
Spotlight on HIV-1 Nef: SERINC3 and SERINC5 Identified as Restriction Factors Antagonized by the Pathogenesis Factor

{kind=link}
Abstract
:1. Introduction
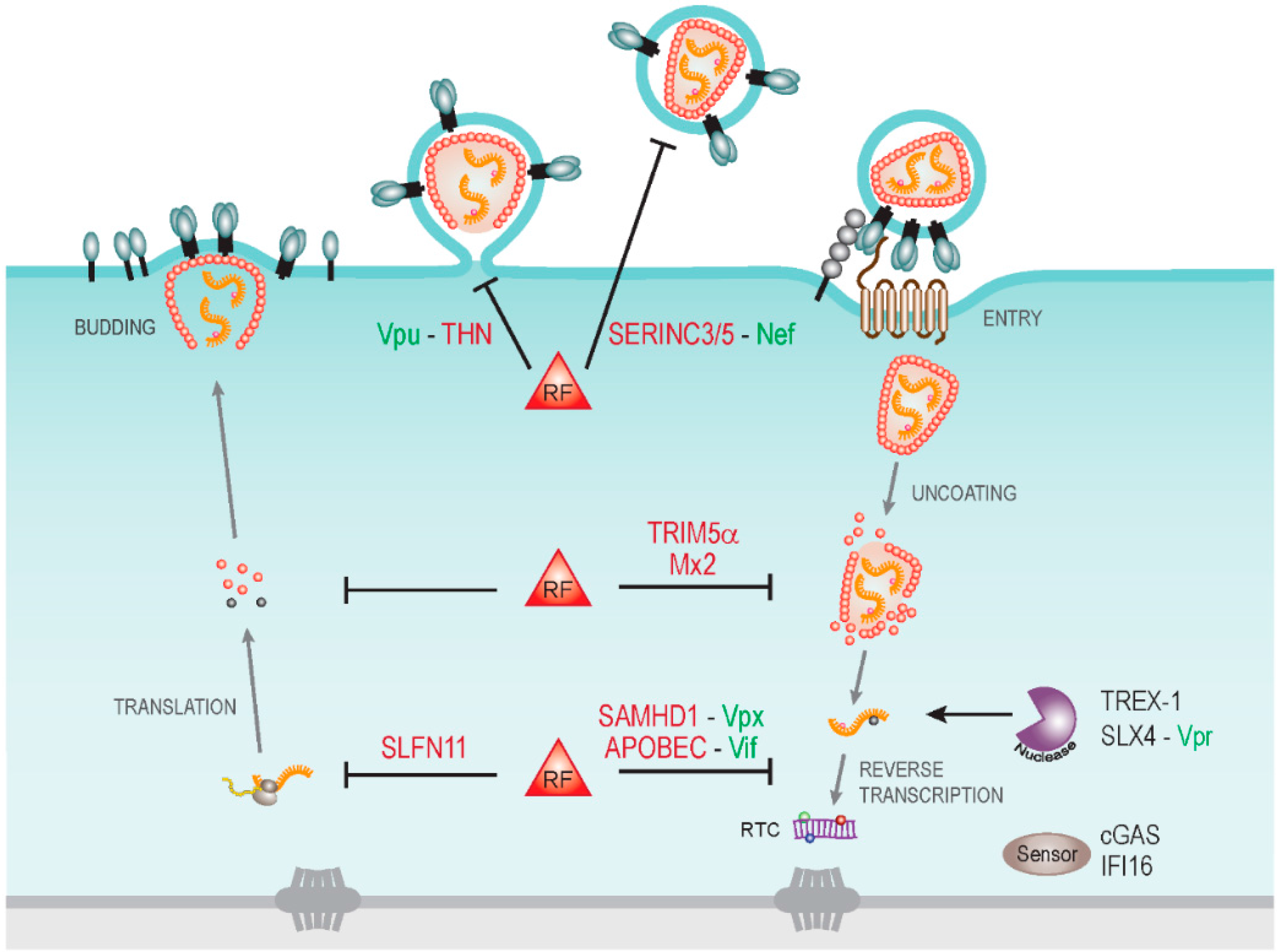
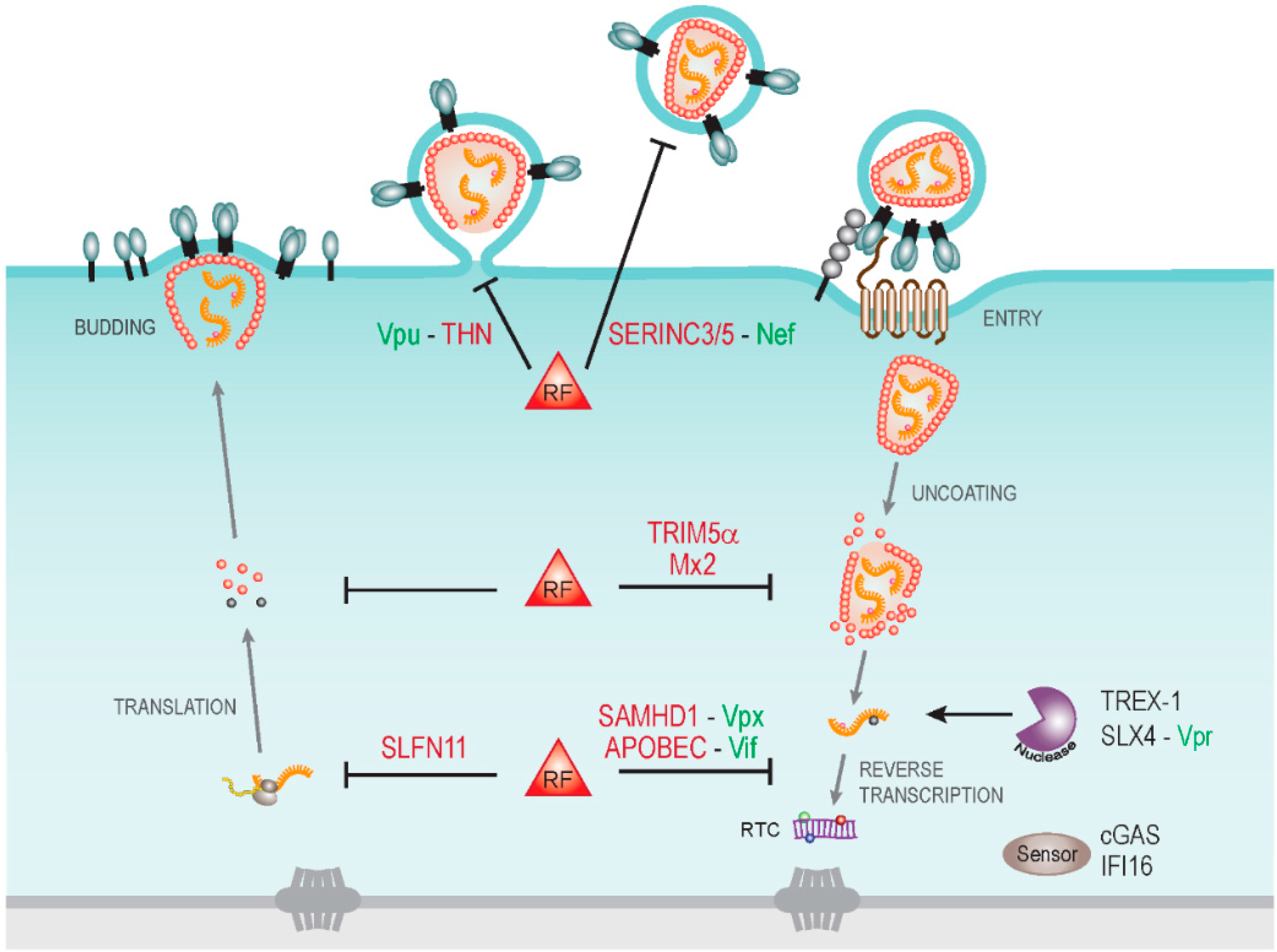
2. HIV-1 Nef: A Multifunctional Adaptor Protein
3. Enhancement of Virion Infectivity by Nef
4. Identification of serine incorporator 3 and 5 (SERINC 3/5) as Nef-Sensitive Restriction Factors
5. Open Questions and Future Directions
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [PubMed]
- Schwartz, O.; Marechal, V.; le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Malim, M.H. APOBEC-mediated editing of viral RNA. Science 2004, 305, 645. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.R.; Lubow, J.; Lukic, Z.; Mashiba, M.; Collins, K.L. Vpr Promotes Macrophage-Dependent HIV-1 Infection of CD4+ T Lymphocytes. PLoS Pathog. 2015, 11, e1005054. [Google Scholar] [CrossRef] [PubMed]
- Mashiba, M.; Collins, D.R.; Terry, V.H.; Collins, K.L. Vpr overcomes macrophage-specific restriction of HIV-1 Env expression and virion production. Cell Host Microbe 2014, 16, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Deacon, N.J.; Tsykin, A.; Solomon, A.; Smith, K.; Ludford-Menting, M.; Hooker, D.J.; McPhee, D.A.; Greenway, A.L.; Ellett, A.; Chatfield, C.; et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 1995, 270, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Kestler, H.W., 3rd; Ringler, D.J.; Mori, K.; Panicali, D.L.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 1991, 65, 651–662. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Greenough, T.C.; Brettler, D.B.; Sullivan, J.L.; Desrosiers, R.C. Brief report: Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 1995, 332, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Abraham, L.; Fackler, O.T. HIV-1 Nef: A multifaceted modulator of T cell receptor signaling. Cell Commun. Signal. CCS 2012, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Bregnard, C.; Benichou, S.; Basmaciogullari, S. Human immunodeficiency virus (HIV) type-1, HIV-2 and simian immunodeficiency virus Nef proteins. Mol. Aspects Med. 2010, 31, 418–433. [Google Scholar] [CrossRef] [PubMed]
- Roeth, J.F.; Collins, K.L. Human immunodeficiency virus type 1 Nef: Adapting to intracellular trafficking pathways. Microbiol. Mol. Biol. Rev. 2006, 70, 548–563. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.V.; Miller, A.D. Downregulation of cell surface CD4 by nef. Res. Virol. 1992, 143, 52–55. [Google Scholar] [CrossRef]
- Haller, C.; Rauch, S.; Michel, N.; Hannemann, S.; Lehmann, M.J.; Keppler, O.T.; Fackler, O.T. The HIV-1 pathogenicity factor Nef interferes with maturation of stimulatory T-lymphocyte contacts by modulation of N-Wasp activity. J. Biol. Chem. 2006, 281, 19618–19630. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Rudolph, J.M.; Abraham, L.; Habermann, A.; Haller, C.; Krijnse-Locker, J.; Fackler, O.T. HIV-1 Nef compensates for disorganization of the immunological synapse by inducing trans-Golgi network-associated Lck signaling. Blood 2012, 119, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Thoulouze, M.I.; Sol-Foulon, N.; Blanchet, F.; Dautry-Varsat, A.; Schwartz, O.; Alcover, A. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 2006, 24, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Imle, A.; Coelho, F.M.; Hons, M.; Gorina, R.; Lyck, R.; Stein, J.V.; Fackler, O.T. HIV-1 Nef interferes with T-lymphocyte circulation through confined environments in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18541–18546. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef Interferes with Host Cell Motility by Deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Muratori, C.; Cavallin, L.E.; Kratzel, K.; Tinari, A.; de Milito, A.; Fais, S.; D’Aloja, P.; Federico, M.; Vullo, V.; Fomina, A.; et al. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4 T Cells. Traffic 2009, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Munch, J.; Kutsch, O.; Li, H.; Santiago, M.L.; Bibollet-Ruche, F.; Muller-Trutwin, M.C.; Novembre, F.J.; Peeters, M.; Courgnaud, V.; et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 2006, 125, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Chowers, M.Y.; Spina, C.A.; Kwoh, T.J.; Fitch, N.J.; Richman, D.D.; Guatelli, J.C. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 1994, 68, 2906–2914. [Google Scholar] [PubMed]
- Basmaciogullari, S.; Pizzato, M. The activity of Nef on HIV-1 infectivity. Front. Microbiol. 2014, 5, 232. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Trono, D. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J. Virol. 1995, 69, 5048–5056. [Google Scholar] [PubMed]
- Schwartz, O.; Dautry-Varsat, A.; Goud, B.; Marechal, V.; Subtil, A.; Heard, J.M.; Danos, O. Human immunodeficiency virus type 1 Nef induces accumulation of CD4 in early endosomes. J. Virol. 1995, 69, 528–533. [Google Scholar] [PubMed]
- Laguette, N.; Benichou, S.; Basmaciogullari, S. Human immunodeficiency virus type 1 Nef incorporation into virions does not increase infectivity. J. Virol. 2009, 83, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Aiken, C. Nef enhances HIV-1 infectivity via association with the virus assembly complex. Virology 2008, 373, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Helander, A.; Popova, E.; Calistri, A.; Zamborlini, A.; Palu, G.; Gottlinger, H.G. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc. Natl. Acad. Sci. USA 2007, 104, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Popova, E.; Gottlinger, H.G. Nef can enhance the infectivity of receptor-pseudotyped human immunodeficiency virus type 1 particles. J. Virol. 2008, 82, 10811–10819. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Popov, S.; Gottlinger, H.G. The Nef-like effect of murine leukemia virus glycosylated gag on HIV-1 infectivity is mediated by its cytoplasmic domain and depends on the AP-2 adaptor complex. J. Virol. 2014, 88, 3443–3454. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pizzato, M. MLV glycosylated-Gag is an infectivity factor that rescues Nef-deficient HIV-1. Proc. Natl. Acad. Sci. USA 2010, 107, 9364–9369. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; de Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, S.; Grivel, J.C.; Suryanarayana, K.; Meylan, P.; Lifson, J.D.; Desrosiers, R.; Margolis, L. Nef enhances human immunodeficiency virus replication and responsiveness to interleukin-2 in human lymphoid tissue ex vivo. J. Virol. 1999, 73, 3968–3974. [Google Scholar] [PubMed]
- Homann, S.; Tibroni, N.; Baumann, I.; Sertel, S.; Keppler, O.T.; Fackler, O.T. Determinants in HIV-1 Nef for enhancement of virus replication and depletion of CD4+ T lymphocytes in human lymphoid tissue ex vivo. Retrovirology 2009, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.D.; Warmerdam, M.T.; Gaston, I.; Greene, W.C.; Feinberg, M.B. The human immunodeficiency virus-1 nef gene product: A positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med. 1994, 179, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Spina, C.A.; Kwoh, T.J.; Chowers, M.Y.; Guatelli, J.C.; Richman, D.D. The importance of nef in the induction of human immunodeficiency virus type 1 replication from primary quiescent CD4 lymphocytes. J. Exp. Med. 1994, 179, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Rucker, E.; Grivel, J.C.; Munch, J.; Kirchhoff, F.; Margolis, L. Vpr and Vpu are important for efficient human immunodeficiency virus type 1 replication and CD4+ T-cell depletion in human lymphoid tissue ex vivo. J. Virol. 2004, 78, 12689–12693. [Google Scholar] [CrossRef] [PubMed]
- Sourisseau, M.; Sol-Foulon, N.; Porrot, F.; Blanchet, F.; Schwartz, O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 2007, 81, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Tibroni, N.; Rudolph, J.M.; Grosse, R.; Fackler, O.T. Nef does not inhibit F-actin remodelling and HIV-1 cell-cell transmission at the T lymphocyte virological synapse. Eur. J. Cell Biol. 2010, 90, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Malbec, M.; Sourisseau, M.; Guivel-Benhassine, F.; Porrot, F.; Blanchet, F.; Schwartz, O.; Casartelli, N. HIV-1 Nef promotes the localization of Gag to the cell membrane and facilitates viral cell-to-cell transfer. Retrovirology 2013, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Brugger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Krausslich, H.G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef] [PubMed]
- Brugger, B.; Krautkramer, E.; Tibroni, N.; Munte, C.E.; Rauch, S.; Leibrecht, I.; Glass, B.; Breuer, S.; Geyer, M.; Krausslich, H.G.; et al. Human immunodeficiency virus type 1 Nef protein modulates the lipid composition of virions and host cell membrane microdomains. Retrovirology 2007, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Gottlinger, H. HIV-1 Nef responsiveness is determined by Env variable regions involved in trimer association and correlates with neutralization sensitivity. Cell Rep. 2013, 5, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Douglas, J.L.; Livingston, R.L.; Garcia, J.V. Infectivity enhancement by HIV-1 Nef is dependent on the pathway of virus entry: Implications for HIV-based gene transfer systems. Virology 1998, 241, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J. Virol. 1997, 71, 5871–5877. [Google Scholar] [PubMed]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fackler, O.T. Spotlight on HIV-1 Nef: SERINC3 and SERINC5 Identified as Restriction Factors Antagonized by the Pathogenesis Factor. Viruses 2015, 7, 6730-6738. https://doi.org/10.3390/v7122970
Fackler OT. Spotlight on HIV-1 Nef: SERINC3 and SERINC5 Identified as Restriction Factors Antagonized by the Pathogenesis Factor. Viruses. 2015; 7(12):6730-6738. https://doi.org/10.3390/v7122970
Chicago/Turabian StyleFackler, Oliver T. 2015. "Spotlight on HIV-1 Nef: SERINC3 and SERINC5 Identified as Restriction Factors Antagonized by the Pathogenesis Factor" Viruses 7, no. 12: 6730-6738. https://doi.org/10.3390/v7122970
APA StyleFackler, O. T. (2015). Spotlight on HIV-1 Nef: SERINC3 and SERINC5 Identified as Restriction Factors Antagonized by the Pathogenesis Factor. Viruses, 7(12), 6730-6738. https://doi.org/10.3390/v7122970