batches, which has been suggested before [24]. Another possibility could be a difference in the homogenization efficiency of the different virus batches, which could result in a variable number and size of virus aggregates. Aggregation is a well-known factor, which affects inactivation efficiency of many disinfectants [38] and could be the reason for the observed residual infectivity in few of the treated Vaccinia virus suspensions which became apparent only at higher dilutions in the plaque test. Higher dilution could have promoted disintegration of aggregates and release of infectious particles. Further experiments must consider these variables and should provide an estimate regarding their effects.
- (5)
- For an application of the suggested inactivation protocols in emergency diagnostics, the inactivation efficiency of the protocols must be tested also for bacteria, since a pre-screening of the samples for bacteria is not possible in any case. From the published data on FA inactivation, at least a significant reduction of vegetative bacteria can be expected [39,40]. Bacterial spores are much more resistant to FA inactivation than their vegetative forms (for a review see [41]), but FA is able to penetrate even the spore core, which houses the DNA [42]. Increased incubation temperature may help to improve inactivation efficiency. Addition of glutaraldehyde may also be considered, because it is not only efficient against spores [43] but also against viruses and vegetative bacteria [39]. Low concentration of GA will minimize GA interference with the detection of viruses by negative staining EM [3,20]. However, it cannot be excluded that addition of GA may interfere with the FA inactivation. Further experiments are necessary to clearly prove efficiency of the proposed protocols for bacteria.
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Biel, S.S.; Gelderblom, H.R. Diagnostic electron microscopy is still a timely and rewarding method. J. Clin. Virol. 1999, 13, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.S.; Madeley, D. Diagnostic virology—The need for electron microscopy: A discussion paper. J. Clin. Virol. 2001, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hazelton, P.R.; Gelderblom, H.R. Electron microscopy for rapid diagnosis of infectious agents in emergent situations. Emerg. Infect. Dis. 2003, 9, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Roingeard, P. Viral detection by electron microscopy: Past, present and future. Biol. Cell. 2008, 100, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Laue, M. Electron microscopy of viruses. Methods Cell. Biol. 2010, 96, 1–20. [Google Scholar] [PubMed]
- Biel, S.S.; Gelderblom, H.R. Electron microscopy of viruses. In Virus Cell Culture-A Practical Approach, 208th ed.; Cann, A., Ed.; Oxford University Press: Oxford, UK, 1999; pp. 111–147. [Google Scholar]
- Goldsmith, C.S.; Miller, S.E. Modern uses of electron microscopy for detection of viruses. Clin. Microbiol. Rev. 2009, 22, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.R. Structure and Classification of Viruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Madeley, C.R.; Field, A.M. Virus Morphology, 2nd ed.; Churchill Livingstone: London, UK, 1988. [Google Scholar]
- Doane, F.W.; Anderson, N. Electron Microscopy in Diagnostic Virology: A Practical Guide and Atlas; Cambridge University Press: Cambridge, UK, 1987. [Google Scholar]
- Madeley, C.R.; Biel, S.S. For debate: Is disinfection of specimens, which may contain unknown or bio-terrorist organisms, essential before electron microscopic examination? J. Infect. 2006, 53, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.R.; Bannert, N.; Pauli, G. Arguments pro disinfection in diagnostic electron microscopy: A response to Madeley and Biel. J. Infect. 2007, 54, 307–308, author reply 308–309. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.R.; Bannert, N.; Pauli, G. Reply to: Disinfection in diagnostic electron microscopy prior to preparation? J. Infect. 2007, 54, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.H.; Johnson, F.B.; Whiting, J.; Roller, P.P. Formaldehyde fixation. J. Histochem. Cytochem. 1985, 33, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Salk, J.E.; Gori, J.B. A review of theoretical, experimental, and practical considerations in the use of formaldehyde for the inactivation of poliovirus. Ann. NY Acad. Sci. 1960, 83, 609–637. [Google Scholar] [CrossRef] [PubMed]
- Barteling, S.J.; Woortmeyer, R. Formaldehyde inactivation of foot-and-mouth disease virus. Conditions for the preparation of safe vaccine. Arch. Virol. 1984, 80, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Gard, S. Inactivation of poliovirus by formaldehyde: Theoretical and practical aspects. Bull. World Health Organ. 1957, 17, 979–989. [Google Scholar] [PubMed]
- Haas, R.; Thomssen, R.; Dostal, V.; Ruschmann, E. Studies on the mechanism of formaldehyde inactivation of poliomyelitis viruses. Arch. Gesamte Virusforsch. 1959, 9, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Charney, J.; Fischer, W.P.; Sagin, J.F.; Tytell, A.A. Inactivation of concentrated purified poliovirus suspensions. Ann. NY Acad. 1960, 83, 649–653. [Google Scholar] [CrossRef]
- Rodgers, F.G.; Hufton, P.; Kurzawska, E.; Molloy, C.; Morgan, S. Morphological response of human rotavirus to ultra-violet radiation, heat and disinfectants. J. Med. Microbiol. 1985, 20, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.L.; Jaeger, R.F. Inactivation of yellow fever virus by glutaraldehyde. Appl. Microbiol. 1968, 16, 177–177. [Google Scholar] [PubMed]
- Saitanu, K.; Lund, E. Inactivation of enterovirus by glutaraldehyde. Appl. Microbiol. 1975, 29, 571–574. [Google Scholar] [PubMed]
- Howard, C.R.; Dixon, J.; Young, P.; van Eerd, P.; Schellekens, H. Chemical inactivation of hepatitis B virus: The effect of disinfectants on virus-associated DNA polymerase activity, morphology and infectivity. J. Virol. Methods. 1983, 7, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, C.W. Kinetics of the inactivation of viruses. Bacteriol. Rev. 1964, 28, 150–163. [Google Scholar] [PubMed]
- Kurth, A.; Wibbelt, G.; Gerber, H.P.; Petschaelis, A.; Pauli, G.; Nitsche, A. Rat-to-elephant-to-human transmission of cowpox virus. Emerg. Infect. Dis. 2008, 14, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Blümel, J.G.; Glebe, D.; Neumann-Haefelin, D.; Rabenau, H.F.; Rapp, I.; von Rheinbaben, F.; Ruf, B.; Sauerbrei, A.; Schwebke, I.; Steinmann, J.; et al. Guideline of “Deutsche Vereinigung zur Bekämpfung der Viruskrankheiten e.V.” (DVV; German Association for the Control of Virus Diseases) and Robert Koch-Institute (RKI; German Federal Health Authority) for testing the virucidal efficacy of chemical disinfectants in the human medical area. Hyg. Med. 2009, 34, 293–299. [Google Scholar]
- Kramski, M.; Matz-Rensing, K.; Stahl-Hennig, C.; Kaup, F.J.; Nitsche, A.; Pauli, G.; Ellerbrok, H. A novel highly reproducible and lethal nonhuman primate model for orthopox virus infection. PLOS ONE 2010, 5, e10412. [Google Scholar] [CrossRef] [PubMed]
- Mahy, B.W.J.; Kangro, H.O. Virology Methods Manual; Academic Press: London, UK, 1996. [Google Scholar]
- Rabenau, H.F.; Rapp, I.; Steinmann, J. Can vaccinia virus be replaced by MVA virus for testing virucidal activity of chemical disinfectants? BMC Infect. Dis. 2010, 10, 185–185. [Google Scholar] [CrossRef] [PubMed]
- Spearman, C. The method of “right and wrong cases” (“constant stimuli”) without Gauss’s formulae. Br. J. Psychol. 1908, 2, 227–242. [Google Scholar]
- Kaerber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Path. Pharmako. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Bozzola, J.J.; Russell, L.D. Electron Microscopy: Principles and Techniques for Biologists, 2nd ed.; Jones and Bartlett: Boston, London, 1999. [Google Scholar]
- Griffiths, G.; Burke, B.; Lucocq, J. Fine Structure Immunocytochemistry; Springer: Berlin, Germany, 1993. [Google Scholar]
- McDonnell, G.; Russell, A.D. Antiseptics and disinfectants: Activity, action, and resistance. Clin. Microbiol. Rev. 1999, 12, 147–179. [Google Scholar] [PubMed]
- Tan, J.A.; Schnagl, R.D. Inactivation of a rotavirus by disinfectants. Med. J. Aust. 1981, 1, 19–23. [Google Scholar] [PubMed]
- Kap, M.; Arron, G.I.; Loibner, M.; Hausleitner, A.; Siaulyte, G.; Zatloukal, K.; Murk, J.L.; Riegman, P. Inactivation of influenza A virus, adenovirus, and cytomegalovirus with PAXgene tissue fixative and formalin. Biopreserv. Biobank. 2013, 11, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Rabenau, H.F.; Steinmann, J.; Rapp, I.; Schwebke, I.; Eggers, M. Evaluation of a virucidal quantitative carrier test for surface disinfectants. PLOS ONE 2014, 9, e86128. [Google Scholar] [CrossRef] [PubMed]
- Mattle, M.J.; Crouzy, B.; Brennecke, M.; Wigginton, K.R.; Perona, P.; Kohn, T. Impact of virus aggregation on inactivation by peracetic acid and implications for other disinfectants. Environ. Sci. Technol. 2011, 45, 7710–7717. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, S.D.; Gardner, J.F.; Webb, R.L. Biocidal activities of glutaraldehyde and related compounds. J. Appl. Bacteriol. 1967, 30, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Spicher, G.; Peters, J. Microbial resistance to formaldehyde. III> dependence of the microbial effect on Staphylococcus aureus, Enterococcus faecium and spores of Bacillus stearothermophilus on temperature. Zentralbl. Hyg. Umweltmed. 1995, 196, 545–561. [Google Scholar] [PubMed]
- Setlow, P. Spores of Bacillus subtilis: Their resistance to and killing by radiation, heat and chemicals. J. Appl. Microbiol. 2006, 101, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Cortezzo, D.E.; Setlow, B.; Setlow, P. Analysis of the action of compounds that inhibit the germination of spores of Bacillus species. J. Appl. Microbiol. 2004, 96, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Spotts Whitney, E.A.; Beatty, M.E.; Taylor, T.H., Jr.; Weyant, R.; Sobel, J.; Arduino, M.J.; Ashford, D.A. Inactivation of Bacillus anthracis spores. Emerg. Infect. Dis. 2003, 9, 623–627. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möller, L.; Schünadel, L.; Nitsche, A.; Schwebke, I.; Hanisch, M.; Laue, M. Evaluation of Virus Inactivation by Formaldehyde to Enhance Biosafety of Diagnostic Electron Microscopy. Viruses 2015, 7, 666-679. https://doi.org/10.3390/v7020666
Möller L, Schünadel L, Nitsche A, Schwebke I, Hanisch M, Laue M. Evaluation of Virus Inactivation by Formaldehyde to Enhance Biosafety of Diagnostic Electron Microscopy. Viruses. 2015; 7(2):666-679. https://doi.org/10.3390/v7020666
Chicago/Turabian StyleMöller, Lars, Livia Schünadel, Andreas Nitsche, Ingeborg Schwebke, Manuela Hanisch, and Michael Laue. 2015. "Evaluation of Virus Inactivation by Formaldehyde to Enhance Biosafety of Diagnostic Electron Microscopy" Viruses 7, no. 2: 666-679. https://doi.org/10.3390/v7020666
APA StyleMöller, L., Schünadel, L., Nitsche, A., Schwebke, I., Hanisch, M., & Laue, M. (2015). Evaluation of Virus Inactivation by Formaldehyde to Enhance Biosafety of Diagnostic Electron Microscopy. Viruses, 7(2), 666-679. https://doi.org/10.3390/v7020666